Structural Evolution Mechanism of Crystalline Polymers in the Isothermal Melt-Crystallization Process: A Proposition Based on Simultaneous WAXD/SAXS/FTIR Measurements

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
2.1. Polymers
2.2. Measurement Systems
- (1)
- Simultaneous Measurement of WAXD and SAXS: The WAXD and SAXS data were collected simultaneously by setting the two detectors at different distances from the sample cell, as illustrated in Figure 1a [14]. The detectors used were a flat panel detector (C9728-DK, Hamamatsu Photonics K.K., Shizuoka, Japan) for the WAXD data and an image-intensified CCD detector (C4742-98, Hamamatsu Photonics K.K.) for the SAXS data. The exposure time was 1–10 s per shot.
- (2)
- Simultaneous Measurement of WAXD/SAXS/FTIR: The WAXD/SAXS data were collected using a similar system as illustrated in Figure 1b. The detectors used were a flat panel for WAXD data and an image-intensifier CCD camera or Pilatus 100k (Dectris, Baden-Daettwil, Switzerland) for SAXS data. A miniature FTIR spectrometer (Bruker α) was set around the sample cell to measure the transmission-type IR spectra [12]. The WAXD and SAXS data were collected at every 1–10 s. The FTIR spectra were collected at the resolution power of 2 cm−1 at every 7 s.
- (3)
- Temperature Jump Cell: In the isothermal crystallization experiment, the temperature of a sample must be changed from the melting temperature (Tm) to the crystallization temperature (Tc) as quickly as possible and kept at Tc as stably as possible. For this purpose, the temperature jump cells were developed. The jump cell of