Water Soluble Responsive Polymer Brushes

Abstract
: Responsive polymer brushes possess many interesting properties that enable them to control a range of important interfacial behaviours, including adhesion, wettability, surface adsorption, friction, flow and motility. The ability to design a macromolecular response to a wide variety of external stimuli makes polymer brushes an exciting class of functional materials, and has been made possible by advances in modern controlled polymerization techniques. In this review we discuss the physics of polymer brush response along with a summary of the techniques used in their synthesis. We then review the various stimuli that can be used to switch brush conformation; temperature, solvent quality, pH and ionic strength as well as the relatively new area of electric field actuation We discuss examples of devices that utilise brush conformational change, before highlighting other potential applications of responsive brushes in real world devices.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction. Why Polymer Brushes? The Physics of Brush Response
Polymer brushes are formed when polymer chains are tethered at one end to a surface at sufficient density to overlap and stretch away from the surface. These nanoscale surface layers are “smart”, i.e., their properties can be switched in response to external triggers [1], and are prime candidate materials for soft nanotechnology [2,3]. Polymer brushes exhibit conformational changes when environmental properties such as temperature or pH are varied, allowing the properties of the surface to be remotely tuned. Polymer brushes are truly biomimetic in the sense that they exploit conformational changes in macromolecules in order to produce useful effects, and are increasingly recognized as being biocompatible due to the tendency of some brushes to resist protein adsorption.
Owing to a large body of research into surface-initiated polymerization during the last two decades, robust and intricately designed polymer brush layers may be grown from a diverse range of surfaces. The subject of polymer brush research has already been the subject of numerous detailed reviews [4–10], demonstrating fundamental research into their synthesis and characterization as well as a number of applications including their use as switchable surfaces, non-fouling coatings, sensors and actuators. In this review, we will give a wide-ranging introduction to this large topic before focusing on a developing area of responsive polymer brush research: the use of applied voltages to achieve remote control of polymer brush structure.
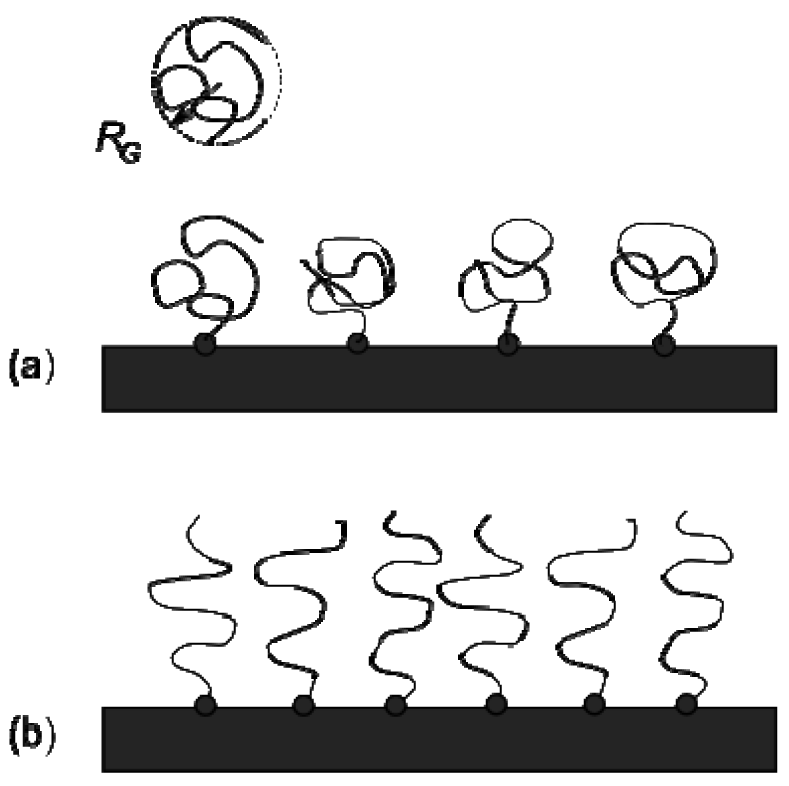
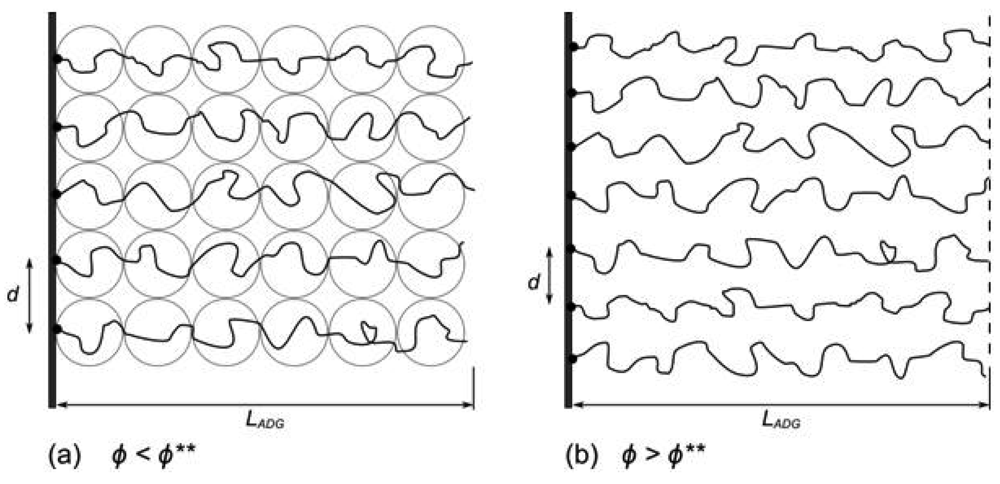
