Temperature-Controlled Assembly/Reassembly of Two Dicarboxylate-Based Three-Dimensional Co(II) Coordination Polymers with an Antiferromagnetic Metallic Layer and a Ferromagnetic Metallic Chain


Abstract
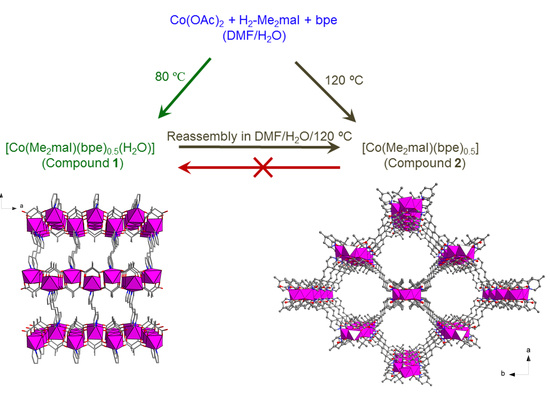
:
1. Introduction
2. Experimental
2.1. Materials and Methods
2.2. The Synthesis of [Co(Me2mal)(bpe)0.5(H2O)]n (1)
2.3. The Synthesis of [Co(Me2mal)(bpe)0.5]n (2)
2.4. X-ray Crystallography
2.5. Physical Measurements
3. Results and Discussion
3.1. Synthesis and Characterization of Compounds 1 and 2
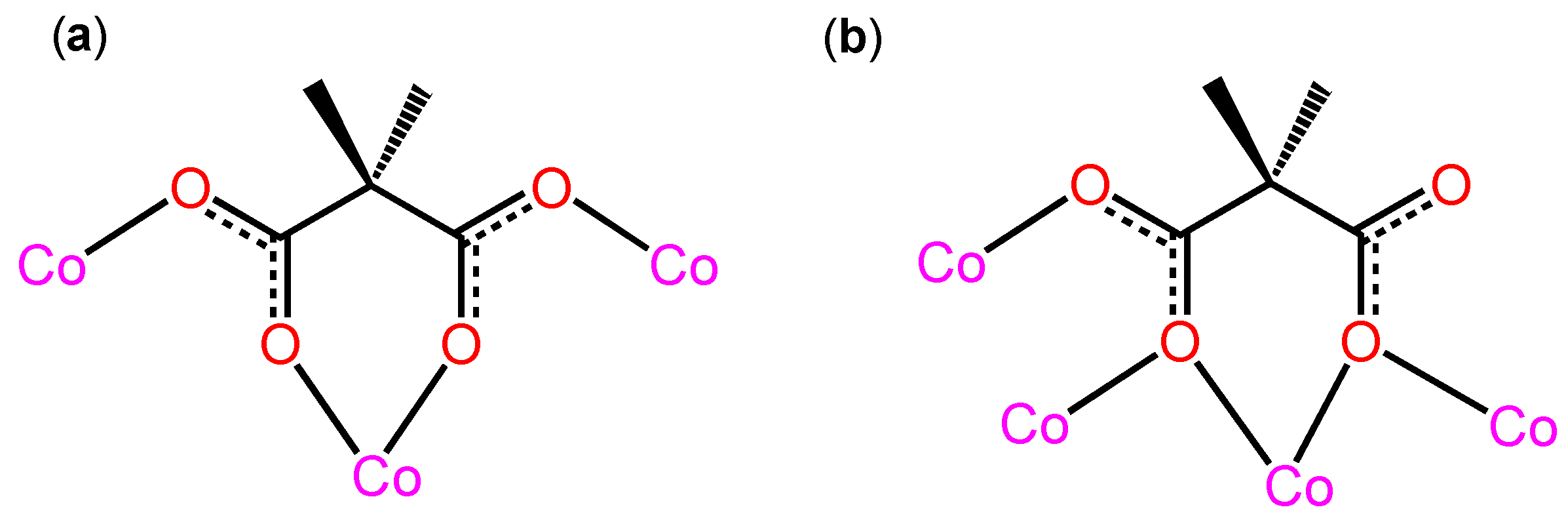
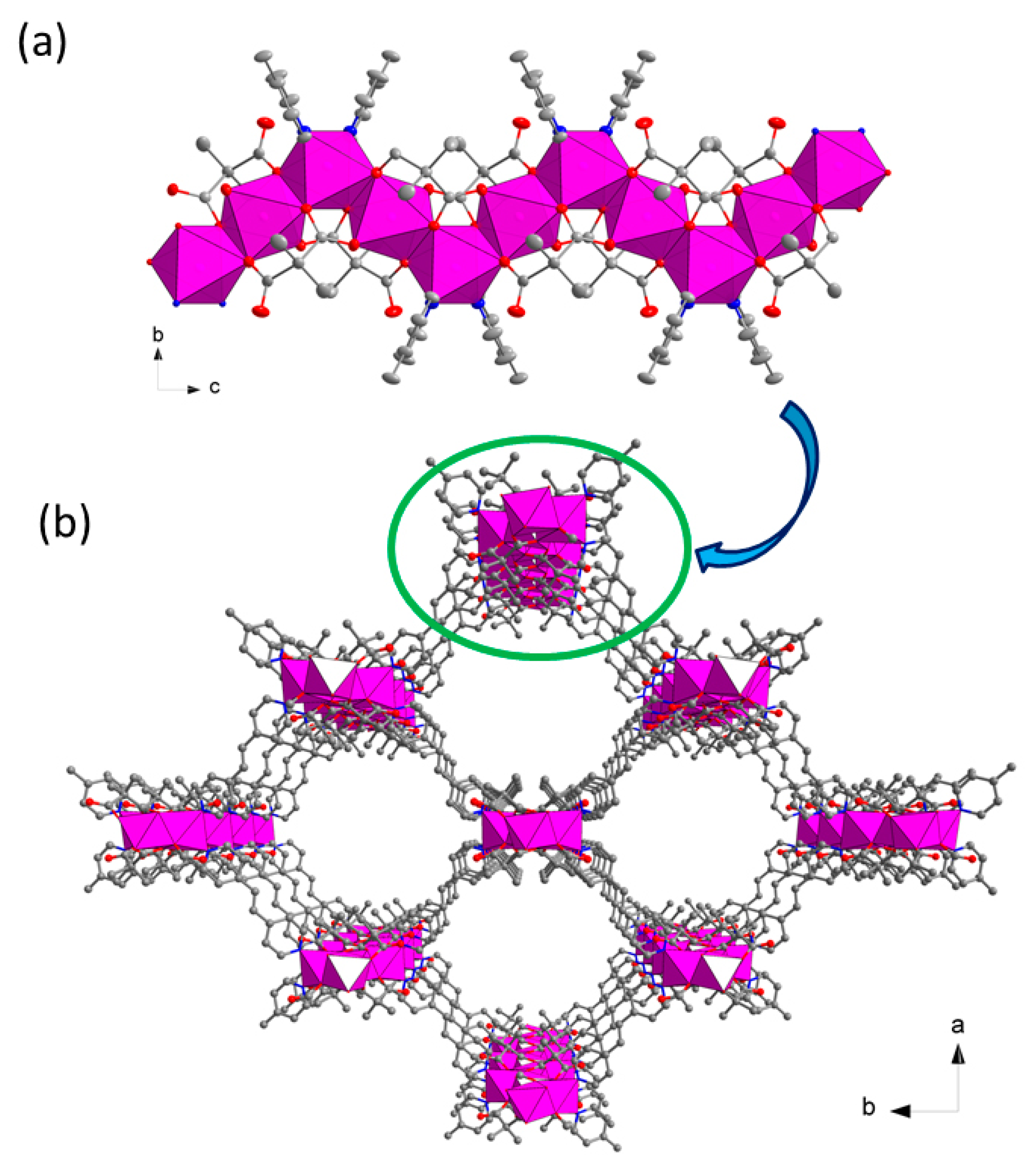
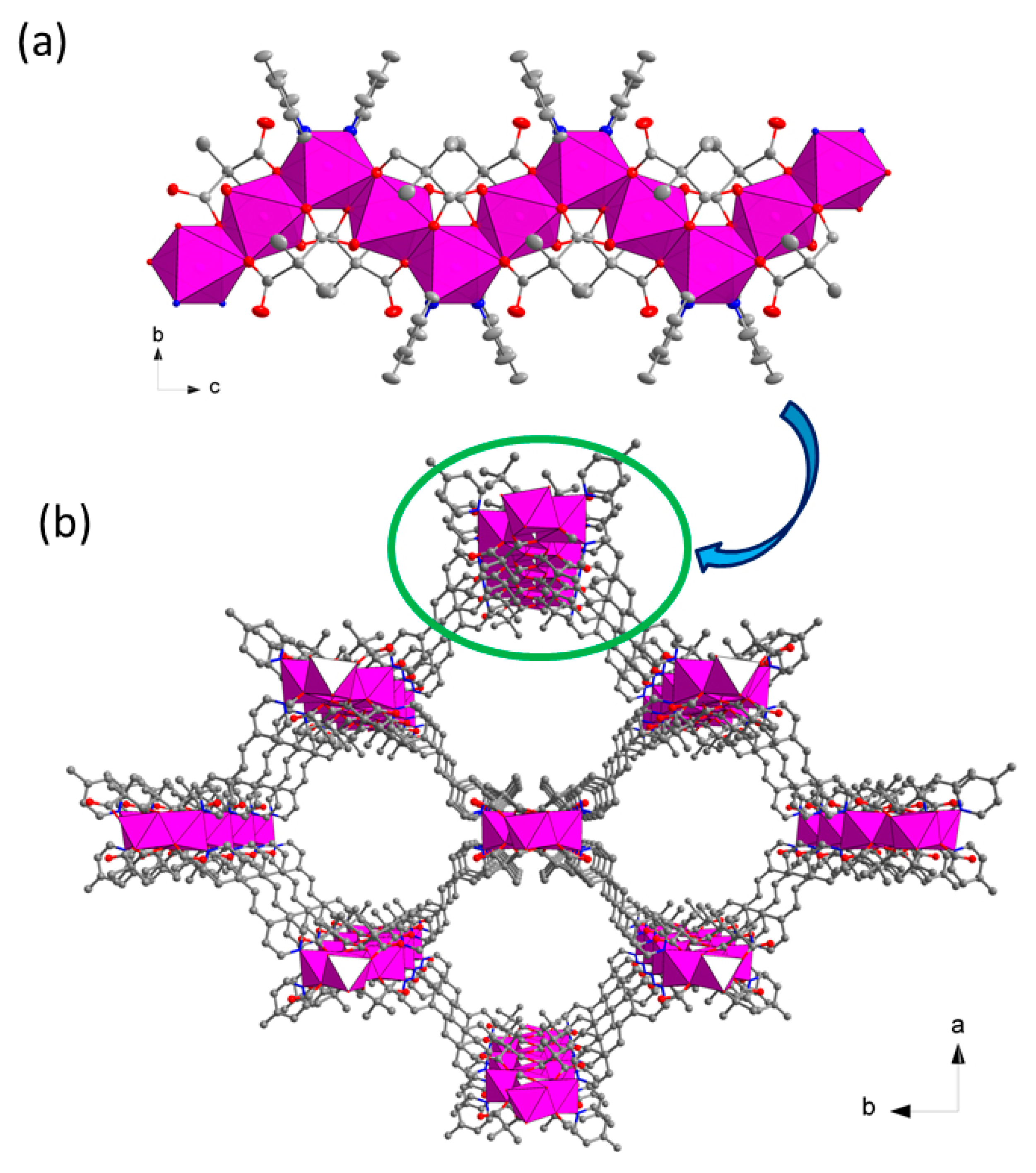
3.2. Description of the Structure
Crystal Structures of Compound 1
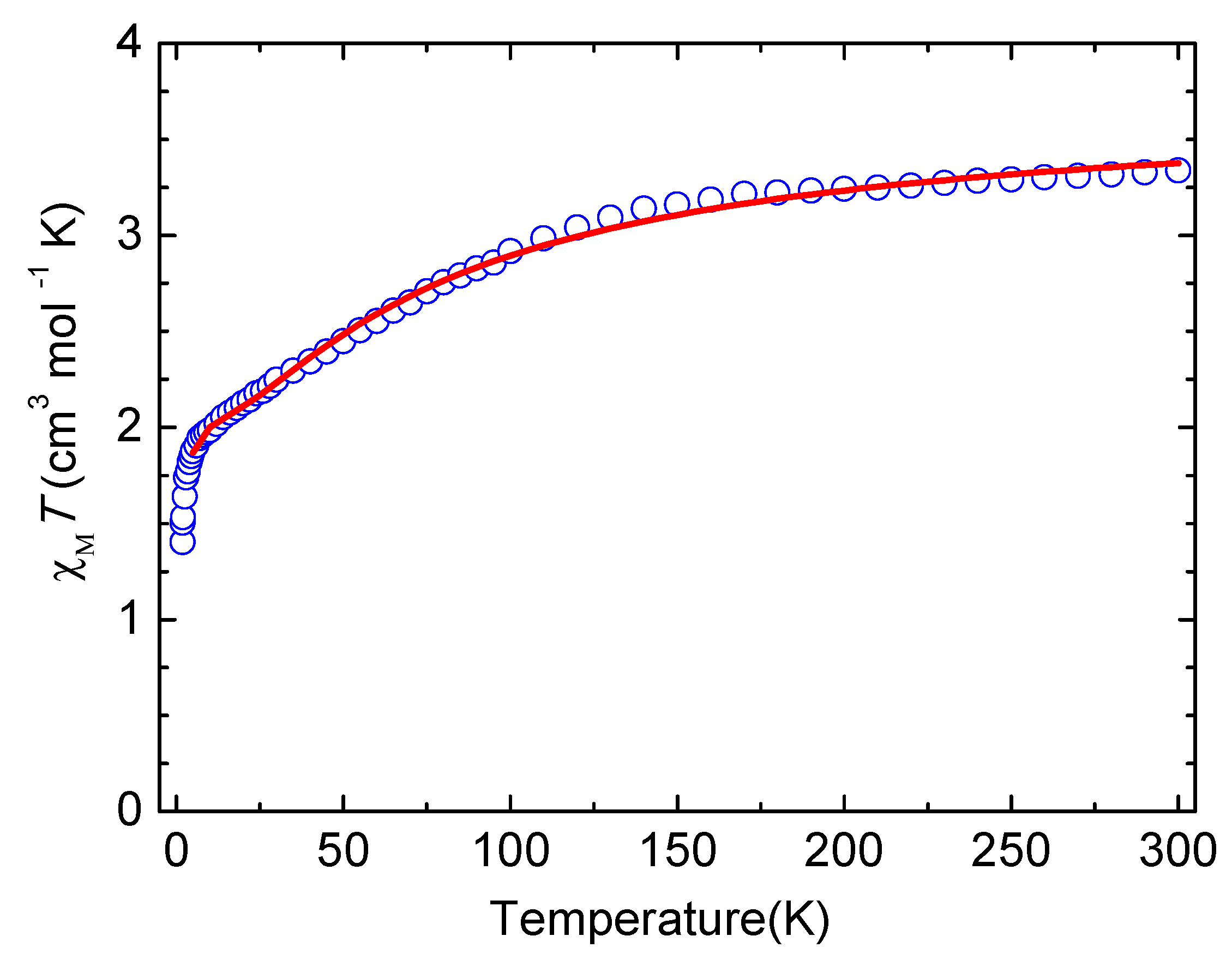
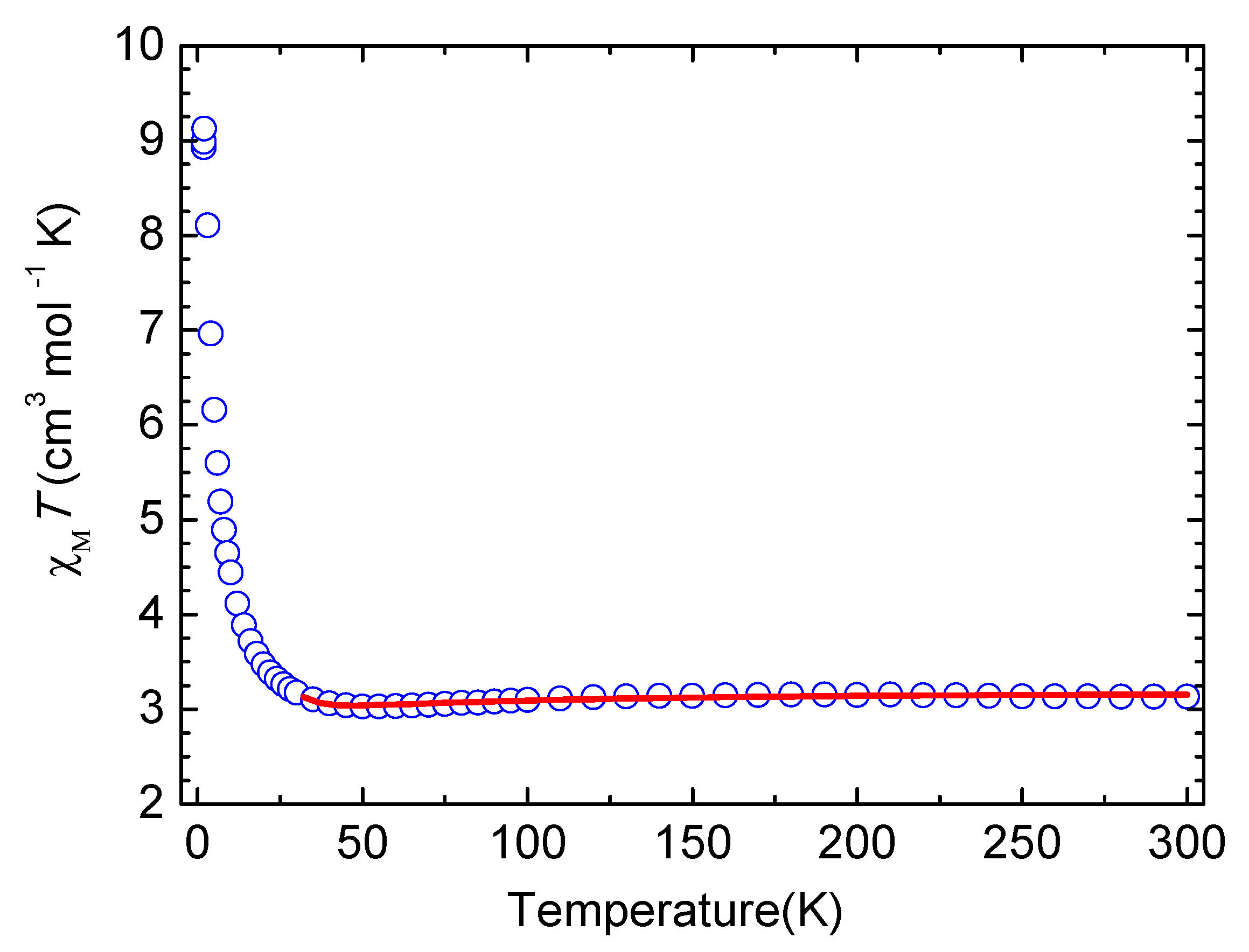
3.3. Magnetic Properties
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Eddaoudi, M.; Kim, J.; Rosi, N.; Vodak, D.; Wachter, J.; O’Keeffe, M.; Yaghi, O.M. Systematic design of pore size and functionality in isoreticular MOFs and their application in methane storage. Science 2002, 295, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, S.; Kitaura, R.; Noro, S. Functional porous coordination polymers. Angew. Chem. Int. Ed. 2004, 43, 2334–2375. [Google Scholar] [CrossRef]
- Ferey, G.; Mellot-Draznieks, C.; Serre, C.; Millange, F. Crystallized frameworks with giant pores: Are there limits to the possible? Acc. Chem. Res. 2005, 38, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.; Wu, X.; Zhang, J.; Fu, R.; Hu, S.; Zhang, X. 3D Canted Antiferromagnetic Porous Metal−Organic Framework with Anatase Topology through Assembly of an Analogue of Polyoxometalate. J. Am. Chem. Soc. 2005, 127, 16352–16353. [Google Scholar] [CrossRef]
- Ockwig, N.W.; Delgado-Friedrichs, O.; O’Keeffe, M.; Yaghi, O.M. Reticular chemistry: Occurrence and taxonomy of nets and grammar for the design of frameworks. Acc. Chem. Res. 2005, 38, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Ferey, G. Hybrid porous solids: Past, present, future. Chem. Soc. Rev. 2008, 37, 191–214. [Google Scholar] [CrossRef]
- Moulton, B.; Zaworotko, M.J. From Molecules to Crystal Engineering: Supramolecular Isomerism and Polymorphism in Network Solids. Chem. Rev. 2001, 101, 1629–1658. [Google Scholar] [CrossRef] [PubMed]
