

Chitosan (CS)/Hydroxyapatite (HA)/Tricalcium Phosphate (β-TCP)-Based Composites as a Potential Material for Pulp Tissue Regeneration

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Natural Hydroxyapatite
2.3. Preparation of the CS/HA/β-TCP Blocks
2.4. Characterization of the CS/HA/β-TCP Blocks
2.4.1. Fourier Transform Infrared Spectroscopy FT-IR
2.4.2. X-ray Diffraction (DRX)
2.4.3. Scanning Electron Spectroscopy (SEM) and Energy Dispersive Spectroscopy (EDS) for the CS/HA/β-TCP Blocks
2.4.4. In Vitro Viability of the CS/HA/β-TCP Blocks
2.4.5. In Vivo Biocompatibility Study of CS/HA/β-TCP Blocks
2.4.6. Statistical Analysis
3. Results and Discussion
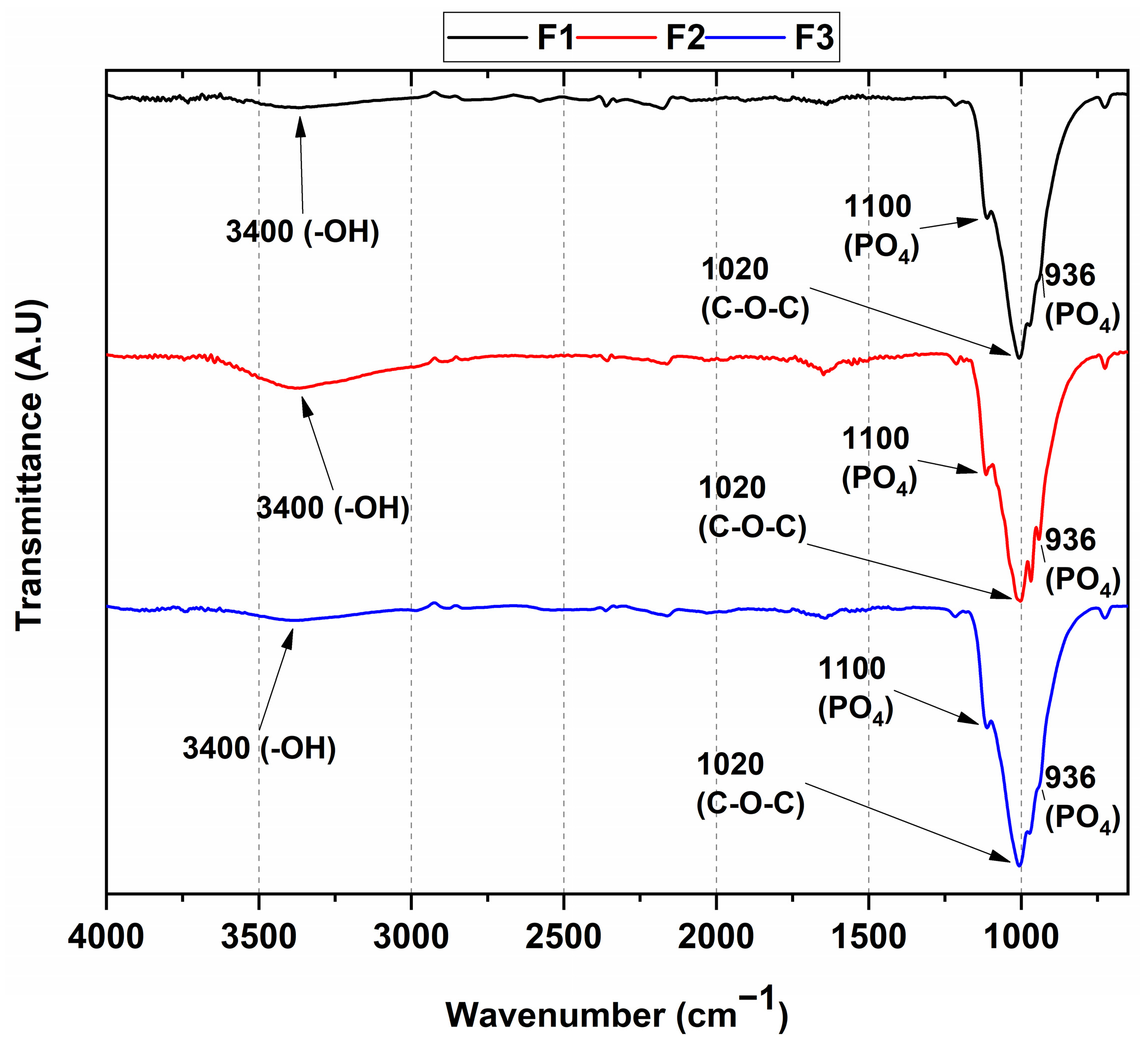
3.1. FT-IR Spectroscopy
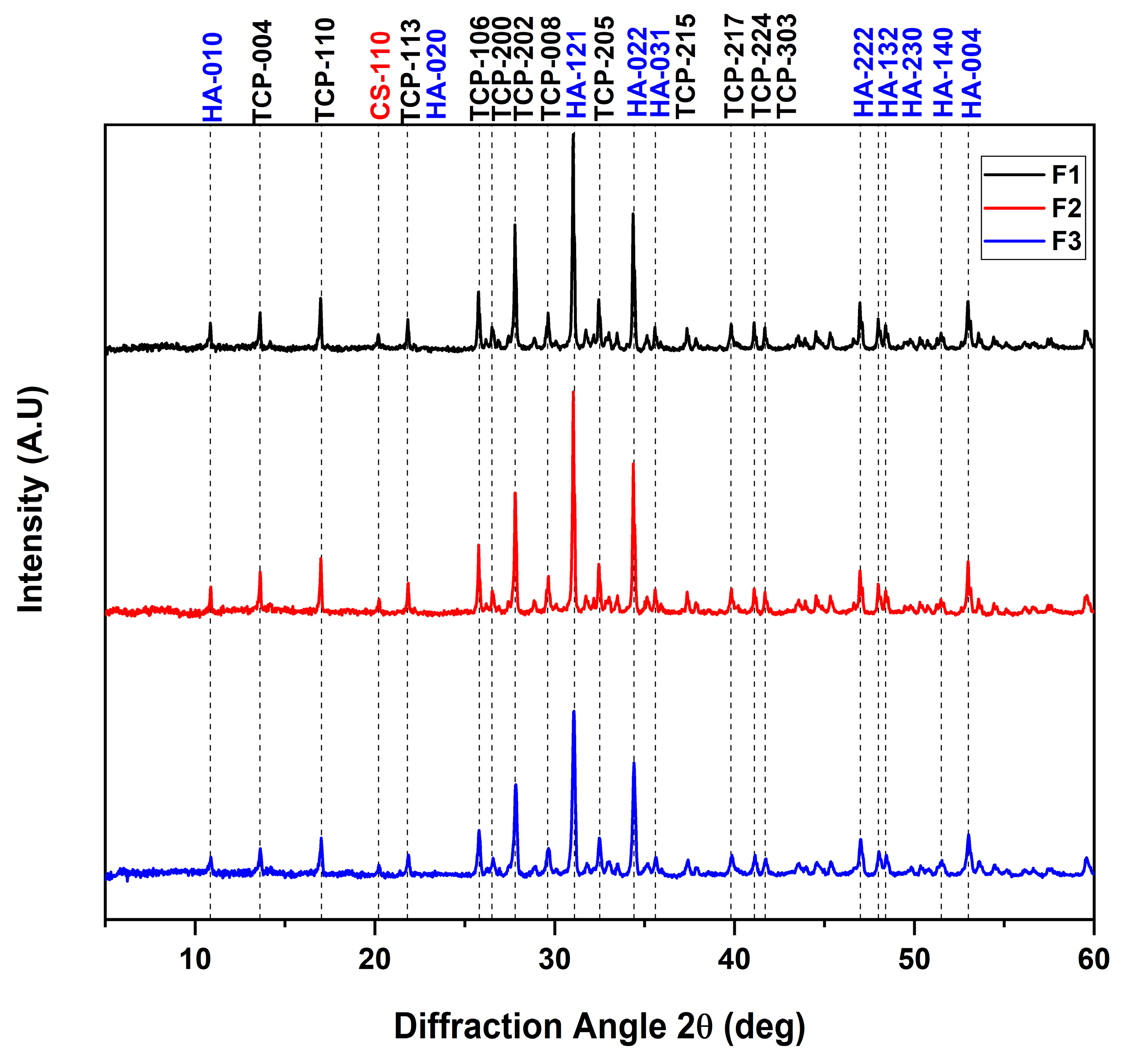
3.2. X-ray Diffraction
3.3. SEM-EDS
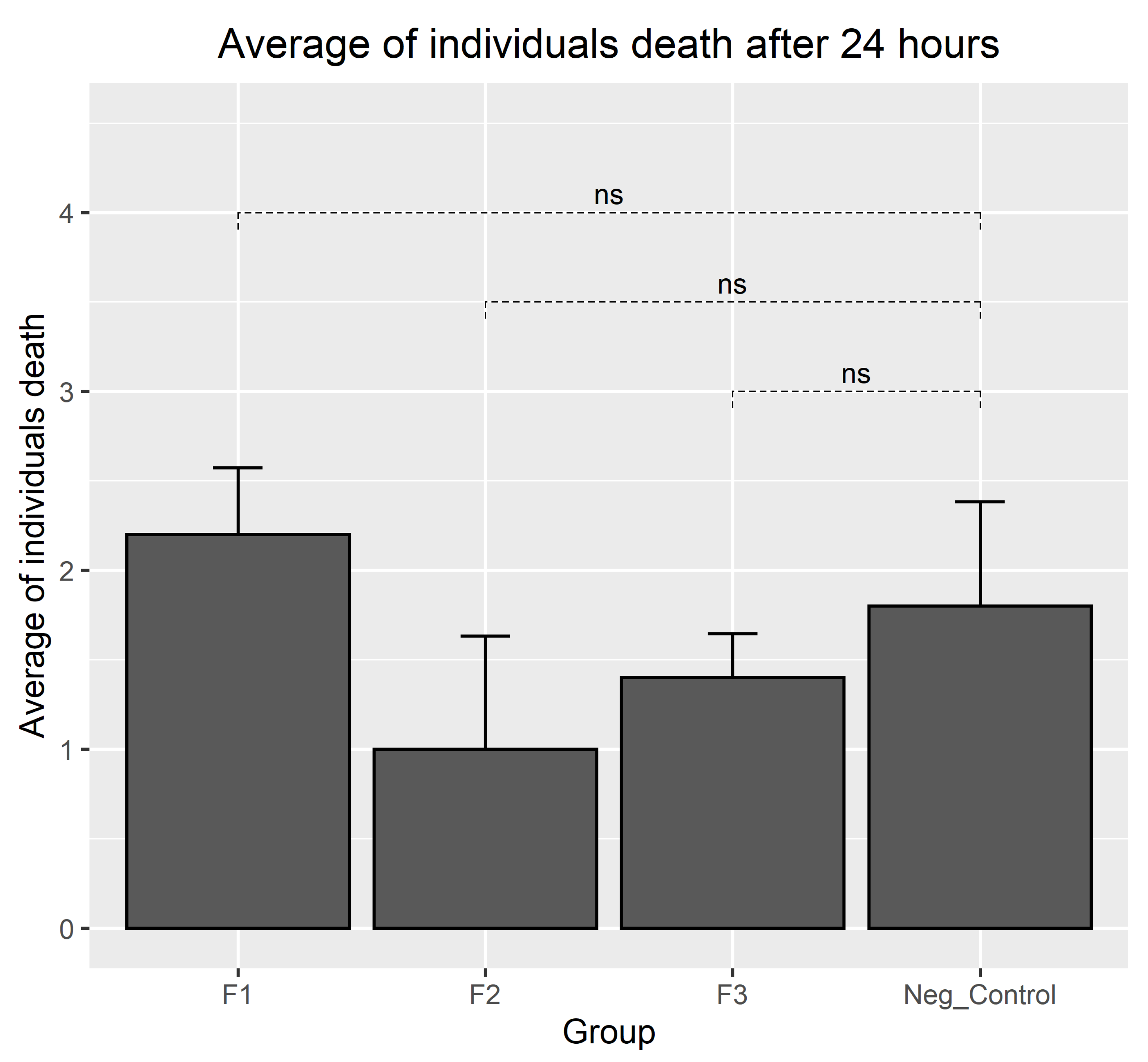
3.4. Results of Biocompatibility Test with A. salina
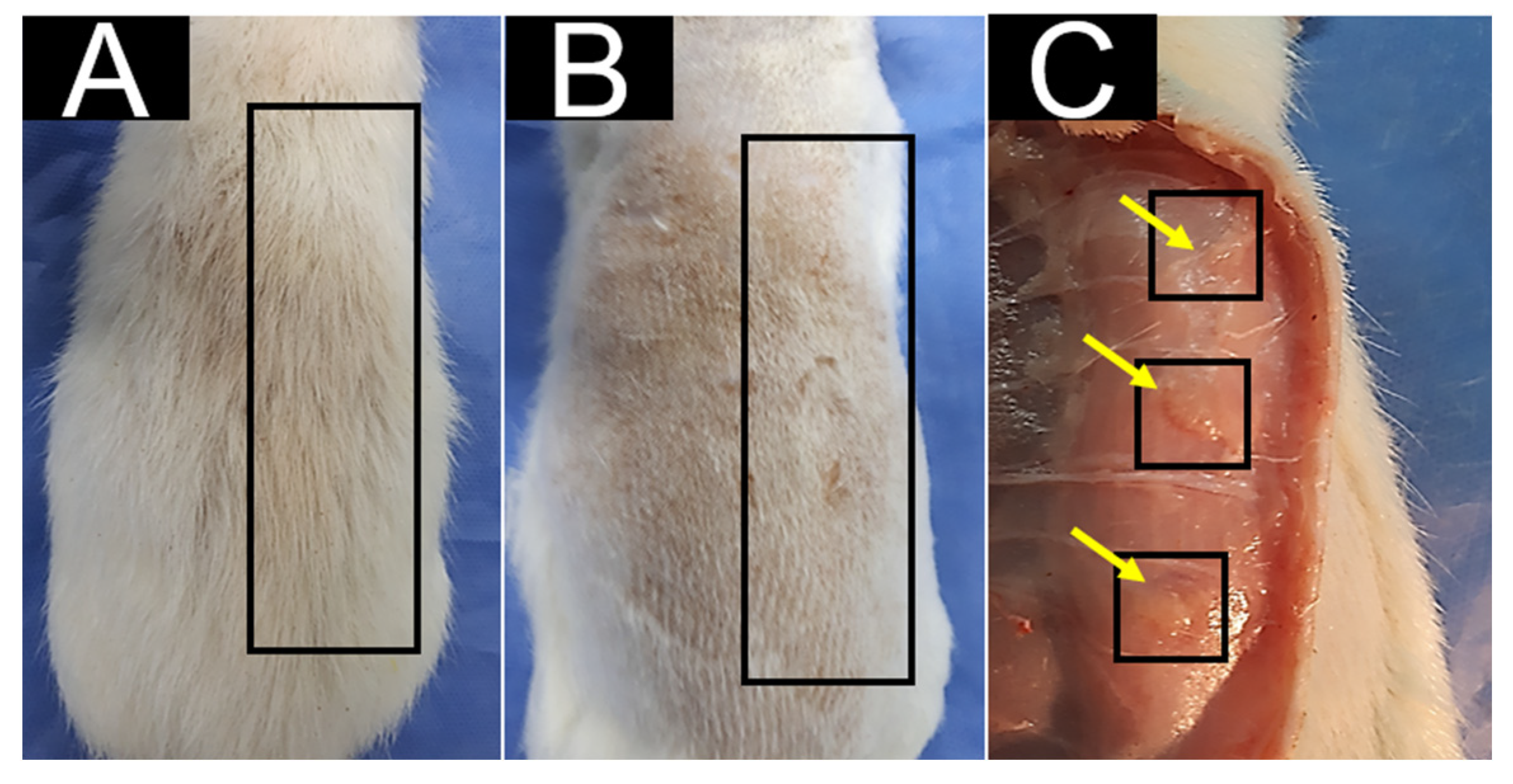
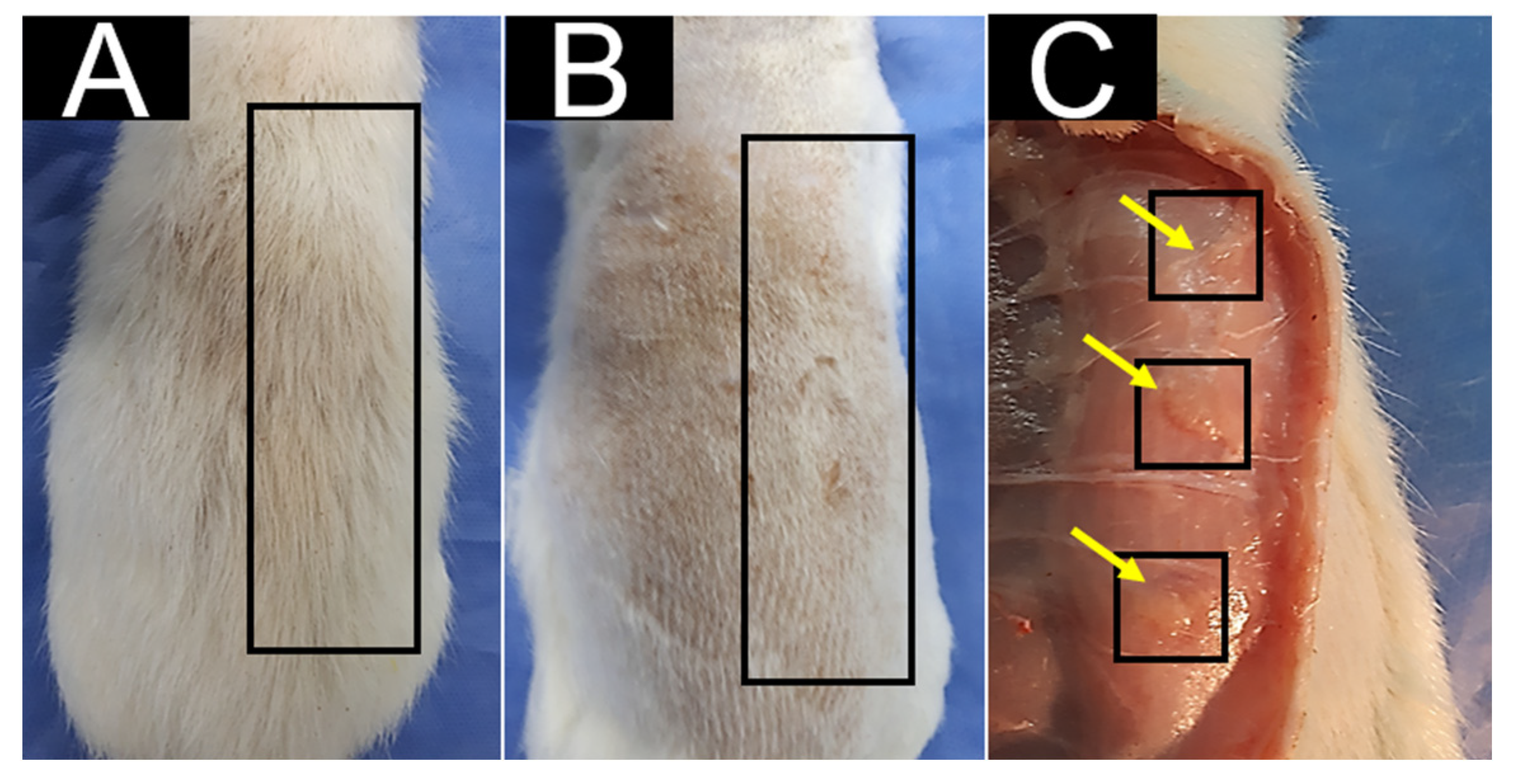
3.5. In Vivo Biocompatibility Test for CS/HA/β-TCP Composites
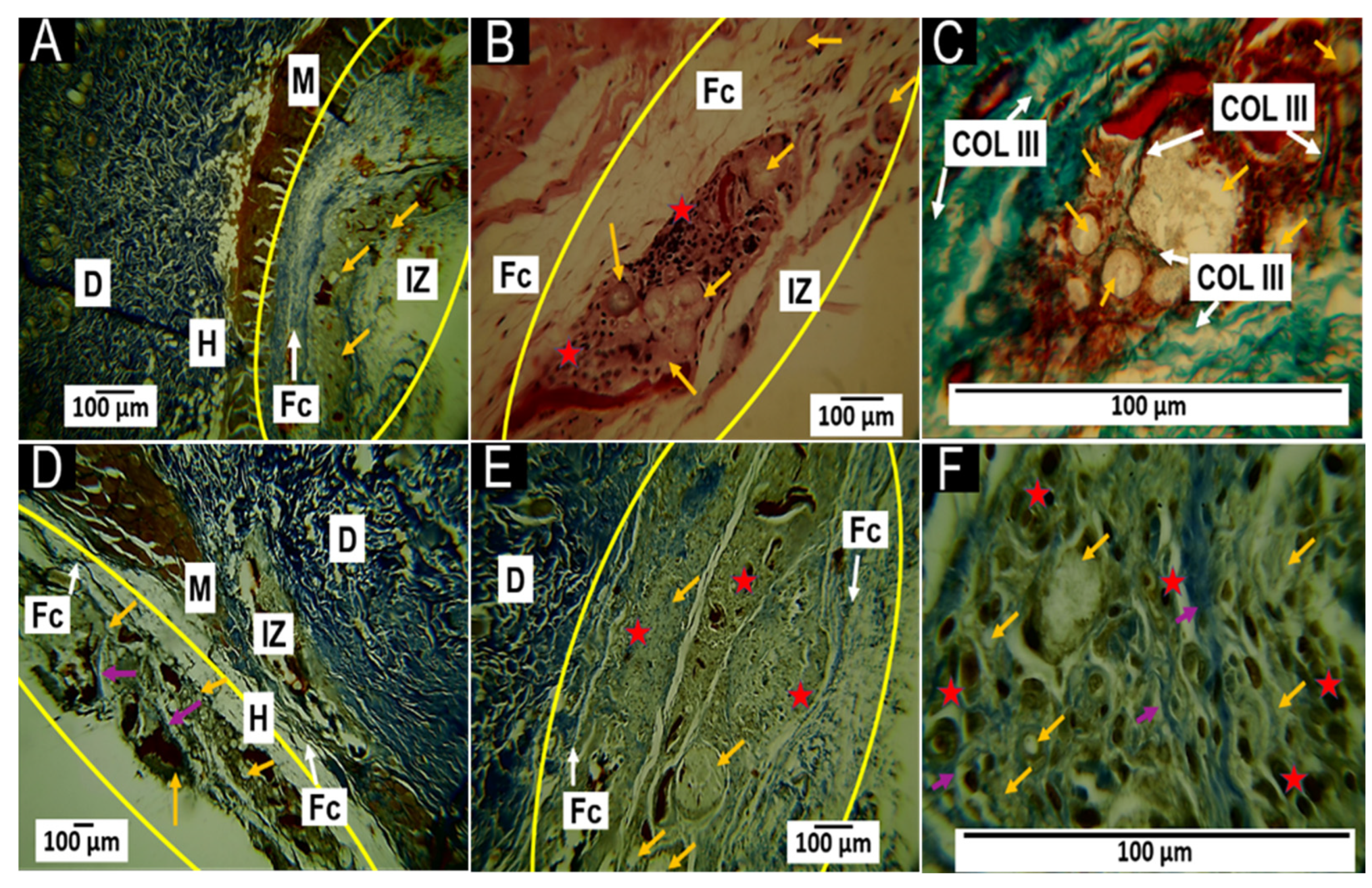
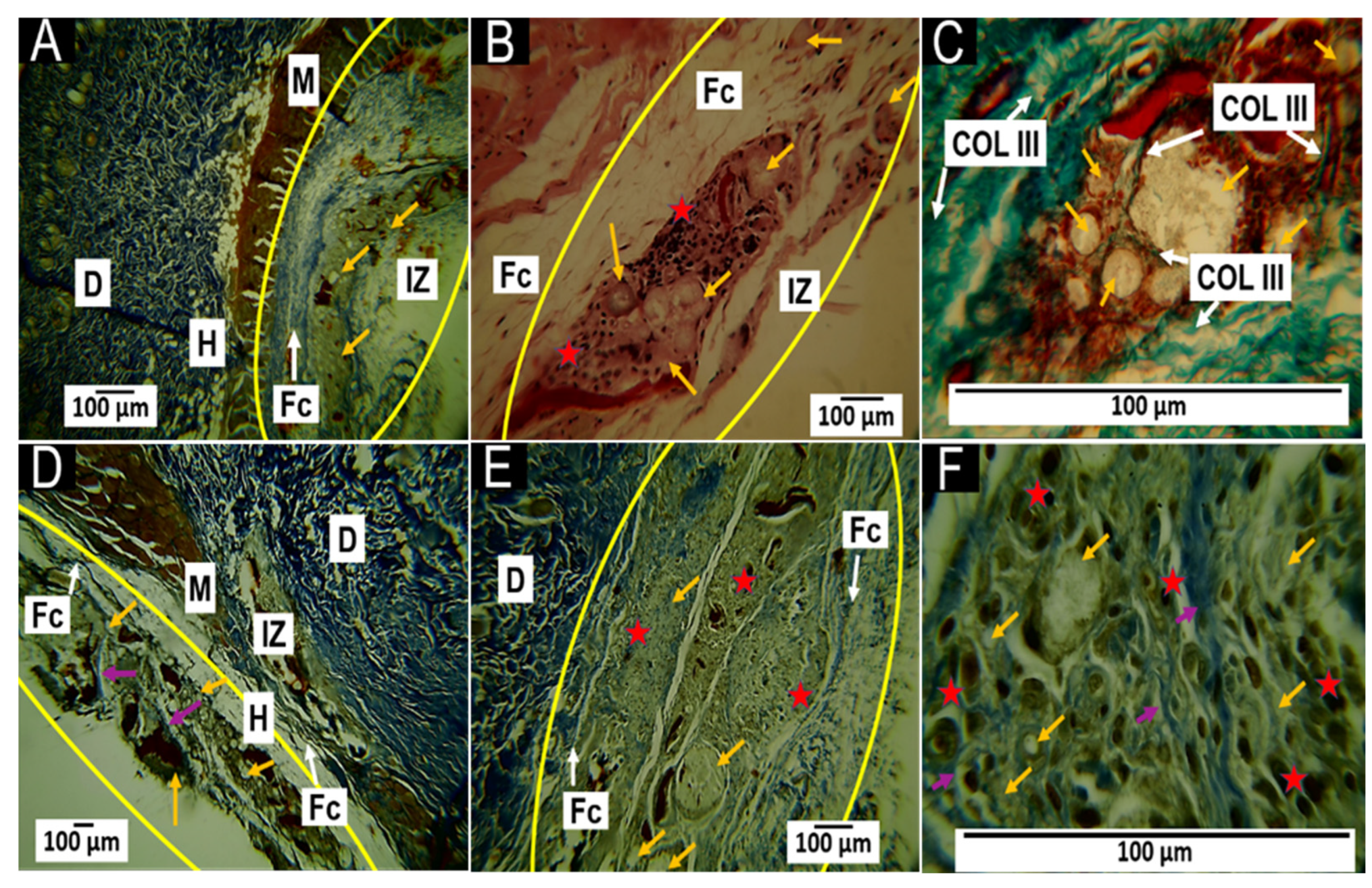
3.5.1. Histological Results for the F1 Formulation at 30 and 60 Days
3.5.2. Histological Results for the F2 Formulation at 30 and 60 Days
3.5.3. Histological Results for Formulation F3 at 30 and 60 Days
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, W.-J.; Tuli, R.; Okafor, C.; Derfoul, A.; Danielson, K.G.; Hall, D.J.; Tuan, R.S. A three-dimensional nanofibrous scaffold for cartilage tissue engineering using human mesenchymal stem cells. Biomaterials 2005, 26, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Amrollahi, P.; Shah, B.; Seifi, A.; Tayebi, L. Recent advancements in regenerative dentistry: A review. Mater. Sci. Eng. C 2016, 69, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Colombo, J.S.; Moore, A.N.; Hartgerink, J.D.; D’Souza, R.N. Scaffolds to control inflammation and facilitate dental pulp regeneration. J. Endod. 