

Phase Equilibria and Structure Formation in the Polylactic-co-Glycolic Acid/Tetraglycol/Water Ternary System
 , , and
, , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Investigation Methods
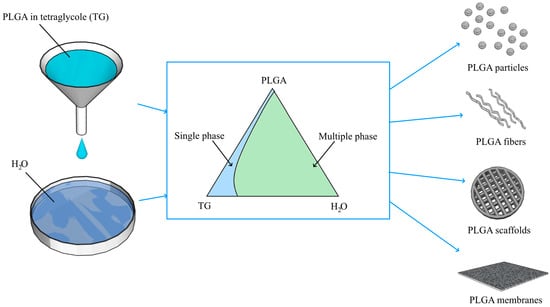
2.2.1. Constructing the Phase Diagram of the Ternary PLGA/TG/Water Mixture
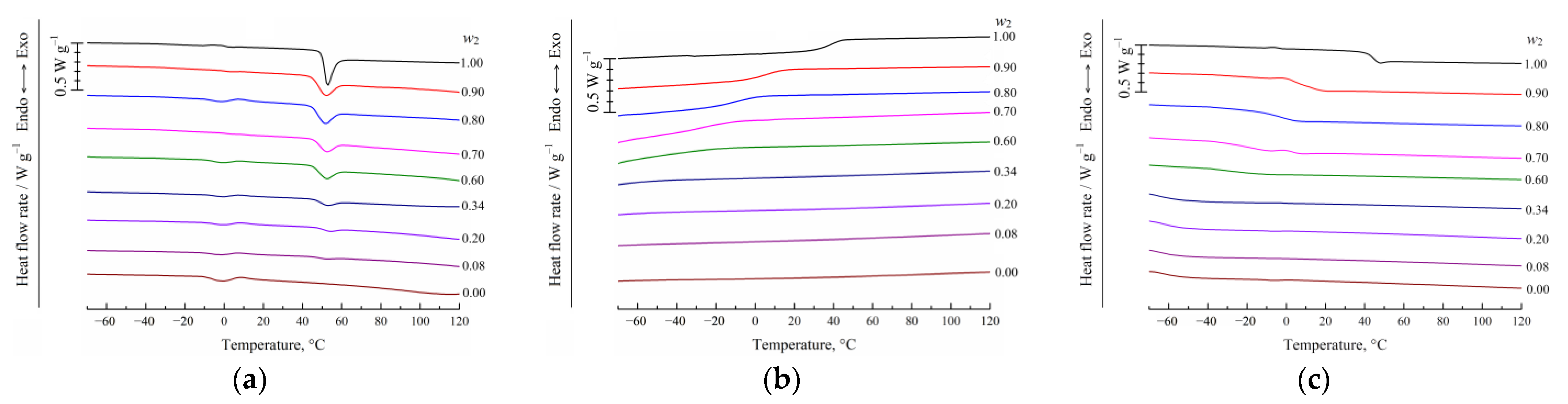
2.2.2. Differential Scanning Calorimetry (DSC) of the Binary PLGA/TG Mixtures
2.2.3. Studying the Evolution of the Structures Formed from PLGA/TG Mixtures upon Their Contact with a Nonsolvent
2.2.4. Methodology for Obtaining Fibers
2.2.5. Studying the Morphology of the Polymer Structures and Fibers Being Formed
3. Results and Discussion
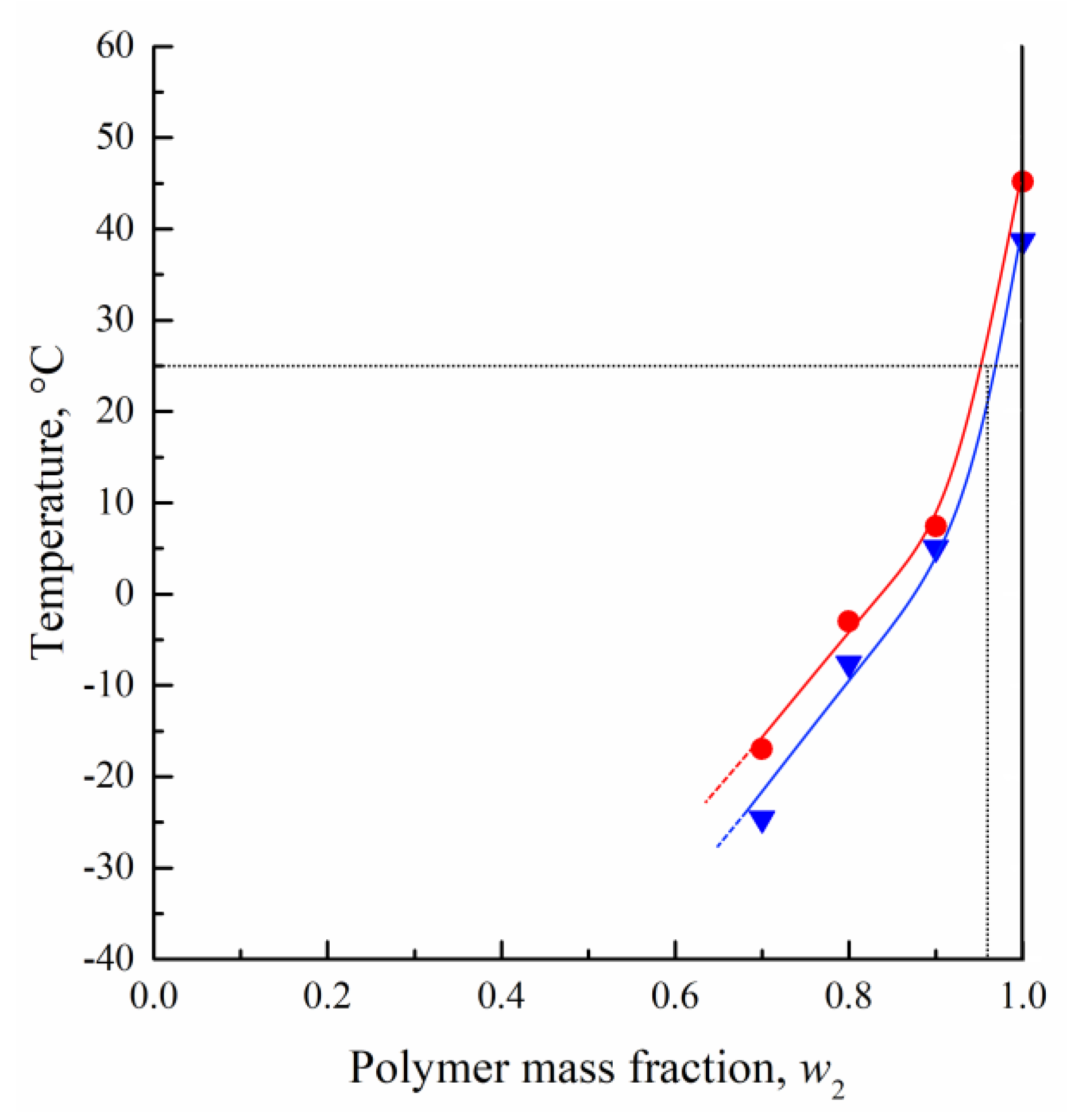
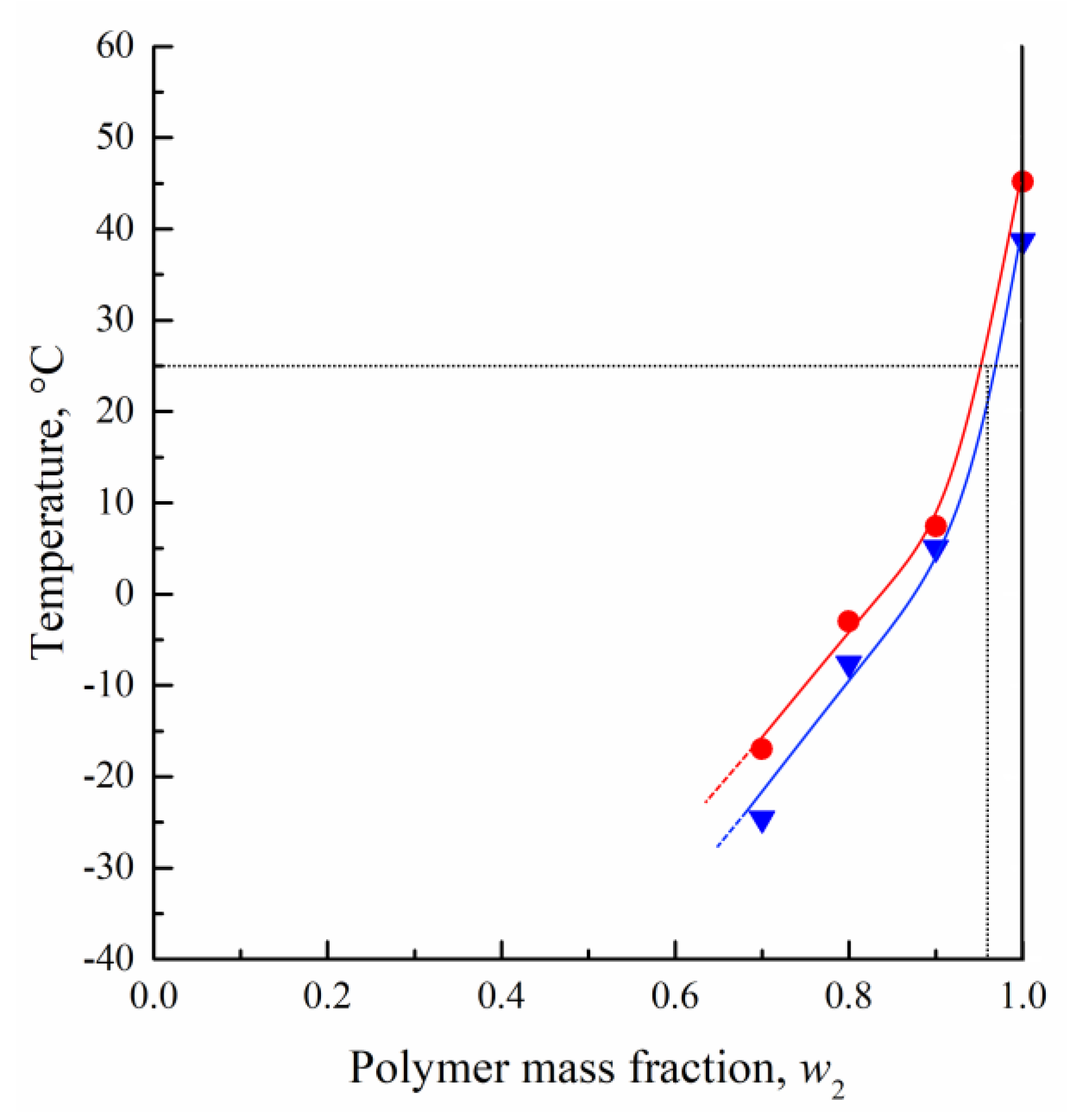
3.1. DSC Studies of PLGA/TG Mixtures
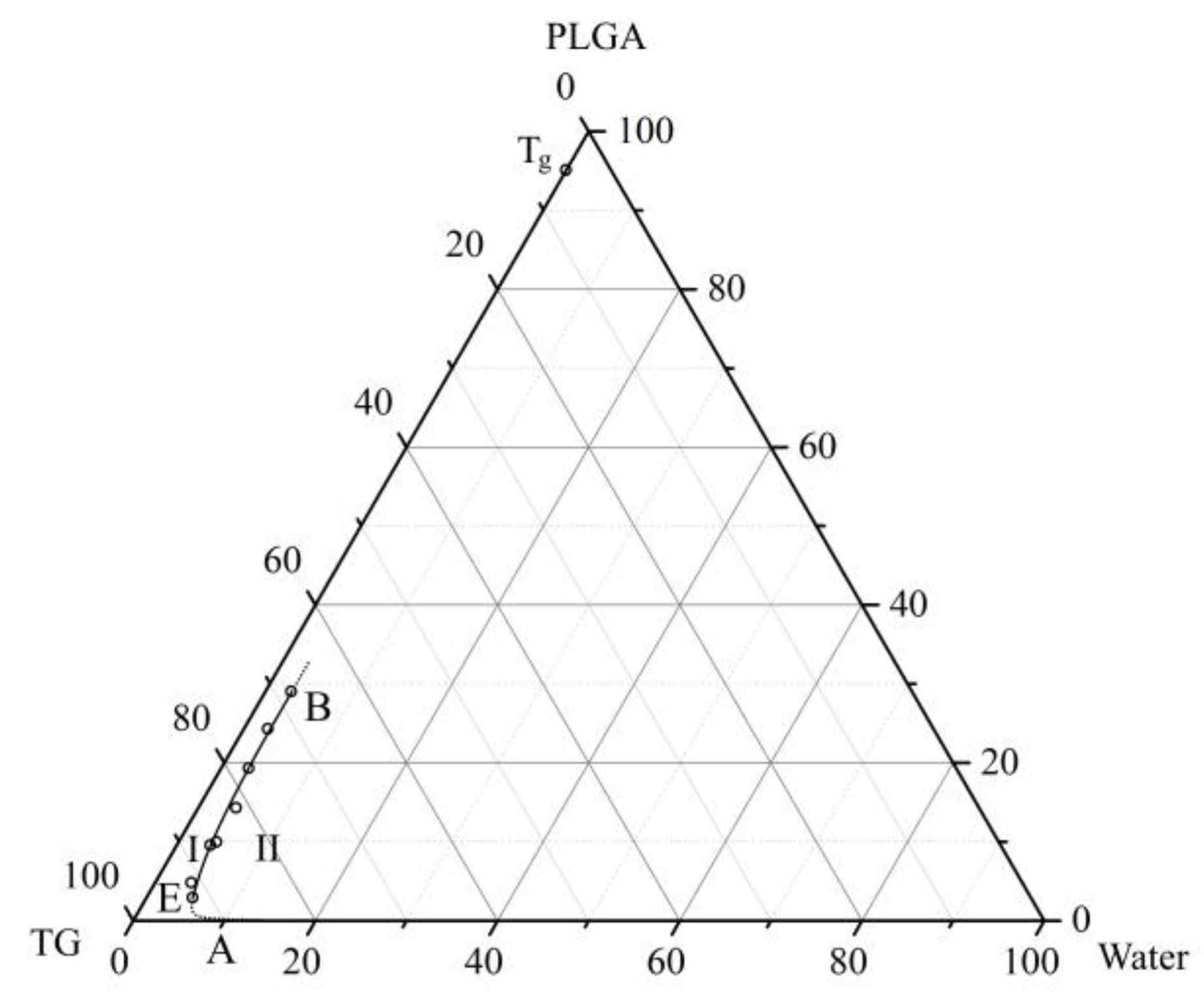
3.2. Phase Diagram of the Ternary PLGA/TG/Water Mixture