- Welte, L.; Calzolari, A.; Di Felice, R.; Zamora, F.; Gomez-Herrero, J. Highly conductive self-assembled nanoribbons of coordination polymers. Nat. Nanotechnol. 2010, 5, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Barth, J.V.; Costantini, G.; Kern, K. Engineering atomic and molecular nanostructures at surfaces. Nature 2005, 437, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Guijarro, A.; Castillo, O.; Welte, L.; Calzolari, A.; Miguel, P.J.S.; Gómez-García, C.J.; Olea, D.; di Felice, R.; Gómez-Herrero, J.; Zamora, F. Conductive Nanostructures of MMX Chains. Adv. Funct. Mater. 2010, 20, 1451–1457. [Google Scholar] [CrossRef]
- Coronado, E.; Minguez Espallargas, G. Dynamic magnetic MOFs. Chem. Soc. Rev. 2013, 42, 1525–1539. [Google Scholar] [CrossRef] [PubMed]
- Chaemchuen, S.; Kabir, N.A.; Zhou, K.; Verpoort, F. Metal–organic frameworks for upgrading biogas via CO2 adsorption to biogas green energy. Chem. Soc. Rev. 2013, 42, 9304. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.; Solomon, E.I. Electronic structures of exchange coupled trigonal trimeric Cu(II) complexes: Spin frustration, antisymmetric exchange, pseudo-A terms, and their relation to O2 activation in the multicopper oxidases. Coordin. Chem. Rev. 2007, 251, 379–400. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, B.; Chen, L. First-Principles Study of Microporous Magnets M-MOF-74 (M = Ni, Co, Fe, Mn): The Role of Metal Centers. Inorg. Chem. 2013, 52, 9356–9362. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, A.; Miyasaka, H.; Yamashita, M.; Clérac, R. Direct evidence of exchange interaction dependence of magnetization relaxation in a family of ferromagnetic-type single-chain magnets. J. Mater. Chem. 2007, 17, 2002–2012. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, A.; Li, Z.; Garcia, H. Catalysis and photocatalysis by metal organic frameworks. Chem. Soc. Rev. 2018, 47, 8134–8172. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, A.; Asiri, A.M.; Garcia, H. Formation of C–C and C–Heteroatom Bonds by C–H Activation by Metal Organic Frameworks as Catalysts or Supports. ACS Catal. 2018, 9, 1081–1102. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, A.; Santiago-Portillo, A.; Asiri, A.M.; Garcia, H. Engineering UiO-66 Metal Organic Framework for Heterogeneous Catalysis. ChemCatChem 2019, 11, 899–923. [Google Scholar] [CrossRef]
- Kurmoo, M. Magnetic metal-organic frameworks. Chem. Soc. Rev. 2009, 38, 1353–1379. [Google Scholar] [CrossRef]