2014, 40, S6–S12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jazayeri, H.E.; Lee, S.-M.; Kuhn, L.; Fahimipour, F.; Tahriri, M.; Tayebi, L. Polymeric scaffolds for dental pulp tissue engineering: A review. Dent. Mater. 2020, 36, e47–e58. [Google Scholar] [CrossRef]
- Kontakiotis, E.G.; Filippatos, C.G.; Tzanetakis, G.N.; Agrafioti, A. Regenerative endodontic therapy: A data analysis of clinical protocols. J. Endod. 2015, 41, 146–154. [Google Scholar] [CrossRef]
- Rodrigues, E.M.; Cornélio, A.L.G.; Mestieri, L.B.; Fuentes, A.S.C.; Salles, L.P.; Rossa-Junior, C.; Faria, G.; Guerreiro-Tanomaru, J.M.; Tanomaru-Filho, M. Human dental pulp cells response to mineral trioxide aggregate (MTA) and MTA Plus: Cytotoxicity and gene expression analysis. Int. Endod. J. 2017, 50, 780–789. [Google Scholar] [CrossRef]
- Tawil, P.Z.; Trope, M.; Curran, A.E.; Caplan, D.J.; Kirakozova, A.; Duggan, D.J.; Teixeira, F.B. Periapical microsurgery: An in vivo evaluation of endodontic root-end filling materials. J. Endod. 2009, 35, 357–362. [Google Scholar] [CrossRef] [Green Version]
- Faraco Jr, I.M.; Holland, R. Response of the pulp of dogs to capping with mineral trioxide aggregate or a calcium hydroxide cement. Dent. Traumatol. 2001, 17, 163–166. [Google Scholar] [CrossRef]
- Parirokh, M.; Torabinejad, M.; Dummer, P.M.H. Mineral trioxide aggregate and other bioactive endodontic cements: An updated overview—Part I: Vital pulp therapy. Int. Endod. J. 2018, 51, 177–205. [Google Scholar]
- Fihri, A.; Len, C.; Varma, R.S.; Solhy, A. Hydroxyapatite: A review of syntheses, structure and applications in heterogeneous catalysis. Coord. Chem. Rev. 2017, 347, 48–76. [Google Scholar] [CrossRef]