the first generation was the manually operated system illustrated in Figure 2a [14]. A sample was put into a small metal holder. The holder was first set at position (A) and was heated above Tm for a fixed time to melt the sample. Then, the holder was immediately moved to position (B), where the sample holder was stopped for a while, during which the air was blown to the sample in order to erase the heat stored in the sample. As a result, the sample temperature decreased steeply to a value between Tm and Tc, as detected by a thermocouple embedded inside the sample. Then, the sample holder was moved quickly to position (C) at the temperature Tc. The temporary stop at position (B) is key to producing a sharp temperature jump.
2.3. X-ray Scattering Data Analyses
2.3.1. Domains
2.3.2. Correlated Domains
2.3.3. Stacked Lamellae
3. Results and Discussion
3.1. Isotactic Polypropylene
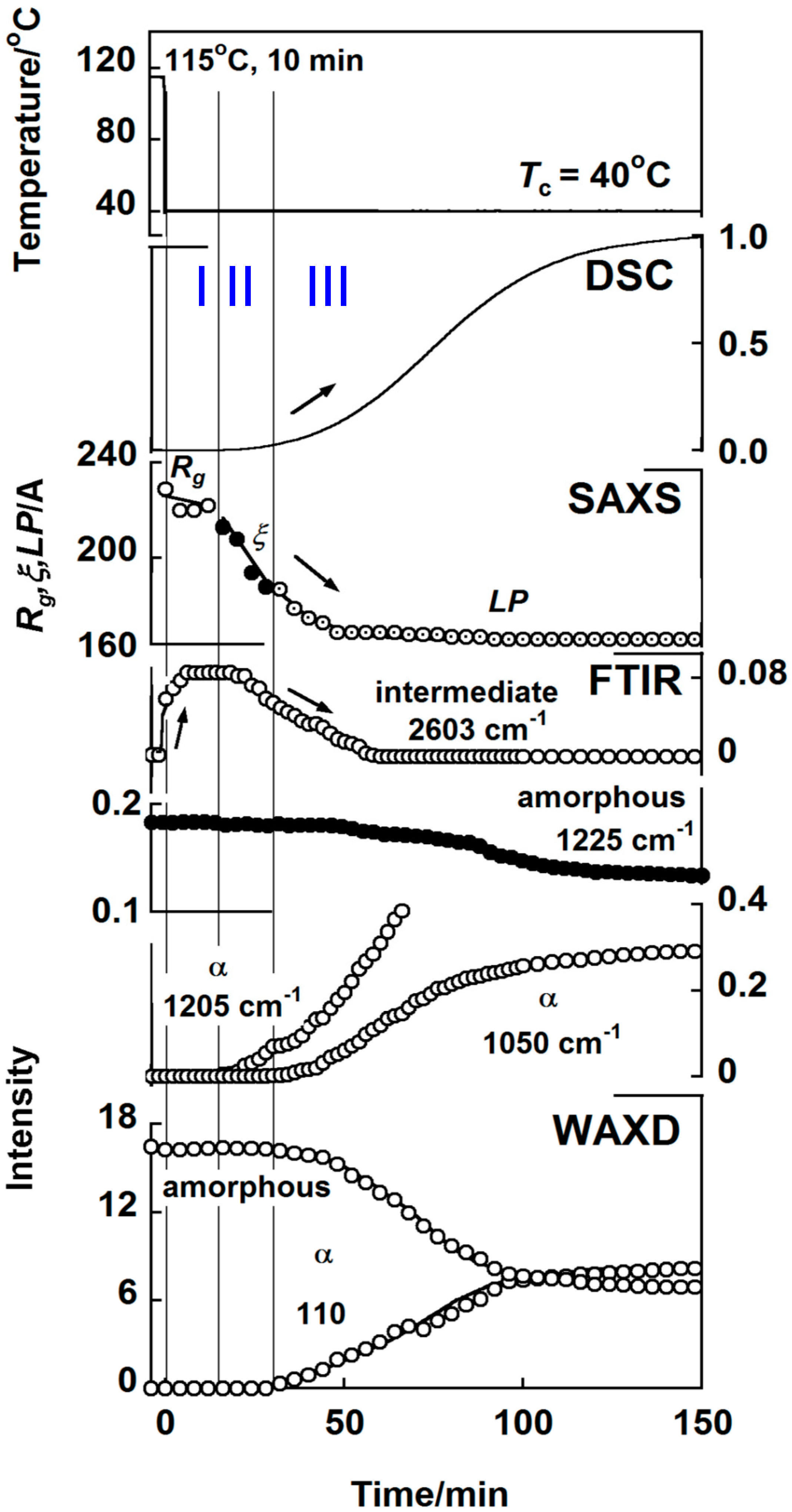
3.2. Polyoxymethylene


3.3. trans-1,4-Polyisoprene
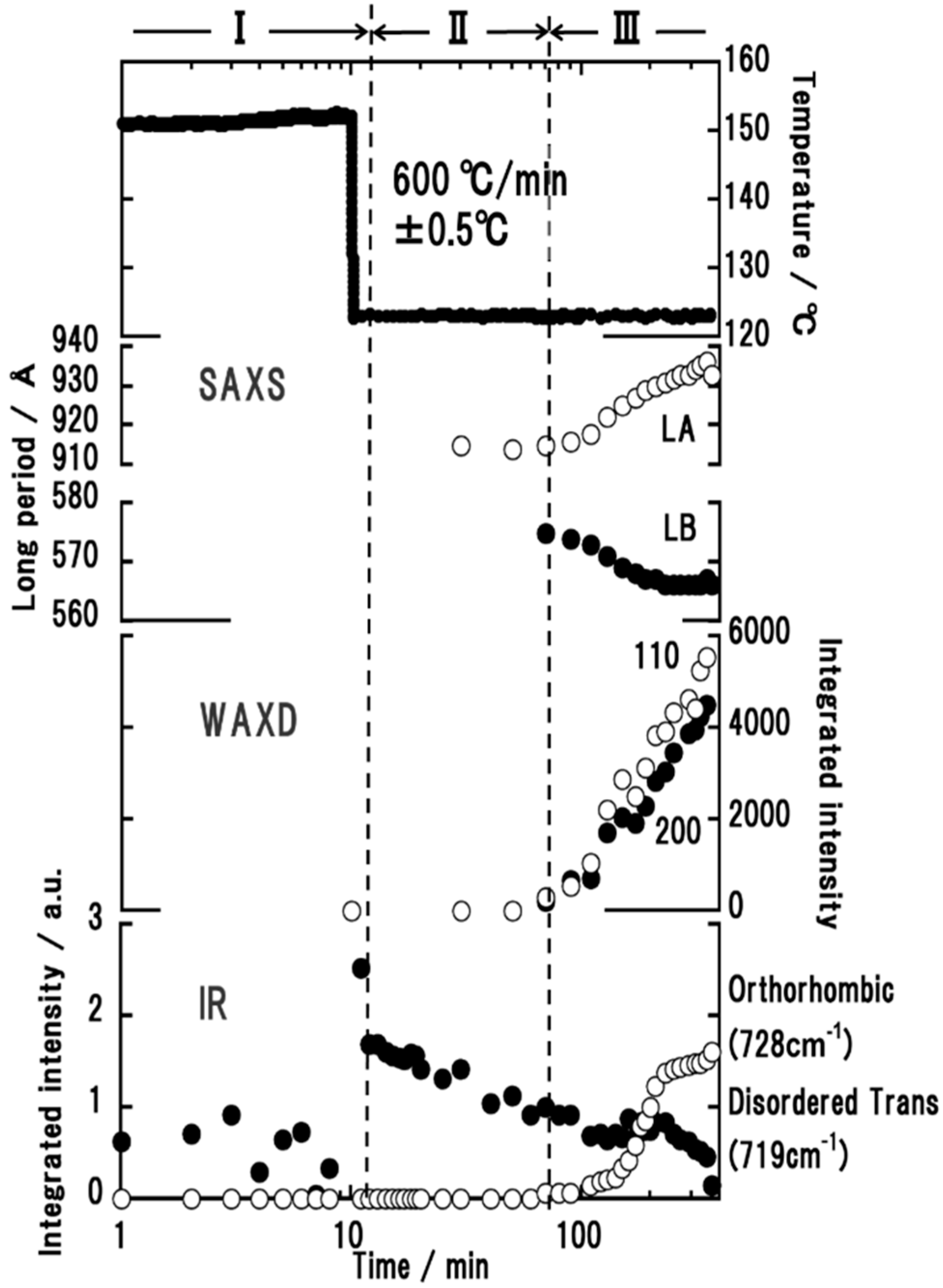
3.4. High-Density Polyethylene
3.5. Nylon 1010
3.6. VDF–TrFE Copolymers
3.7. Poly(Alkylene Terephthalate)
4. Interpretation
- (i)
- The thermally-induced fluctuation in the melt [50] caused the heterogeneous distribution of the density and the domains were formed, which were the aggregation of the more or less conformationally disordered chain segments.
- (ii)
- The domains became larger and increased in number. As a result, the correlation became stronger among the neighboring domains.
- (iii)