- Zeng, Y.F.; Hu, X.; Liu, F.C.; Bu, X.H. Azido-mediated systems showing different magnetic behaviors. Chem. Soc. Rev. 2009, 38, 469–480. [Google Scholar] [CrossRef]
- Gatteschi, D.; Kahn, O.; Miller, J.S.; Palacio, F. Magnetic Molecular Materials; Kluwer Academic: Dordrecht, The Netherlands, 1991. [Google Scholar]
- Willet, R.D.; Gatteschi, D.; Kahn, O. Magneto-Structural Correlations in Exchange Coupled Systems; Reidel Publishing: Dordrecht, The Netherlands, 1985. [Google Scholar]
- Weng, D.F.; Wang, Z.M.; Gao, S. Framework-structured weak ferromagnets. Chem. Soc. Rev. 2011, 40, 3157–3181. [Google Scholar] [CrossRef]
- Wang, X.P.; Chen, W.M.; Qi, H.; Li, X.Y.; Rajnak, C.; Feng, Z.Y.; Kurmoo, M.; Boca, R.; Jia, C.J.; Tung, C.H.; et al. Solvent-Controlled Phase Transition of a Co(II)-Organic Framework: From Achiral to Chiral and Two to Three Dimensions. Chemistry 2017, 23, 7990–7996. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Chen, Z.; Cao, F.; Wang, S.; Huang, X.; Li, Y.; Lu, J.; Li, D.; Dou, J. Two 2-D multifunctional cobalt(ii) compounds: Field-induced single-ion magnetism and catalytic oxidation of benzylic C-H bonds. Dalton Trans. 2017, 46, 2137–2145. [Google Scholar] [CrossRef]
- Zheng, T.; Ren, M.; Bao, S.-S.; Zheng, L.-M. M2(pbtcH)(phen)2(H2O)2 [M(II)=Co, Ni]: Mixed-ligated metal phosphonates based on 5-phosphonatophenyl-1,2,4-tricarboxylic acid showing double chain structures. Chin. Chem. Lett. 2014, 25, 835–838. [Google Scholar] [CrossRef]
- Wang, S.; Cao, T.; Yan, H.; Li, Y.; Lu, J.; Ma, R.; Li, D.; Dou, J.; Bai, J. Functionalization of Microporous Lanthanide-Based Metal-Organic Frameworks by Dicarboxylate Ligands with Methyl-Substituted Thieno[2,3-b]thiophene Groups: Sensing Activities and Magnetic Properties. Inorg. Chem. 2016, 55, 5139–5151. [Google Scholar] [CrossRef]
- Liu, Y.N.; Su, H.F.; Li, Y.W.; Liu, Q.Y.; Jaglicic, Z.; Wang, W.G.; Tung, C.H.; Sun, D. Space Craft-like Octanuclear Co(II)-Silsesquioxane Nanocages: Synthesis, Structure, Magnetic Properties, Solution Behavior, and Catalytic Activity for Hydroboration of Ketones. Inorg Chem 2019, 58, 4574–4582. [Google Scholar] [CrossRef]
- Chen, Z.; Yin, L.; Mi, X.; Wang, S.; Cao, F.; Wang, Z.; Li, Y.; Lu, J.; Dou, J. Field-induced slow magnetic relaxation of two 1-D compounds containing six-coordinated cobalt(ii) ions: Influence of the coordination geometry. Inorg. Chem. Front. 2018, 5, 2314–2320. [Google Scholar] [CrossRef]