- Kwon, S.H.; Jun, Y.K.; Hong, S.H.; Kim, H.E. Synthesis and dissolution behavior of β-TCP and HA/β-TCP composite powders. J. Eur. Ceram. Soc. 2003, 23, 1039–1045. [Google Scholar] [CrossRef]
- Xie, L.; Yu, H.; Deng, Y.; Yang, W.; Liao, L.; Long, Q. Preparation, characterization and in vitro dissolution behavior of porous biphasic α/β-tricalcium phosphate bioceramics. Mater. Sci. Eng. C 2016, 59, 1007–1015. [Google Scholar] [CrossRef]
- Turan, Y.; Kalkandelen, C.; Palaci, Y.; Sahin, A.; Gokce, H.; Gunduz, O.; Ben-Nissan, B. Synthesis and cytotoxicity analysis of porous β-TCP/starch bioceramics. J. Aust. Ceram. Soc. 2022, 58, 487–494. [Google Scholar] [CrossRef]
- Noordin, N.N.F.N.M.; Ahmad, N.; Mariatti, M.; Yahaya, B.H.; Sulaiman, A.R.; Hamid, Z.A.A. A review on bioceramics scaffolds for bone defect in different types of animal models: HA and β-TCP. Biomed. Phys. Eng. Express 2022, 8, 052002. [Google Scholar]
- Helaehil, J.V.; Lourenço, C.B.; Huang, B.; Helaehil, L.V.; de Camargo, I.X.; Chiarotto, G.B.; Santamaria-Jr, M.; Bártolo, P.; Caetano, G.F. In vivo investigation of polymer-ceramic PCL/HA and PCL/β-TCP 3D composite scaffolds and electrical stimulation for bone regeneration. Polymers 2021, 14, 65. [Google Scholar] [CrossRef]
- Rinaudo, M. Chitin and chitosan: Properties and applications. Prog. Polym. Sci. 2006, 31, 603–632. [Google Scholar] [CrossRef]
- Kim, I.-Y.; Seo, S.-J.; Moon, H.-S.; Yoo, M.-K.; Park, I.-Y.; Kim, B.-C.; Cho, C.-S. Chitosan and its derivatives for tissue engineering applications. Biotechnol. Adv. 2008, 26, 1–21. [Google Scholar] [CrossRef]
- Nasrin, R.; Biswas, S.; Rashid, T.U.; Afrin, S.; Jahan, R.A.; Haque, P.; Rahman, M.M. Preparation of Chitin-PLA laminated composite for implantable application. Bioact. Mater. 2017, 2, 199–207. [Google Scholar] [CrossRef]