- The domains fused into large lamellae. In these lamellae, the chain segments were arrayed in parallel, but they were not necessarily totally ordered. For the polymer with long methylene segments, the methylene conformation was still disordered more or less. In the case of nylon, the molecular chain stems in the early stage of lamellar formation possessed the regular methylene segments, but this was only in the CO-side, and the methylene segments in the NH side were still in the disordered state, although the intermolecular hydrogen bonds were already created.
- (iv)
- The size of thus created lamellae increased with time. The random coils in the melt approached the front surface of the growing lamella and they were adsorbed into the lamellar inside to become the stems with the folded parts.
- (v)
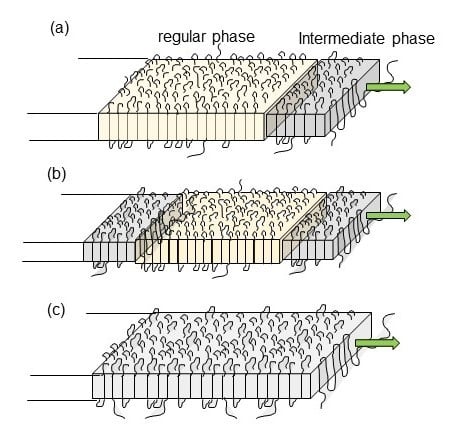
- The thus adsorbed and regularized stems were existent in the various states. For example, as seen in the case of VDF–TrFE copolymers, the newly attached stems were in the HT phase and coexisted with the LT phase region, or they transform totally to the more regular structure of the LT phase. It depends on the crystallization temperature. In the case of PE, the attached segments may be in the conformationally disordered trans form and exist in the hexagonal lattice for a while. This hexagonal phase is unstable and transforms to the orthorhombic phase, since only the latter is thermodynamically possible at atmospheric pressure, different from the case of VDF–TrFE copolymer. In the case of it-PP and POM, the random coils adsorbed on the lamellar surface changed immediately to the regular helices because of the relatively rigid helices.