- De Munno, G.; Viterbo, D.; Caneschi, A.; Lloret, F.; Julve, M. A Giant Antiferromagnetic Interaction through the Bihydroxide Bridge (H3O2-). Inorg. Chem. 1994, 33, 1585–1586. [Google Scholar] [CrossRef]
- Kar, P.; Drew, M.G.; Gomez-Garcia, C.J.; Ghosh, A. Coordination polymers containing manganese(II)-azido layers connected by dipyridyl-tetrazine and 4,4’-azobis(pyridine) linkers. Inorg. Chem. 2013, 52, 1640–1649. [Google Scholar] [CrossRef]
- Cheng, X.N.; Xue, W.; Huang, J.H.; Chen, X.M. Spin canting and/or metamagnetic behaviours of four isostructural grid-type coordination networks. Dalton. Trans. 2009, 5701–5707. [Google Scholar] [CrossRef]
- De Munno, G.; Poerio, T.; Julve, M.; Lloret, F.; Faus, J.; Caneschi, A. Syntheses, crystal structures and magnetic properties of one-, two- and three-dimensional 2,2′-bipyrimidine-containing copper(II) complexes. J. Chem. Soc. Dalton Trans. 1998, 1679–1686. [Google Scholar] [CrossRef]
- Okawa, H.; Shigematsu, A.; Sadakiyo, M.; Miyagawa, T.; Yoneda, K.; Ohba, M.; Kitagawa, H. Oxalate-bridged bimetallic complexes {NH(prol)3}[MCr(ox)3] (M = Mn(II), Fe(II), Co(II); NH(prol)3(+) = tri(3-hydroxypropyl)ammonium) exhibiting coexistent ferromagnetism and proton conduction. J. Am. Chem. Soc. 2009, 131, 13516–13522. [Google Scholar] [CrossRef]
- Toma, L.M.; Ruiz-Perez, C.; Pasan, J.; Wernsdorfer, W.; Lloret, F.; Julve, M. Molecular engineering to control the magnetic interaction between single-chain magnets assembled in a two-dimensional network. J. Am. Chem. Soc. 2012, 134, 15265–15268. [Google Scholar] [CrossRef]
- Miyasaka, H. Control of charge transfer in donor/acceptor metal-organic frameworks. Acc. Chem. Res. 2013, 46, 248–257. [Google Scholar] [CrossRef]
- Yang, T.; Cui, H.; Zhang, C.; Zhang, L.; Su, C.Y. Porous metal-organic framework catalyzing the three-component coupling of sulfonyl azide, alkyne, and amine. Inorg. Chem. 2013, 52, 9053–9059. [Google Scholar] [CrossRef]
- Lin, R.B.; Chen, D.; Lin, Y.Y.; Zhang, J.P.; Chen, X.M. A zeolite-like zinc triazolate framework with high gas adsorption and separation performance. Inorg. Chem. 2012, 51, 9950–9955. [Google Scholar] [CrossRef]
- Wriedt, M.; Yakovenko, A.A.; Halder, G.J.; Prosvirin, A.V.; Dunbar, K.R.; Zhou, H.C. Reversible switching from antiferro- to ferromagnetic behavior by solvent-mediated, thermally-induced phase transitions in a trimorphic MOF-based magnetic sponge system. J. Am. Chem. Soc. 2013, 135, 4040–4050. [Google Scholar] [CrossRef]