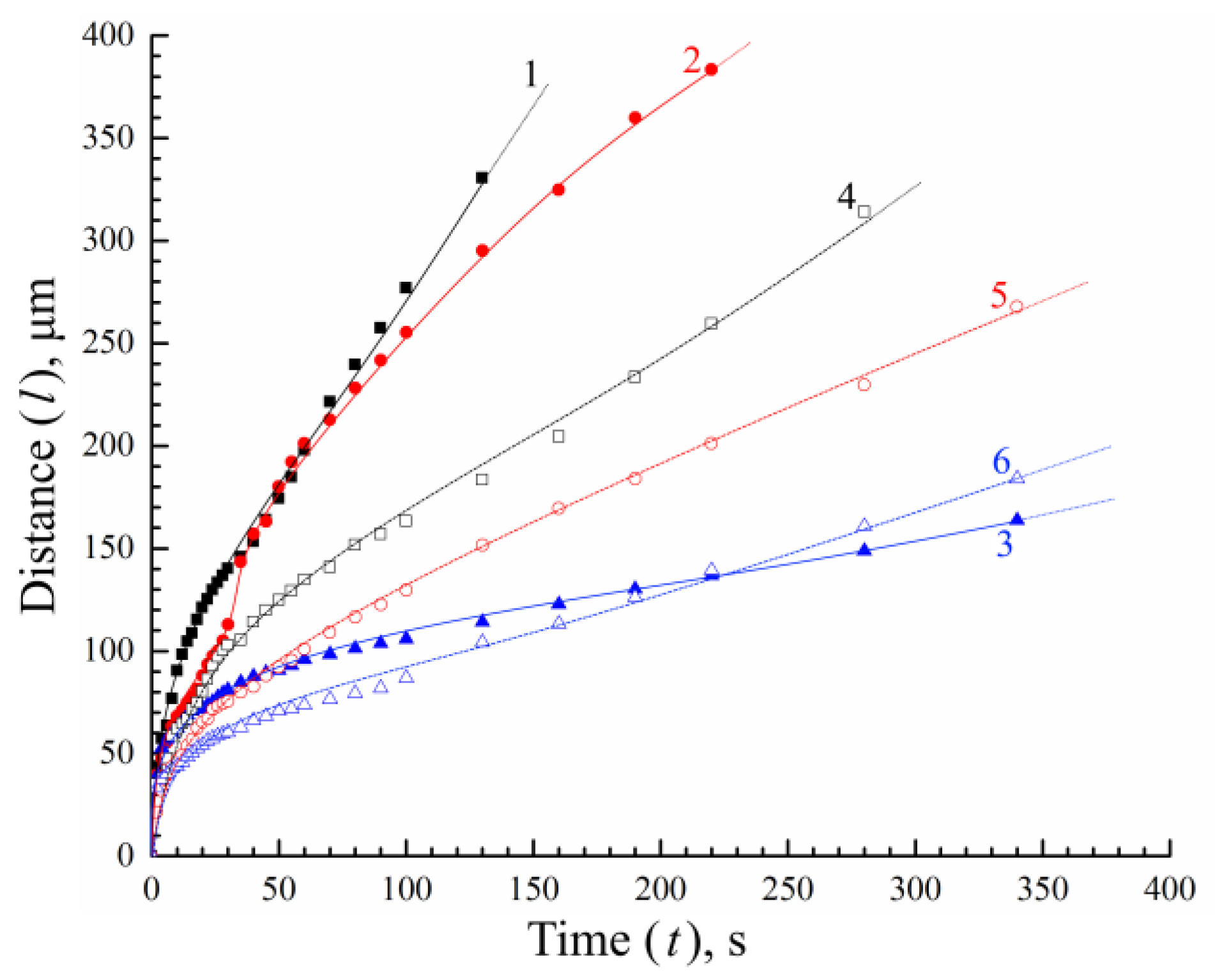
3.3. Structure Formation in PLGA/TG Mixtures Induced by Nonsolvents Differing in Activity
- When the amount of PLGA in the structure-forming mixture is increased, as well as when water is replaced with a mixed nonsolvent (i.e., the activity of the nonsolvent is reduced), the rate of structure formation tends to decrease, with the rate of progress of the structure formation front being more susceptible to the polymer concentration than to the precipitators’ virtue.
- The lowering of the rate of progress of the structure formation front in the case of the application of the mixed nonsolvent is accompanied by the slowing down of the tempo of the growth of the viscosity of the layer in contact with the fluid. A decrease in the rate of structure formation and a change in the volumetric distribution of the resulting phases with an