When polymer chains are grafted at one end to a surface at sufficient density to allow the chains to overlap, they stretch away from the surface into a brush-like conformation [11]. When free in solution, the polymer chain is in the coil conformation and has a size characterised by its radius of gyration, RG. When chains are grafted to the surface, the distance between grafting points is referred to as the grafting distance d, and the number of chains per unit area is the grafting density σg = d−2 (measured as the number of chains per nm2, however care must be taken in the literature as σ is sometimes used as the area per grafted chain [12]). At low grafting density, where RG << d, the chains are isolated upon the surface and adopt the so-called mushroom regime [13] (Figure 1). At grafting densities where d becomes comparable to RG, neighbouring polymer chains begin to overlap and are forced to stretch away from the surface due to repulsive excluded volume interactions. The equilibrium height of the polymer brush layer is dictated by the competition between the stretching of chains away from the surface and the entropic elasticity that resists such stretching.
As the grafting density increases, and the grafting distance decreases, the polymer molecules are increasingly stretched away from the surface in a brush-like equilibrium conformation. Unlike the bristles of a sweeping brush, whose stiffness is the dominant factor in keeping them perpendicular to the grafting surface, in a polymer brush it is the repulsive interactions between neighbouring chains that keep them stretched away from the surface, while the polymer molecules remain flexible on length scales comparable to the thickness of the brush (∼10 nm). In neutral polymer brushes, the monomer-monomer excluded volume interactions are responsible for the repulsive interactions. In polyelectrolyte brushes, where electrical charges are present upon the polymer chains, electrostatic interactions become important either directly through repulsive interactions between charged monomers, or indirectly since they dictate the distribution and hence the osmotic pressure of the counterion cloud which remains bound to the brush to preserve electroneutrality.
1.1. Alexander—de Gennes Brush Model
The simplest model of a polymer brush assumes a step-function volume fraction profile, comprising monodisperse chains stretching away from the surface with each chain end located the same distance away from the grafting surface giving a layer thickness L, every chain having the same degree of stretching. Alexander and De Gennes refined this into the blob model for a polymer brush [14,15] which divides the chains of the brush into a series of blobs as depicted in Figure 2(a). Assuming that the distance between the grafted chains d dictates the dimension of each independent blob of polymer, and that the brush is in the semi-dilute regime in a good solvent a relation between the brush height L, the monomer size a, and the grafting density σg, is derived (Equation (1))
The relation in Equation (1) describes the very interesting point about polymer brushes, which is that they have a linear dependence of brush height L with N. This is very different from the result for free chains, in which excluded volume interactions result in . It also tells us that grafted brush layers are stretched, and that this is the origin of their interesting behaviour.