- Zeng, M.H.; Feng, X.L.; Chen, X.M. Crystal-to-crystal transformations of a microporous metal-organic laminated framework triggered by guest exchange, dehydration and readsorption. Dalton Trans. 2004, 2217–2223. [Google Scholar] [CrossRef]
- Choi, H.J.; Suh, M.P. Dynamic and redox active pillared bilayer open framework: Single-crystal-to-single-crystal transformations upon guest removal, guest exchange, and framework oxidation. J. Am. Chem. Soc. 2004, 126, 15844–15851. [Google Scholar] [CrossRef]
- Chen, C.L.; Goforth, A.M.; Smith, M.D.; Su, C.Y.; zur Loye, H.C. [Co2(ppca)2(H2O)(V4O12)0.5]: A framework material exhibiting reversible shrinkage and expansion through a single-crystal-to-single-crystal transformation involving a change in the cobalt coordination environment. Angew. Chem. Int. Ed. 2005, 44, 6673–6677. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Englert, U. Crystal-to-crystal transformation from a chain polymer to a two-dimensional network at low temperatures. Angew. Chem. Int. Ed. 2005, 44, 2281–2283. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Moorefield, C.N.; Panzner, M.; Newkome, G.R. TerpyridineCuIIPolycarboxylate Crystal Reorganization to One- and Two-Dimensional Nanostructures: Crystal Disassembly and Reassembly. Cryst. Growth Des. 2006, 6, 1563–1565. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Program for the Refinement of Crystal Structures; University of Göttingen: Göttingen, Germany, 1993. [Google Scholar]
- Sheldrick, G.M. SHELXL2014; University of Goëttingen: Goëttingen, Germany, 2014. [Google Scholar]
- Mulay, L.N.; Boudreaux, E.A. Theory and Applications of Molecular Diamagnetism; Wiley-VCH: New York, NY, USA, 1976; pp. 491–494. [Google Scholar]
- Kang, X.M.; Wang, W.M.; Yao, L.H.; Ren, H.X.; Zhao, B. Solvent-dependent variations of both structure and catalytic performance in three manganese coordination polymers. Dalton Trans. 2018, 47, 6986–6994. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Srirambalaji, R.; Patra, S.; Anantharaman, G. Anion triggered and solvent assisted structural diversity and reversible single-crystal-to-single-crystal (SCSC) transformation between 1D and 2D coordination polymers. CrystEngComm 2015, 17, 8876–8887. [Google Scholar] [CrossRef]
- Schneemann, A.; Bon, V.; Schwedler, I.; Senkovska, I.; Kaskel, S.; Fischer, R.A. Flexible metal-organic frameworks. Chem. Soc. Rev. 2014, 43, 6062–6096. [Google Scholar] [CrossRef]