increase in polymer concentration is associated with an increase in the viscosity of the mixture under study, which varies from ~1.0 Pa·s to more than 50 Pa·s. [26]. This gives us reason to believe that use can be made, in antisolvent 3D printing, of more concentrated structure-forming solutions, compared to those previously used [26] by us. This, in particular, will allow for improving the strength of the finished products and reducing the porosity of their components (fibers). That this supposition is valid is supported by the SEM photographs of the surface and the cleavage of a fiber obtained from a PLGA/TG mixture containing 30 wt.% PLGA (Figure 8) in a bath of a mixed nonsolvent.
- The intersection of curves 4 and 5 at the 50th second, as well as that of curves 5 and 6 at the 220th second, indicates that, prior to the intersection, the rate of progress of the structure formation front is governed largely by the driving force of the mass-exchange process, which weakens when water is replaced with a mixed nonsolvent, while after the intersection, the barrier properties of the surface layer are more expressed in the case where the polymer solution is immersed in water (Figure 5).
- The results of our “optical” experiments using nonsolvents stained with Grade E133 dye show that it takes around 60 s for the concentrations of the LM components in the nonsolvent bath and in the pores of the structure being formed to equalize (although the structure formation front proper progresses deep into the sample rather rapidly—at an average speed of the order of a few microns per second). This means that though PLGA fibers are formed in the antisolvent 3D-printing process quite quickly, they should be kept in the nonsolvent bath long enough to free them completely from TG.