- Islam, M.M.; Shahruzzaman, M.; Biswas, S.; Nurus Sakib, M.; Rashid, T.U. Chitosan based bioactive materials in tissue engineering applications-A review. Bioact. Mater. 2020, 5, 164–183. [Google Scholar] [CrossRef]
- Mahoney, C.; McCullough, M.B.; Sankar, J.; Bhattarai, N. Nanofibrous structure of chitosan for biomedical applications. J Nanomedic Biother. Discov. 2012, 2, 102. [Google Scholar]
- Shavandi, A.; Bekhit, A.E.D.A.; Sun, Z.; Ali, A.; Gould, M. A novel squid pen chitosan/hydroxyapatite/β-tricalcium phosphate composite for bone tissue engineering. Mater. Sci. Eng. C 2015, 55, 373–383. [Google Scholar] [CrossRef]
- Shavandi, A.; Bekhit, A.E.D.A.; Ali, M.A.; Sun, Z.; Gould, M. Development and characterization of hydroxyapatite/β-TCP/chitosan composites for tissue engineering applications. Mater. Sci. Eng. C 2015, 56, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Kasaai, M.R. Calculation of Mark–Houwink–Sakurada (MHS) equation viscometric constants for chitosan in any solvent–temperature system using experimental reported viscometric constants data. Carbohydr. Polym. 2007, 68, 477–488. [Google Scholar] [CrossRef]
- Rinaudo, M.; Milas, M.; Le Dung, P. Characterization of chitosan. Influence of ionic strength and degree of acetylation on chain expansion. Int. J. Biol. Macromol. 1993, 15, 281–285. [Google Scholar] [CrossRef]
- Guerrero Ibarguen, H.J. Preparación y Caracterización de Cementos Óseos Acrílicos Modificados con un Fosfato Calcico y Metacrilato de Dietilamino Etilo (DEAEM); Universidad del Valle: Valle del Cauca, Colombia, 2014. [Google Scholar]
- Lima, L.R.; Andrade, F.K.; Alves, D.R.; De Morais, S.M.; Vieira, R.S. Anti-acetylcholinesterase and toxicity against Artemia salina of chitosan microparticles loaded with essential oils of Cymbopogon flexuosus, Pelargonium x ssp and Copaifera officinalis. Int. J. Biol. Macromol. 2021, 167, 1361–1370. [Google Scholar] [CrossRef] [PubMed]