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zachmann, H.G.; Wutz, G. Studies of the mechanism of crystallization by means WAXS and SAXS employing synchrotron radiation. Cryst. Polym. 1993, 405, 403–414. [Google Scholar]
- Schultz, J.M. Polymer crystallization. In The Development of Crystalline Order in Thermoplastic Polymers; Oxford University Press: New York, NY, USA, 2001. [Google Scholar]
- Tashiro, K. Growth of polymer crystals. In Handbook of Polymer Crystallization, Chapter 6; Piorkowska, E., Rutledge, G., Eds.; Wiley: Hoboken, NJ, USA, 2013; pp. 165–195. [Google Scholar]
- Hu, W. Polymer Crystallization. In Polymer Physics; Springer: Berlin/Heidelberg, Germany, 2012; pp. 187–221. [Google Scholar]
- Janeschitz-Kriegl, H. Crystallization Modalities in Polymer Melt Processing: Fundamental Aspects of Structure Formation; Springer: New York, NY, USA, 2010. [Google Scholar]
- Allegra, G. (Ed.) Interephases and mesophases in polymer crystallization, I. In Advances in Polymer Science; Springer: Berlin/Heidelberg, Germany, 2005; Volume 180. [Google Scholar]
- Allegra, G. (Ed.) Interephases and mesophases in polymer crystallization II. In Advances in Polymer Science; Springer: Berlin/Heidelberg, Germany, 2005; Volume 181. [Google Scholar]
- Allegra, G. (Ed.) Interephases and mesophases in polymer crystallization III. In Advances in Polymer Science; Springer: Berlin/Heidelberg, Germany, 2005; Volume 191. [Google Scholar]
- Tashiro, K.; Kariyo, S.; Nishimori, A.; Fujii, T.; Saragai, S.; Nakamoto, S.; Kawaguchi, T.; Matsumoto, A.; Rangsiman, O. Development of a simultaneous measurement system of X-ray diffraction and Raman spectra: Application to structural study of crystalline-phase transitions of chain molecules. J. Polym. Sci. Polym. Phys. 2002, 40, 495–506. [Google Scholar] [CrossRef]
- Hirose, R.; Yoshioka, T.; Yamamoto, H.; Reddy, R.K.; Tahara, D.; Hamada, K.; Tashiro, K. In-house simultaneous collection of SAXS, WAXD and Raman scattering data from polymeric materials. J. Appl. Cryst. 2014, 47, 922–930. [Google Scholar] [CrossRef]
- Tashiro, K.; Yamamoto, H.; Yoshioka, T.; Ninh, T.H.; Shimada, S.; Nakatani, T.; Iwamoto, H.; Ohta, N.; Masunaga, H. Development of simultaneous measurement system of wide- and small-angle X-ray scattering and vibrational spectra for the static and dynamic analyses of hierarchical structures of polymers. Kobunshi Ronbunshu. 2012, 69, 213–227. [Google Scholar] [CrossRef]
- Tashiro, K.; Yamamoto, H.; Yoshika, T.; Ninh, T.H.; Tasaki, M.; Shimada, S.; Nakatani, T.; Iwamoto, H.; Ohta, N.; Masunaga, H. Hierarchical structural change in the stress-induced phase transition of poly (tetramethylene terephthalate) as studied by the simultaneous measurement of FTIR spectra and 2D synchrotron undulator WAXD/SAXSdata. Macromolecules 2014, 47, 2052–2061. [Google Scholar] [CrossRef]