3.4. Structure Formation in Amorphous Polymer/Solvent/Nonsolvent Mixtures via NIPS
- In Figure 5a,b, one can see that the growth front of the finger-like pores outpaces the clouding front that reflexes the microscopic phase separation of the ternary mixture. This means that the formation of the finger-like pores cannot stem from the coalescence of the drops of the polymer-poor phase already in existence.
- It is evident from Figure 5a,b that with the rates of progress of the structure formation front being nearly the same and relatively high, different structures, in principle, can form.
- In Figure 5a, it is seen that the structure formation process in the ternary mixture in hand continues even after the “fingers” have stopped growing. This means that the composition of the ternary mixture in the layer following that where the “fingers” have stopped growing just reaches that which corresponds to the liquid–liquid equilibrium binodal on which microscopic liquid–liquid phase separation takes place and not that which conforms to the fixing of the polymer.
3.5. Morphology of Fibers Formed from PLGA/TG Mixtures via NIPS
4. Conclusions
- A phase diagram of the above mixture at room temperature containing only a single boundary line—the liquid–liquid equilibrium binodal—is constructed for the first time.
- Using the DSC analysis technique, the PLGA/TG mixture composition, with which the polymer turns glassy at room temperature, is determined. The point corresponding to this composition (~95 wt.% PLGA + 5 wt.% TG) is plotted on the phase diagram.
- Using an improved methodology, the evolution of the structure of PLGA/TG mixtures containing 10, 20, and 30 wt.% PLGA immersed either in pure water or in a mixed water/TG (50:50) nonsolvent is studied for the first time. It is found that as the polymer content of the structure-forming mixture is increased, the structure formed transforms from a matrix with finger-like pores concentrated near its surface layer to matrices wherein such pores are located at some distance from this layer and, finally, to structures altogether devoid of such pores. Where use is made of the mixed nonsolvent instead of pure water, mixtures containing over 25 wt.% PLGA develop the only type of structure—a spongy (interconnected) structure—free from “fingers”.
- A new structure formation mechanism of the mixtures under study is suggested, according to which the architectonics of the structure formed as a consequence of the microscopic liquid–liquid phase separation of the PLGA/TG/water mixture depends on the barrier properties (morphology) of the surface layer that comes into existence at the interface between the contacting phases, with these properties being, in turn, dependent on the thermodynamic affinity of the solvent for the nonsolvent and the polymer content of the structure-forming mixture.
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Adamkiewicz, M.; Rubinsky, B. Cryogenic 3D printing for tissue engineering. Cryobiology 2015, 71, 518–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamali, A.; Shamloo, A. Fabrication and evaluation of a bilayer hydrogel-electrospinning scaffold prepared by the freeze-gelation method. J. Biomech. 2020, 98, 109466. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, D.R.; Kim, S.S.; Kinzer, K.E. Microporous membrane formation via thermally-induced phase separation. II. Liquid-liquid phase separation. J. Membr. Sci. 1991, 64, 1–11. [Google Scholar] [CrossRef]
- Nam, Y.S.; Park, T.G. Porous biodegradable polymeric scaffolds prepared by thermally induced phase separation. J. Biomed. Mater. Res. 1999, 47, 8–17. [Google Scholar] [CrossRef]
- Ji, G.L.; Du, C.H.; Zhu, B.K.; Xu, Y.Y. Preparation of porous PVDF membrane via thermally induced phase separation with diluent mixture of DBP and DEHP. J. Appl. Polym. Sci. 2007, 105, 1496–1502. [Google Scholar] [CrossRef]
- Xu, K.; Cai, Y.; Hassankiadeh, N.T.; Cheng, Y.; Li, X.; Wang, X.; Wang, Z.; Drioli, E.; Cui, Z. ECTFE membrane fabrication via TIPS method using ATBC diluent for vacuum membrane distillation. Desalination 2019, 456, 13–22. [Google Scholar] [CrossRef]
- Pochivalov, K.V.; Basko, A.V.; Lebedeva, T.N.; Ilyasova, A.N.; Yurov, M.Y.; Golovanov, R.Y.; Artemov, V.V.; Volkov, V.V.; Volkov, A.V.; Kudryavtsev, Y.V. Thermally induced phase separation in semicrystalline polymer solutions: How does the porous structure actually arise? Mater. Today Commun. 2021, 28, 102558. [Google Scholar] [CrossRef]