- Okumu, M.O.; Mbaria, J.M.; Gikunju, J.K.; Mbuthia, P.G.; Madadi, V.O.; Ochola, F.O.; Jepkorir, M.S. Artemia salina as an animal model for the preliminary evaluation of snake venom-induced toxicity. Toxicon X 2021, 12, 100082. [Google Scholar] [CrossRef]
- Pecoraro, R.; Scalisi, E.M.; Messina, G.; Fragalà, G.; Ignoto, S.; Salvaggio, A.; Zimbone, M.; Impellizzeri, G.; Brundo, M.V. Artemia salina: A microcrustacean to assess engineered nanoparticles toxicity. Microsc. Res. Tech. 2020, 84, 531–536. [Google Scholar] [CrossRef]
- ISO 10993-6; Biological Evaluation of Medical Devices—Part 6: Tests for Local Effects after Implantation. ISO: Geneva, Switzerland, 1995.
- Hubrecht, R.C.; Carter, E. The 3Rs and humane experimental technique: Implementing change. Animals 2019, 9, 754. [Google Scholar] [CrossRef] [Green Version]
- Grande-Tovar, C.D.; Castro, J.I.; Valencia Llano, C.H.; Tenorio, D.L.; Saavedra, M.; Zapata, P.A.; Chaur, M.N. Polycaprolactone (PCL)-Polylactic acid (PLA)-Glycerol (Gly) Composites Incorporated with Zinc Oxide Nanoparticles (ZnO-NPs) and Tea Tree Essential Oil (TTEO) for Tissue Engineering Applications. Pharmaceutics 2023, 15, 43. [Google Scholar] [CrossRef]
- Castro, J.I.; Valencia-llano, C.H.; Eliana, M.; Zapata, V.; Restrepo, Y.J.; Mina, H.; Navia-porras, D.P.; Valencia, Y.; Valencia, C.; Grande-tovar, C.D. Chitosan/Polyvinyl Alcohol/Tea Tree Essential Oil Composite Films for Biomedical Applications. Polymers 2021, 13, 3753. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, S.H.; Kim, H.K.; Akaike, T.; Kim, S.C. Synthesis and characterization of hydroxyapatite crystals: A review study on the analytical methods. J. Biomed. Mater. Res. 2002, 62, 600–612. [Google Scholar] [CrossRef]