- Liu, Y.-H.; Lee, S.-H.; Chiang, J.-C.; Chen, P.-C.; Chien, P.-H.; Yang, C.-I. Dehydration induced 2D-to-3D crystal-to-crystal network re-assembly and ferromagnetism tuning within two chiral copper(II)–tartrate coordination polymers. Dalton Trans. 2013, 42, 16857–16867. [Google Scholar] [CrossRef]
- Tsao, J.Y.; Tsai, J.D.; Yang, C.I. Azide-bridged Cu(ii), Mn(ii) and Co(ii) coordination polymers constructed with a bifunctional ligand of 6-(1H-tetrazol-5-yl)-2,2′-bipyridine. Dalton Trans. 2016, 45, 3388–3397. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-Z.; Chang, H.-T.; Kuo, Y.; Lee, G.-H.; Yang, C.-I. Two New Three-Dimensional Pillared-Layer Co(II) and Cu(II) Frameworks Involving a [M2(EO-N3)2] Motif from a Semi-Flexible N-Donor Ligand, 5,5′-Bipyrimidin: Syntheses, Structures and Magnetic Properties. Polymers 2018, 10, 229. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja, C.M.; Haldar, R.; Maji, T.K.; Rao, C.N.R. Chiral Porous Metal–Organic Frameworks of Co(II) and Ni(II): Synthesis, Structure, Magnetic Properties, and CO2 Uptake. Inorg. Chem. 2012, 12, 975–981. [Google Scholar] [CrossRef]
- Rodríguez-Martín, Y.; Hernández-Molina, M.; Sanchiz, J.; Ruiz-Pérez, C.; Lloret, F.; Julve, M. Crystal structures and magnetic properties of two- and three-dimensional malonato-bridged manganese(ii) complexes. Dalton Trans. 2003, 2359–2365. [Google Scholar] [CrossRef]
- Déniz, M.; Hernández-Rodríguez, I.; Pasán, J.; Fabelo, O.; Cañadillas-Delgado, L.; Yuste, C.; Julve, M.; Lloret, F.; Ruiz-Pérez, C. Pillaring Role of 4,4′-Azobis(pyridine) in Substituted Malonate-Containing Manganese(II) Complexes: Syntheses, Crystal Structures, and Magnetic Properties. Cryst. Growth Des. 2012, 12, 4505–4518. [Google Scholar] [CrossRef]
- O’Keeffe, M.; Peskov, M.A.; Ramsden, S.J.; Yaghi, O.M. The Reticular Chemistry Structure Resource (RCSR) Database of, and Symbols for, Crystal Nets. Acc. Chem. Res. 2008, 41, 1782–1789. [Google Scholar] [CrossRef]
- Zeng, M.H.; Wu, M.C.; Liang, H.; Zhou, Y.L.; Chen, X.M.; Ng, S.W. 3D homometallic carboxylate ferrimagnet constructed from a manganese(II) succinate carboxylate layer motif pillared by isonicotinate spacers. Inorg. Chem. 2007, 46, 7241–7243. [Google Scholar] [CrossRef] [PubMed]
- Rueff, J.-M.; Masciocchi, N.; Rabu, P.; Sironi, A.; Skoulios, A. Synthesis, Structure and Magnetism of Homologous Series of Polycrystalline Cobalt Alkane Mono- and Dicarboxylate Soaps. Chem. Eur. J. 2002, 8. [Google Scholar] [CrossRef]