- Sun, H.; Zhao, Q.; Zheng, L.W.; Lu, W.W.; Wang, M. Cell-Incorporated Bioactive Tissue Engineering Scaffolds made by Concurrent Cell Electrospinning and Emulsion Electrospinning. Nano Life 2021, 11, 2141005. [Google Scholar] [CrossRef]
- Wang, H.H.; Jung, J.T.; Kim, J.F.; Kim, S.; Drioli, E.; Lee, Y.M. A novel green solvent alternative for polymeric membrane preparation via nonsolvent-induced phase separation (NIPS). J. Membr. Sci. 2019, 574, 44–54. [Google Scholar] [CrossRef]
- Xin, Y.; Fujimoto, T.; Uyama, H. Facile fabrication of polycarbonate monolith by non-solvent induced phase separation method. Polymer 2012, 53, 2847–2853. [Google Scholar] [CrossRef]
- Wijmans, J.G.; Kant, J.; Mulder, M.H.V.; Smolders, C.A. Phase separation phenomena in solutions of polysulfone in mixtures of a solvent and a nonsolvent: Relationship with membrane formation. Polymer 1985, 26, 1539–1545. [Google Scholar] [CrossRef] [Green Version]
- Kahrs, C.; Schwellenbach, J. Membrane formation via non-solvent induced phase separation using sustainable solvents: A comparative study. Polymer 2020, 186, 122071. [Google Scholar] [CrossRef]
- Kahrs, C.; Gühlstorf, T.; Schwellenbach, J. Influences of different preparation variables on polymeric membrane formation via nonsolvent induced phase separation. J. Appl. Polym. Sci. 2020, 137, 48852. [Google Scholar] [CrossRef] [Green Version]
- Kuo, C.Y.; Lin, H.N.; Tsai, H.A.; Wang, D.M.; Lai, J.Y. Fabrication of a high hydrophobic PVDF membrane via nonsolvent induced phase separation. Desalination 2008, 233, 40–47. [Google Scholar] [CrossRef]
- Zhao, J.; Chong, J.Y.; Shi, L.; Wang, R. Explorations of combined nonsolvent and thermally induced phase separation (N-TIPS) method for fabricating novel PVDF hollow fiber membranes using mixed diluents. J. Membr. Sci. 2019, 572, 210–222. [Google Scholar] [CrossRef]
- Wang, H.H.; Jung, J.T.; Kim, J.F.; Lee, J.; Kim, J.S.; Drioli, E.; Lee, Y.M. Tailoring nonsolvent-thermally induced phase separation (N-TIPS) effect using triple spinneret to fabricate high performance PVDF hollow fiber membranes. J. Membr. Sci. 2018, 559, 117–126. [Google Scholar] [CrossRef]
- Park, H.C.; Kim, Y.P.; Kim, H.Y.; Kang, Y.S. Membrane formation by water vapor induced phase inversion. J. Membr. Sci. 1999, 156, 169–178. [Google Scholar] [CrossRef]
- Mou, Z.L.; Zhao, L.J.; Zhang, Q.A.; Zhang, J.; Zhang, Z.Q. Preparation of porous PLGA/HA/collagen scaffolds with supercritical CO2 and application in osteoblast cell culture. J. Supercrit. Fluids 2011, 58, 398–406. [Google Scholar] [CrossRef]
- Yabuno, Y.; Mihara, K.; Miyagawa, N.; Komatsu, K.; Nakagawa, K.; Shintani, T.; Matsuyama, H.; Yoshioka, T. Preparation of polyamide–PVDF composite hollow fiber membranes with well-developed interconnected bicontinuous structure using high-temperature rapid NIPS for forward osmosis. J. Membr. Sci. 2020, 612, 118468. [Google Scholar] [CrossRef]
- Rezabeigi, E.; Wood-Adams, P.M.; Drew, R.A.L. Production of porous polylactic acid monoliths via nonsolvent induced phase separation. Polymer 2014, 55, 6743–6753. [Google Scholar] [CrossRef]
- Barzin, J.; Sadatnia, B. Correlation between macrovoid formation and the ternary phase diagram for polyethersulfone membranes prepared from two nearly similar solvents. J. Membr. Sci. 2008, 325, 92–97. [Google Scholar] [CrossRef]
- Fashandi, H.; Ghomi, A. Interplay of phase separation and physical gelation in morphology evolution within nanoporous fibers electrospun at high humidity atmosphere. Ind. Eng. Chem. Res. 2015, 54, 240–253. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, Z.; Zhao, L.; Yang, X.; Zhang, J.; Song, X.; Liu, B.; Ding, J. PEGylated stereocomplex polylactide coating of stent for upregulated biocompatibility and drug storage. Mater. Sci. Eng. C 2017, 81, 443–451. [Google Scholar] [CrossRef]
- Van De Witte, P.; Esselbrugge, H.; Dijkstra, P.J.; Van Den Berg, J.W.A.; Feijen, J. Phase transitions during membrane formation of polylactides. I. A morphological study of membranes obtained from the system polylactide-chloroform-methanol. J. Membr. Sci. 1996, 113, 223–236. [Google Scholar] [CrossRef] [Green Version]
- Sawalha, K.; Schroën, K.; Boom, R. Polylactide films formed by immersion precipitation: Effects of additives, nonsolvent, and temperature. J. Appl. Polym. Sci. 2007, 104, 959–971. [Google Scholar] [CrossRef]
- Mironov, A.V.; Mironova, O.A.; Syachina, M.A.; Popov, V.K. 3D printing of polylactic-co-glycolic acid fiber scaffolds using an antisolvent phase separation process. Polymer 2019, 182, 121845. [Google Scholar] [CrossRef]