- Tashiro, K.; Reddy, K.R.; Hanesaka, M.; Tsuji, S.; Hashida, T.; Cao, W.; Masunaga, H.; Sasaki, S.; Takata, M. Evolution process of regular structure in isothermal crystalline polymers viewed from synchrotron small-and wide-angle X-ray scatterings and vibrational spectroscopy. Kobunshi Ronbunshu 2009, 66, 536–549. [Google Scholar] [CrossRef]
- Tashiro, T.; Sasaki, S. Structural changes in the ordering process of polymers as studied by an organized combination of the various measurement techniques. Progr. Polym. Sci. 2003, 28, 451–519. [Google Scholar] [CrossRef]
- Kobayashi, M.; Sakashita, M. Morphology dependent anomalous frequency shifts of infrared absorption bands of polymer crystals: Interpretation in terms of transition dipole-dipole coupling theory. J. Chem. Phys. 1992, 96, 748–760. [Google Scholar] [CrossRef]
- Sasaki, S.; Tashiro, K.; Kobayashi1, M.; Izumi, Y.; Kobayashi, K. Microscopically viewed structural change of PE during the isothermal crystallization from the melt II. conformational ordering and lamellar formation mechanism derived from the coupled interpretation of time-resolved SAXS and FTIR data. Polymer 1999, 40, 7125–7135. [Google Scholar] [CrossRef]
- Sasaki, S.; Tashiro, K.; Gose, N.; Imanishi, K.; Izuchi, M.; Kobayashi, M.; Imai, M.; Ohashi, M.; Yamaguchi, Y.; Ohyama, K. Spatial distribution of chain stems and chain folding mode in polyethylene lamellae as revealed by coupled information of DSC, FTIR, SANS and WANS. Polym. J. 1999, 31, 677–686. [Google Scholar] [CrossRef]
- Tashiro, K.; Sasaki, S.; Kobayashi, M. Structural investigation of orthorhombic-to-hexagonal phase transition in polyethylene crystal: The experimental confirmation of the conformationally-disordered structure by X-ray diffraction and infrared/Raman spectroscopic measurements. Macromolecules 1996, 29, 7460–7469. [Google Scholar] [CrossRef]
- Tashiro, K.; Sasaki, S.; Gose, N.; Kobayashi, M. Microscopically-viewed structural change of polyethylene during isothermal crystallization from the melt 1 time-resolved FTIR Spectral Measurements. Polym. J. 1998, 6, 485–491. [Google Scholar] [CrossRef]
- Strobl, G. A thermodynamic multiphase scheme treating polymer crystallization and melting. Eur. Phys. J. E 2005, 18, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Strobl, G. From the melt via mesomorphic and granular crystalline layers to lamellar crystallites: A major route followed in polymer crystallization? Eur. Phys. J. E 2000, 3, 165–183. [Google Scholar] [CrossRef]
- Organization of Macromolecules in the Condensed Phase. Faraday Discuss. Chem. Soc. 1979, 68, 1–520.