- Huang, F.-P.; Li, H.-Y.; Yu, Q.; Bian, H.-D.; Tian, J.-L.; Yan, S.-P.; Liao, D.-Z.; Cheng, P. Co(ii)/Ni(ii) coordination polymers incorporated with a bent connector: Crystal structures and magnetic properties. CrystEngComm 2012, 14, 4756–4766. [Google Scholar] [CrossRef]
- Beghidja, A.; Rogez, G.; Rabu, P.; Welter, R.; Drillon, M. An approach to chiral magnets using α-hydroxycarboxylates. J. Mater. Chem. 2006, 16, 2715–2728. [Google Scholar] [CrossRef]
- Boonmak, J.; Nakanob, M.; Youngme, S. Structural diversity and magnetic properties in 1D and 2D azido-bridged cobalt(II) complexes with 1,2-bis(2-pyridyl)ethylene. Dalton Trans. 2011, 40, 1254–1260. [Google Scholar] [CrossRef]
- Fabelo, O.; Canadillas-Delgado, L.; Pasan, J.; Delgado, F.S.; Lloret, F.; Cano, J.; Julve, M.; Ruiz-Perez, C. Study of the influence of the bridge on the magnetic coupling in cobalt(II) complexes. Inorg. Chem. 2009, 48, 11342–11351. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 | 2 |
|---|---|---|
| Formula | C11H13CoNO5 | C11H11CoNO4 |
| Fw | 298.15 | 280.14 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/n | P2/c |
| a/Å | 7.4005(18) | 9.352(4) |
| b/Å | 21.647(5) | 12.278(5) |
| c/Å | 7.4050(19) | 9.880(4) |
| α/° | 90 | 90 |
| β/° | 92.473(5) | 93.296(7) |
| γ/° | 90 | 90 |
| V/Å3 | 1185.2(5) | 1132.6(8) |
| Z | 4 | 4 |
| T/K | 150(2) | 150(2) |
| Dc/g cm−3 | 1.671 | 1.643 |
| µ/mm−1 | 1.460 | 1.516 |
| (Δρ) max, min/e Å−3 | 0.400, −0.393 | 0.602, −0.552 |
| Measured/independent (Rint) reflections | 8905/2832(0.0602) | 7570/2549(0.0577) |
| Observed reflections [I > 2σ (I)] | 2832 | 2549 |
| Goodness-of-fits on F2 | 1.009 | 1.000 |
| R11, wR22 (all data) | 0.0636, 0.0870 | 0.0584, 0.0976 |
| R11, wR22 (I > 2 σ (I)) | 0.0396, 0.0775 | 0.0386, 0.0897 |
| Compound 1 | |||
|---|---|---|---|
| Co(1)-O(2) | 2.0747(18) | Co(1)-O(4) | 2.1147(19) |
| Co(1)-O(1) | 2.0984(19) | Co(1)-O(6) | 2.1247(19) |
| Co(1)-O(3) | 2.1026(18) | Co(1)-N(1) | 2.151(2) |
| O(2)-Co(1)-O(1) | 170.43(7) | O(3)-Co(1)-O(6) | 96.72(8) |
| O(2)-Co(1)-O(3) | 84.76(7) | O(4)-Co(1)-O(6) | 86.47(8) |
| O(1)-Co(1)-O(3) | 87.53(7) | O(2)-Co(1)-N(1) | 88.50(8) |
| O(2)-Co(1)-O(4) | 89.85(7) | O(1)-Co(1)-N(1) | 86.03(8) |
| O(1)-Co(1)-O(4) | 97.55(7) | O(3)-Co(1)-N(1) | 91.42(8) |
| O(3)-Co(1)-O(4) | 173.96(8) | O(4)-Co(1)-N(1) | 85.69(8) |