- Martina, M.; Hutmacher, D.W. Biodegradable polymers applied in tissue engineering research: A review. Polym. Int. 2007, 56, 145–157. [Google Scholar] [CrossRef]
- Wang, D.; Li, K.; Teo, W.K. Phase Separation in Polyetherimide/Solvent/Nonsolvent Systems and Membrane Formation. J. Appl. Polym. Sci. 1999, 71, 1789–1796. [Google Scholar] [CrossRef]
- Müller, M.; Abetz, V. Nonequilibrium Processes in Polymer Membrane Formation: Theory and Experiment. Chem. Rev. 2021, 121, 14189–14231. [Google Scholar] [CrossRef]
- Bulte, A.M.W.; Mulder, M.H.V.; Smolders, C.A.; Strathmann, H. Diffusion induced phase separation with crystallizable nylons. I. Mass transfer processes for nylon 4,6. J. Membr. Sci. 1996, 121, 37–49. [Google Scholar] [CrossRef] [Green Version]
- Reuvers, A.J.; Smolders, C.A. Formation of membranes by means of immersion precipitation: Part II. the mechanism of formation of membranes prepared from the system cellulose acetate-acetone-water. J. Membr. Sci. 1987, 34, 67–86. [Google Scholar] [CrossRef] [Green Version]
- Smolders, C.A.; Reuvers, A.J.; Boom, R.M.; Wienk, I.M. Microstructures in phase-inversion membranes. Part 1. Formation of macrovoids. J. Membr. Sci. 1992, 73, 259–275. [Google Scholar] [CrossRef] [Green Version]
- Matz, R. The structure of cellulose acetate membranes 1. The development of porous structures in anisotropic membranes. Desalination 1972, 10, 1–15. [Google Scholar] [CrossRef]
- Guillen, G.R.; Ramon, G.Z.; Kavehpoor, H.P.; Kaner, R.B.; Hoek, E.M.V. Direct microscopic observation of membrane formation by non-solvent induced phase separation. J. Membr. Sci. 2013, 431, 212–220. [Google Scholar] [CrossRef]
- Hung, W.L.; Wang, D.M.; Lai, J.Y.; Chou, S.C. On the initiation of macrovoids in polymeric membranes–effect of polymer chain entanglement. J. Membr. Sci. 2016, 505, 70–81. [Google Scholar] [CrossRef]
- Kim, H.J.; Tyagi, R.K.; Fouda, A.E.; Ionasson, K. The kinetic study for asymmetric membrane formation via phase-inversion process. J. Appl. Polym. Sci. 1996, 62, 621–629. [Google Scholar] [CrossRef]
- Ren, J.; Li, Z.; Wong, F.S. Membrane structure control of BTDA-TDI/MDI (P84) co-polyimide asymmetric membranes by wetphase inversion process. J. Membr. Sci. 2004, 241, 305–314. [Google Scholar] [CrossRef]
- Yu, L.; Yang, F.; Xiang, M. Phase separation in a PSf/DMF/water system: A proposed mechanism for macrovoid formation. RSC Adv. 2014, 4, 42391–42402. [Google Scholar] [CrossRef]
- Lin, D.J.; Chang, H.H.; Chen, T.C.; Lee, Y.C.; Cheng, L.P. Formation of porous poly(vinylidene fluoride) membranes with symmetric or asymmetric morphology by immersion precipitation in the water/TEP/PVDF system. Eur. Polym. J. 2006, 42, 1581–1594. [Google Scholar] [CrossRef]
- Madaeni, S.S.; Bakhtiari, L. Thermodynamic-based predictions of membrane morphology in water/dimethylsulfoxide/polyethersulfone systems. Polymer 2012, 53, 4481–4488. [Google Scholar] [CrossRef]
- Mazinani, S.; Darvishmanesh, S.; Ehsanzadeh, A.; Van der Bruggen, B. Phase separation analysis of Extem/solvent/non-solvent systems and relation with membrane morphology. J. Memb. Sci. 2017, 526, 301–314. [Google Scholar] [CrossRef]
- Mironov, A.V.; Algebraistova, P.Y.; Komlev, V.S.; Mironova, O.A.; Popov, V.K. An Experimental Device for Studying the 3D Cryoprinting Processes. Instrum. Exp. Tech. 2020, 63, 890–892. [Google Scholar] [CrossRef]
- Lee, S.C.; Il Han, J.; Heo, J.W. Endotherm just above glass transition in uniaxially drawn poly(lactic acid)s films with various d-isomer contents. Polymer 2013, 54, 3624–3632. [Google Scholar] [CrossRef]
- Reich, G. Use of DSC to Study the Degradation Behavior of PLA and PLGA Microparticles. Drug Dev. Ind. Pharm. 1997, 23, 1177–1189. [Google Scholar] [CrossRef]
- Orofino, T.A.; Flory, P.J. Relationship of the Second Virial Coefficient to Polymer Chain Dimensions and Interaction Parameters. J. Chem. Phys. 2004, 26, 1067. [Google Scholar] [CrossRef]
- Surdutovich, L.I.; Tager, A.A.; Ovchinnikova, G.P.; Khomyakova, N.I.; Safonov, Y.A. An investigation of the swelling pressures of three-dimensional polymers. Polym. Sci. U.S.S.R. 1972, 14, 360–370. [Google Scholar] [CrossRef]
- Hildebrand, J.H. An Irregularity in the Solvent Powers of Paraffins. J. Chem. Phys. 2004, 18, 1337. [Google Scholar] [CrossRef]
- Hansen, C.M. The universality of the solubility parameter. Ind. Eng. Chem. Prod. Res. Dev. 1969, 8, 2–11. [Google Scholar] [CrossRef]
- Ahmad, H.; Yaseen, M. Application of a chemical group contribution technique for calculating solubility parameters of polymers. Polym. Eng. Sci. 1979, 19, 858–863. [Google Scholar] [CrossRef]