- Reddy, R.K.; Tashiro, K.; Sakurai, T.; Yamaguchi, N.; Sasaki, S.; Masunaga, H.; Takata, M. Isothermal crystallization behavior of isotactic polypropylene H/D blends as viewed from time-resolved FTIR and synchrotron SAXS/WAXD measurements. Macromolecules 2009, 42, 4191–4199. [Google Scholar] [CrossRef]
- Reddy, R.K.; Tashiro, K.; Sakurai, T.; Yamaguchi, N. Isotope effect on the isothermal crystallization behavior of isotactic polypropylene blends between the deuterated and hydrogenated species. Macromolecules 2009, 42, 1672–1678. [Google Scholar] [CrossRef]
- Feigin, L.A.; Svergun, D.I. Structure Analysis by Small-Angle X-ray and Neutron Scattering; Plenum Press: New York, NY, USA, 1987. [Google Scholar]
- Guinier, A.; Fournet, G. Small-Angle Scattering of X-rays; John Wiley & Sons: New York, NY, USA, 1955. [Google Scholar]
- Glatter, O.; Kratky, O. (Eds.) Small angle X-ray Scattering; Academic Press: London, UK, 1982. [Google Scholar]
- Debye, P.; Bueche, A.M. Scattering by an inhomogeneoous solid. J. Appl. Phys. 1949, 20, 518–525. [Google Scholar] [CrossRef]
- Strobl, G.R.; Schneider, M. Direct evaluation of the electron density correlation function of partially crystalline polymers. J. Polym. Sci. Polym. Ed. 1980, 18, 1343–1359. [Google Scholar] [CrossRef]
- Kobayashi, M.; Akita, K.; Tadokoro, H. Infrared spectra and regular sequence lengths in isotactic polymer chains. Makromol. Chem. 1968, 118, 324–342. [Google Scholar] [CrossRef]
- Kummara, S.; Tashiro, K.; Monma, T.; Horita, K. Isotope effect on the melt−isothermal crystallization of polyoxymethylene D/H random copolymers and D/H blend samples. Macromolecules 2015, 48, 8070–8081. [Google Scholar] [CrossRef]
- Kummara, S.; Tashiro, K. Isotope effect on the structural evolution process in the isothermal crystallization phenomenon of polyoxymethylene. Polymer 2016, 90, 76–88. [Google Scholar] [CrossRef]
- Tashiro, K.; Kummara, S.; Yamamoto, H.; Yoshioka, T.; Tahara, D.; Masunaga, H.; Ohta, N. Study of melt-isothermal crystallization phenomenon of crystalline polymers by utilizing a simultaneous measurement system of synchrotron wide-angle and small-angle X-ray scatterings and transmission FTIR spectra: Application to the case of polyoxymethylene. Spring 8 Res. Rep. (Sect. A) 2017, 5, 20–26. [Google Scholar]
- Hama, H.; Tashiro, K. Structural changes in isothermal crystallization process of polyoxymethylene investigated by time-resolved FTIR, SAXS and WAXS measurements. Polymer 2003, 44, 6973–6988. [Google Scholar] [CrossRef]
- Tashiro, K.; Hama, H. Structural changes in isothermal crystallization processes of synthetic polymers studied by time-resolved measurements of synchrotron-sourced X-ray scatterings and vibrational spectra. Macromol. Res. 2014, 12, 1–10. [Google Scholar] [CrossRef]
- Ratri, P.J.; Tashiro, K. Application of the simultaneous measurement system of WAXD, SAXS and transmission FTIR spectra to the study of structural changes in the cold-and melt-crystallization processes of trans-1,4-polyisoprene. Polym. J. 2013, 45, 1019–1026. [Google Scholar] [CrossRef]
- Ratri, P.J.; Tashiro, K. Phase-transition behavior of a crystalline polymer near the melting point: Case studies of the ferroelectric phase transition of poly (vinylidene fluoride) and the β-to-α transition of trans-1, 4-polyisoprene. Polym. J. 2013, 45, 1107–1114. [Google Scholar] [CrossRef]
- Bunn, C.W.; Garner, E.V. The crystal structures of two polyamides (‘Nylons’). Proc. Roy. Soc. Lond. A Mater. Physic. Sci. 1947, 189, 39–68. [Google Scholar]
- Yoshioka, Y.; Tashiro, K.; Ramesh, C. A new interpretation of progression bands observed in infrared spectra of Nylons m/n. J. Polym. Sci. Part. B Polym. Phys. Ed. 2003, 41, 1294–1307. [Google Scholar] [CrossRef]