| O(2)-Co(1)-O(6) | 95.11(7) | O(6)-Co(1)-N(1) | 171.36(8) |
| O(1)-Co(1)-O(6) | 91.41(8) | ||
| Compound 2 | |||
|---|---|---|---|
| Co(1)-N(1) | 2.067(2) | Co(2)-O(3) | 1.9919(19) |
| Co(1)-N(1)#1 | 2.068(2) | Co(2)-O(3)#2 | 1.9919(19) |
| Co(1)-O(1)#1 | 2.145(2) | Co(2)-O(2)#2 | 2.104(2) |
| Co(1)-O(1) | 2.145(2) | Co(2)-O(2) | 2.104(2) |
| Co(1)-O(3)#1 | 2.163(2) | Co(2)-O(1) | 2.1164(19) |
| Co(1)-O(3) | 2.163(2) | Co(2)-O(1)#2 | 2.1164(19) |
| N(1)-Co(1)-N(1)#1 | 98.11(14) | O(3)-Co(2)-O(3)#2 | 180.0 |
| N(1)-Co(1)-O(1)#1 | 90.27(9) | O(3)-Co(2)-O(2)#2 | 88.90(8) |
| N(1)#1-Co(1)-O(1)#1 | 171.61(8) | O(3)#2-Co(2)-O(2)#2 | 91.10(8) |
| N(1)-Co(1)-O(1) | 171.60(9) | O(3)-Co(2)-O(2) | 91.10(8) |
| N(1)#1-Co(1)-O(1) | 90.27(9) | O(3)#2-Co(2)-O(2) | 88.90(8) |
| O(1)#1-Co(1)-O(1) | 81.35(11) | O(2)#2-Co(2)-O(2) | 180.0 |
| N(1)-Co(1)-O(3)#1 | 88.63(9) | O(3)-Co(2)-O(1) | 84.56(7) |
| N(1)#1-Co(1)-O(3)#1 | 99.80(9) | O(3)#2-Co(2)-O(1) | 95.45(7) |
| O(1)#1-Co(1)-O(3)#1 | 79.86(7) | O(2)#2-Co(2)-O(1) | 97.78(8) |
| O(1)-Co(1)-O(3)#1 | 90.37(7) | O(2)-Co(2)-O(1) | 82.22(8) |
| N(1)-Co(1)-O(3) | 99.80(9) | O(3)-Co(2)-O(1)#2 | 95.45(7) |
| N(1)#1-Co(1)-O(3) | 88.63(9) | O(3)#2-Co(2)-O(1)#2 | 84.55(7) |
| O(1)#1-Co(1)-O(3) | 90.37(7) | O(2)#2-Co(2)-O(1)#2 | 82.22(8) |
| O(1)-Co(1)-O(3) | 79.86(7) | O(2)-Co(2)-O(1)#2 | 97.78(8) |
| O(3)#1-Co(1)-O(3) | 167.17(10) | O(1)-Co(2)-O(1)#2 | 180.0 |
| Co(2)-O(3)-Co(1) | 98.52(8) | Co(2)-O(1)-Co(1) | 95.34(8) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, H.-C.; Lin, C.-H.; Yang, C.-I. Temperature-Controlled Assembly/Reassembly of Two Dicarboxylate-Based Three-Dimensional Co(II) Coordination Polymers with an Antiferromagnetic Metallic Layer and a Ferromagnetic Metallic Chain. Polymers 2019, 11, 795. https://doi.org/10.3390/polym11050795
Yu H-C, Lin C-H, Yang C-I. Temperature-Controlled Assembly/Reassembly of Two Dicarboxylate-Based Three-Dimensional Co(II) Coordination Polymers with an Antiferromagnetic Metallic Layer and a Ferromagnetic Metallic Chain. Polymers. 2019; 11(5):795. https://doi.org/10.3390/polym11050795
Chicago/Turabian StyleYu, Hui-Chen, Chin-Hsuan Lin, and Chen-I Yang. 2019. "Temperature-Controlled Assembly/Reassembly of Two Dicarboxylate-Based Three-Dimensional Co(II) Coordination Polymers with an Antiferromagnetic Metallic Layer and a Ferromagnetic Metallic Chain" Polymers 11, no. 5: 795. https://doi.org/10.3390/polym11050795