- Just, S.; Sievert, F.; Thommes, M.; Breitkreutz, J. Improved group contribution parameter set for the application of solubility parameters to melt extrusion. Eur. J. Pharm. Biopharm. 2013, 85, 1191–1199. [Google Scholar] [CrossRef]
- Stefanis, E.; Panayiotou, C. Prediction of Hansen Solubility Parameters with a New Group-Contribution Method. Int. J. Thermophys. 2008, 292, 568–585. [Google Scholar] [CrossRef]
- Mironov, A.V.; Grigoryev, A.M.; Krotova, L.I.; Skaletsky, N.N.; Popov, V.K.; Sevastianov, V.I. 3D printing of PLGA scaffolds for tissue engineering. J. Biomed. Mater. Res. Part A 2017, 105, 104–109. [Google Scholar] [CrossRef]
- Saljoughi, E.; Sadrzadeh, M.; Mohammadi, T. Effect of preparation variables on morphology and pure water permeation flux through asymmetric cellulose acetate membranes. J. Memb. Sci. 2009, 326, 627–634. [Google Scholar] [CrossRef]
- Ariono, D.; Aryanti, P.T.P.; Subagjo, S.; Wenten, I.G. The effect of polymer concentration on flux stability of polysulfone membrane. AIP Conf. Proc. 2017, 1788, 030048. [Google Scholar] [CrossRef] [Green Version]
- Rezaei, M.; Ismail, A.F.; Hashemifard, S.A.; Matsuura, T. Preparation and characterization of PVDF-montmorillonite mixed matrix hollow fiber membrane for gas–liquid contacting process. Chem. Eng. Res. Des. 2014, 92, 2449–2460. [Google Scholar] [CrossRef]
- Xing, Q.; Dong, X.; Li, R.; Yang, H.; Han, C.C.; Wang, D. Morphology and performance control of PLLA-based porous membranes by phase separation. Polymer 2013, 54, 5965–5973. [Google Scholar] [CrossRef]
- Rabiee, H.; Farahani, M.H.D.A.; Vatanpour, V. Preparation and characterization of emulsion poly(vinyl chloride) (EPVC)/TiO2 nanocomposite ultrafiltration membrane. J. Membr. Sci. 2014, 472, 185–193. [Google Scholar] [CrossRef]
- Emadzadeh, D.; Lau, W.J.; Matsuura, T.; Rahbari-Sisakht, M.; Ismail, A.F. A novel thin film composite forward osmosis membrane prepared from PSf–TiO2 nanocomposite substrate for water desalination. Chem. Eng. J. 2014, 237, 70–80. [Google Scholar] [CrossRef]
- Blanco, J.F.; Sublet, J.; Nguyen, Q.T.; Schaetzel, P. Formation and morphology studies of diferent polysulfones-based membranes made by wet phase inversion process. J. Membr. Sci. 2006, 283, 27–37. [Google Scholar] [CrossRef]
- Huang, X. Cellular porous polyvinylidene fluoride composite membranes for lithium-ion batteries. J. Solid State Electrochem. 2013, 17, 591–597. [Google Scholar] [CrossRef]
- Stropnik, R.; Kaiser, V. Polymeric membranes preparation by wet phase separation: Mechanisms and elementary processes. Desalination 2002, 145, 1–10. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, Z.; Wu, Z. Effects of solvent compositions on physicochemical properties and anti-fouling ability of PVDF microfiltration membranes for wastewater treatment. Desalination 2012, 297, 79–86. [Google Scholar] [CrossRef]
- Zhang, Y.; Shao, H.; Wu, C.; Hu, X. Formation and Characterization of Cellulose Membranes from N-Methylmorpholine-N-oxide Solution. Macromol. Biosci. 2001, 1, 141–148. [Google Scholar] [CrossRef]
- Mohamed, M.A.; Salleh, W.N.W.; Jafaar, J.; Ismail, A.F.; Mutalib, M.A.; Jamil, S.M. Feasibility of recycled newspaper as cellulose source for regenerated cellulose membrane fabrication. J. Appl. Polym. Sci. 2015, 132, 42684. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substance | δd, MPa0.5 | δp, MPa0.5 | δH, MPa0.5 |
|---|---|---|---|
| PLGA | 17.3 | 10.1 | 8.4 |
| TG | 17.8 | 10.0 | 12.7 |
| Water | 15.5 | 16.0 | 42.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Algebraistova, P.Y.; Basko, A.V.; Ilyasova, A.N.; Lebedeva, T.N.; Mironov, A.V.; Pochivalov, K.V.; Popov, V.K. Phase Equilibria and Structure Formation in the Polylactic-co-Glycolic Acid/Tetraglycol/Water Ternary System. Polymers 2023, 15, 1281. https://doi.org/10.3390/polym15051281
Algebraistova PY, Basko AV, Ilyasova AN, Lebedeva TN, Mironov AV, Pochivalov KV, Popov VK. Phase Equilibria and Structure Formation in the Polylactic-co-Glycolic Acid/Tetraglycol/Water Ternary System. Polymers. 2023; 15(5):1281. https://doi.org/10.3390/polym15051281
Chicago/Turabian StyleAlgebraistova, Polina Yu., Andrey V. Basko, Anna N. Ilyasova, Tatyana N. Lebedeva, Anton V. Mironov, Konstantin V. Pochivalov, and Vladimir K. Popov. 2023. "Phase Equilibria and Structure Formation in the Polylactic-co-Glycolic Acid/Tetraglycol/Water Ternary System" Polymers 15, no. 5: 1281. https://doi.org/10.3390/polym15051281