- Yoshioka, Y.; Tashiro, K. Structural changes in phase transitions of Nylon model compounds (1) transition behavior of model compounds of R-NHCO-R’ type. J. Phys. Chem. B. 2003, 107, 11835–11842. [Google Scholar] [CrossRef]
- Yoshioka, Y.; Tashiro, K.; Ramesh, R. Structural change in the Brill transition of Nylon m/n (2) conformational disordering as viewed from the temperature-dependent infrared spectral measurements. Polymer 2003, 44, 6407–6417. [Google Scholar] [CrossRef]
- Tashiro, K.; Nishiyama, A.; Tsuji, S.; Hashida, T.; Hanesaka, M.; Takeda, S.; Cao, W.; Reddy, R.K.; Masunaga, H.; Sasaki, S.; et al. Structural evolution in the isothermal crystallization process of the molten nylon 10/10 traced by time-resolved infrared spectral measurements and synchrotron SAXS/WAXD measurements. J. Phys. Conf. Ser. 2009, 184, 012002. [Google Scholar] [CrossRef]
- Tashiro, K. Crystal structure and phase transition of PVDF and related copolymers. In Ferroelectric Polymers: Chemistry, Physics, and Technology; Nalwa, H.S., Ed.; Marcel Dekker Inc.: New York, NY, USA, 1995; pp. 63–182. [Google Scholar]
- Tashiro, K.; Sasaki, S.; Saragai, S. Time-resolved wide-angle X-ray scattering measurements during the isothermal crystallization and ferroelectric phase transition processes of VDF73%-TrFE copolymer. J. Polym. Sci. Part. B Polym. Phys. 2004, 42, 4175–4181. [Google Scholar] [CrossRef]
- Daubeny, R.; De, O.; Bunn, C.W. The crystal structure of polyethylene terephthalate. Proc. R. Soc. A. 1954, 226, 531–542. [Google Scholar]
- Poulin-Dandurand, S.; Pérez, J.F.; Brisse, R.F. The crystal structure of poly (trimethylene terephthalate) by X-ray and electron diffraction. Polymer 1979, 20, 419–426. [Google Scholar] [CrossRef]
- Tasaki, M.; Yamamoto, H.; Yoshioka, T.; Hanesaka, M.; Ninh, T.H.; Tashiro, K.; Jeon, H.J.; Choi, K.B.; Jeong, H.S.; Song, H.H.; et al. Crystal structure analyses of arylate polyesters with long methylene segments and their model compounds on the basis of 2-D X-ray diffractions and infrared progression bands. Polymer 2014, 55, 1228–1248. [Google Scholar] [CrossRef]
- Tasaki, M.; Yamamoto, H.; Yoshioka, T.; Hanesaka, M.; Ninh, T.H.; Tashiro, K.; Jeon, H.J.; Choi, K.B.; Jeong, H.S.; Song, H.H.; et al. Microscopically-viewed relationship between the chain conformation and ultimate Young’s modulus of a series of acrylate polyesters with long methylene segments. Polymer 2014, 55, 1799–1808. [Google Scholar] [CrossRef]
- Stein, R.S.; Cronauer, J.; Zachmann, H.G. Real time scattering measurements of the crystallization of polymers and their blends 1. J. Mol. Struct. 1996, 383, 19–22. [Google Scholar] [CrossRef]
- Baert, J.; Puyvelde, P.V. Density fluctuations during the early stages of polymer crystallzaition: An overview. Macromol. Mater. Eng. 2008, 293, 255–273. [Google Scholar] [CrossRef]















© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tashiro, K.; Yamamoto, H. Structural Evolution Mechanism of Crystalline Polymers in the Isothermal Melt-Crystallization Process: A Proposition Based on Simultaneous WAXD/SAXS/FTIR Measurements. Polymers 2019, 11, 1316. https://doi.org/10.3390/polym11081316
Tashiro K, Yamamoto H. Structural Evolution Mechanism of Crystalline Polymers in the Isothermal Melt-Crystallization Process: A Proposition Based on Simultaneous WAXD/SAXS/FTIR Measurements. Polymers. 2019; 11(8):1316. https://doi.org/10.3390/polym11081316
Chicago/Turabian StyleTashiro, Kohji, and Hiroko Yamamoto. 2019. "Structural Evolution Mechanism of Crystalline Polymers in the Isothermal Melt-Crystallization Process: A Proposition Based on Simultaneous WAXD/SAXS/FTIR Measurements" Polymers 11, no. 8: 1316. https://doi.org/10.3390/polym11081316