- Tanase, C.E.; Popa, M.I.; Verestiuc, L. Biomimetic chitosan-calcium phosphate composites with potential applications as bone substitutes: Preparation and characterization. J. Biomed. Mater. Res.-Part B Appl. Biomater. 2012, 100B, 700–708. [Google Scholar] [CrossRef]
- Zhu, Q.; Ablikim, Z.; Chen, T.; Cai, Q.; Xia, J.; Jiang, D.; Wang, S. The preparation and characterization of HA/β-TCP biphasic ceramics from fish bones. Ceram. Int. 2017, 43, 12213–12220. [Google Scholar] [CrossRef]
- Ortiz, C.H.; Aperador, W.; Caicedo, J.C. Physical properties evolution of β -tricalcium phosphate / hydroxyapatite heterostructures in relation to the bilayer number. Thin Solid Films 2022, 752, 139256. [Google Scholar] [CrossRef]
- Cárdenas, G.; Cabrera, G.; Taboada, E.; Miranda, S.P. Chitin characterization by SEM, FTIR, XRD, and 13C cross polarization/mass angle spinning NMR. J. Appl. Polym. Sci. 2004, 93, 1876–1885. [Google Scholar] [CrossRef]
- Teng, S.; Lee, E.; Yoon, B.; Shin, D.; Kim, H.; Oh, J. Chitosan/nanohydroxyapatite composite membranes via dynamic filtration for guided bone regeneration. J. Biomed. Mater. Res. Part A Off. J. Soc. Biomater. Japanese Soc. Biomater. Aust. Soc. Biomater. Korean Soc. Biomater. 2009, 88, 569–580. [Google Scholar] [CrossRef]
- Yuan, Y.; Chesnutt, B.M.; Haggard, W.O.; Bumgardner, J.D. Deacetylation of chitosan: Material characterization and in vitro evaluation via albumin adsorption and pre-osteoblastic cell cultures. Materials 2011, 4, 1399–1416. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.; Bano, S.; Poojary, S.; Chaudhary, A.; Kumar, D.; Negi, Y.S. Effect of cellulose nanocrystals on chitosan/PVA/nano β-TCP composite scaffold for bone tissue engineering application. J. Biomater. Sci. Polym. Ed. 2022, 33, 1–19. [Google Scholar] [CrossRef]
- Kumari, S.; Rath, P.; Kumar, A.S.H.; Tiwari, T.N. Extraction and characterization of chitin and chitosan from fishery waste by chemical method. Environ. Technol. Innov. 2015, 3, 77–85. [Google Scholar] [CrossRef]
- Manocha, S.; Joshi, P.; Patel, B.; Manocha, L.M. Synthesis and characterization of hydroxyapatite nanoparticles using sol-gel method. Eurasian Chem. J. 2011, 13, 85–88. [Google Scholar] [CrossRef]
- Huang, Y.; Huang, W.; Sun, L.; Wang, Q.; He, A.; Han, C.C. Phase Transition from α-TCP into β-TCP in TCP/HA Composites. Int. J. Appl. Ceram. Technol. 2010, 7, 184–188. [Google Scholar] [CrossRef]
- de Albuquerque Sarmento, P.; da Rocha Ataíde, T.; Fernandes Barbosa, A.P.; de Araújo-Júnior, J.X.; Lúcio, I.M.L.; de Assis Bastos, M.L. Avaliação do extrato da Zeyheria tuberculosa na perspectiva de um produto para cicatrização de feridas. Rev. Lat. Am. Enfermagem 2014, 22, 165–172. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.M.; Rodriguez, A.; Chang, D.T. Foreign body reaction to biomaterials. Semin. Immunol. 2008, 20, 86–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adib, Y.; Bensussan, A.; Michel, L. Review Article Cutaneous Wound Healing: A Review about Innate Immune Response and Current Therapeutic Applications. Mediat. Inflamm. 2022, 2022, 5344085. [Google Scholar] [CrossRef]
- Luttikhuizen, D.T.; Harmsen, M.C.; Van Luyn, M.J.A. Cellular and molecular dynamics in the foreign body reaction. Tissue Eng. 2006, 12, 1955–1970. [Google Scholar] [CrossRef]
- Mariani, E.; Lisignoli, G.; Borzì, R.M.; Pulsatelli, L. Biomaterials: Foreign bodies or tuners for the immune response? Int. J. Mol. Sci. 2019, 20, 636. [Google Scholar] [CrossRef] [Green Version]
- Sheikh, Z.; Brooks, P.J.; Barzilay, O.; Fine, N.; Glogauer, M. Macrophages, foreign body giant cells and their response to implantable biomaterials. Materials 2015, 8, 5671–5701. [Google Scholar] [CrossRef] [Green Version]
- Eming, S.A.; Brachvogel, B.; Odorisio, T.; Koch, M. Regulation of angiogenesis: Wound healing as a model. Prog. Histochem. Cytochem. 2007, 42, 115–170. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | F1 | F2 | F3 |
|---|---|---|---|
| β-TCP (%) | 87.7 | 87.7 | 87.7 |
| CS (%) | 3.5 | 3.5 | 3.5 |
| Commercial HA (%) | 8.8 | 8.8 | 0 |
| Natural HA (%) | 0 | 0 | 8.8 |
| 5.4% CaCl2 (mL) | 0.8 | 1.0 | 0.8 |
| Sample | C (%) | O (%) | P (%) | Cl (%) | Ca (%) |
|---|---|---|---|---|---|
| F1 | 3.64 ± 4.08 | 51.57 ± 4.62 | 17.66 ± 2.11 | 0 ± 0 | 27.13 ± 1.72 |
| F2 | 2.31 ± 4.01 | 53.29 ± 8.27 | 16.67 ± 1.34 | 0.51 ± 0.89 | 27.22 ± 3.58 |
| F3 | 3.61 ± 4.18 | 48.98 ± 5.72 | 18.46 ± 1.33 | 0 ± 0 | 28.96 ± 2.86 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zamora, I.; Alfonso Morales, G.; Castro, J.I.; Ruiz Rojas, L.M.; Valencia-Llano, C.H.; Mina Hernandez, J.H.; Valencia Zapata, M.E.; Grande-Tovar, C.D. Chitosan (CS)/Hydroxyapatite (HA)/Tricalcium Phosphate (β-TCP)-Based Composites as a Potential Material for Pulp Tissue Regeneration. Polymers 2023, 15, 3213. https://doi.org/10.3390/polym15153213
Zamora I, Alfonso Morales G, Castro JI, Ruiz Rojas LM, Valencia-Llano CH, Mina Hernandez JH, Valencia Zapata ME, Grande-Tovar CD. Chitosan (CS)/Hydroxyapatite (HA)/Tricalcium Phosphate (β-TCP)-Based Composites as a Potential Material for Pulp Tissue Regeneration. Polymers. 2023; 15(15):3213. https://doi.org/10.3390/polym15153213
Chicago/Turabian StyleZamora, Ingrid, Gilbert Alfonso Morales, Jorge Iván Castro, Lina Marcela Ruiz Rojas, Carlos Humberto Valencia-Llano, Jose Herminsul Mina Hernandez, Mayra Eliana Valencia Zapata, and Carlos David Grande-Tovar. 2023. "Chitosan (CS)/Hydroxyapatite (HA)/Tricalcium Phosphate (β-TCP)-Based Composites as a Potential Material for Pulp Tissue Regeneration" Polymers 15, no. 15: 3213. https://doi.org/10.3390/polym15153213