2. Attaching Brushes to a Surface or an Interface
There are many useful texts that describe some of the synthesis routes to attaching polymers to a surface [8,16–18]. The easiest way to attach a polymer to a surface or an interface is to have one end-group that will preferentially attach to the surface. This has many advantages as well as severe limits on the maximal grafting density of the resulting grafted brushes, these constraints are discussed later. Principally the thickness of the brushes is fixed to a very narrow regime, when the brushes are synthesized using the grafted to route.
2.1. Brushes via “Grafting To”
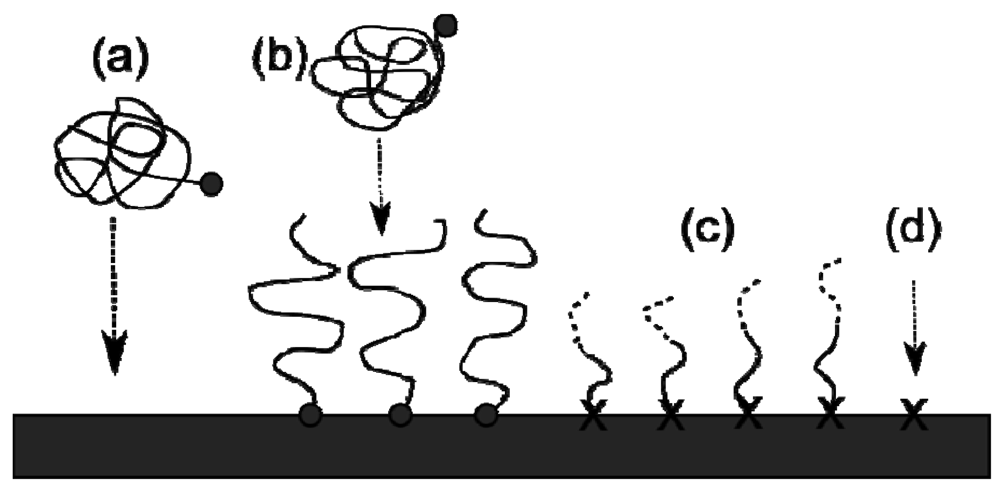
The most straightforward mechanism of making a grafted polymer brush layer is the “grafting to” route, as the attachment is from an end-functionalised group, e.g., a thiol to a gold substrate as shown in Figure 3(a,b). The synthesis of the chain is carried out before attachment, making it a simple self-assembly route, although the grafting density (number of chains per unit area) is lower for this technique and the resulting thickness is low, as the surface attachment becomes more difficult due to steric effects.
Increasing the solution concentration will increase the adsorbed amount to a point until a non-grafted wetting layer is formed on top of the end-adsorbed grafted layer [15]. It has been shown that judicious use of excess non-functionalized homopolymer can increase the maximum grafting density [19]. The great advantage of the grafting to method is that extensive characterisation of an existing batch of polymer can take place before it is attached to the surface, meaning that accurate knowledge of molecular weight and polydispersity is possible without the need to first remove polymer from the surface.
2.2. Brushes via “Grafting From”
The “grafting from” route [20–23] uses a self-assembled monolayer (SAM) to initiate a living polymerisation generating the brush as it “grows” from the substrate. As the chains are growing from the surface, the only limit to the chain growth is the diffusion of the monomer to the growing ends. This makes “grafting from” the only way to make thick brushes with high grafting densities. Comparable grafting densities can be achieved using Langmuir-Blodgett methodology but this can restrict the type of brush grown and in practice is not as reliable at producing homogeneous brushes over large surface areas. The two main planar substrates used are gold [24] and silicon [20] as these allow for thiol and trichlorosilane attachable groups, respectively. Both of these functional groups have been used extensively as surface anchoring groups. Section 2.4 focuses on Atom Transfer Radical Polymerisation, which is one technique commonly used in such surface-initiated polymerisations.
2.3. Is Polydispersity Important?
Control over polydispersity (or at least the ability to measure polydispersity) is important in understanding the system under study. An accurate description of a polymer brush (in addition to the chemical structure of the monomer residues) includes information about its grafting density, molecular weight and polydispersity. Early experiments in surface-initiated polymerisation valued control over polydispersity because this allowed the production of well-characterised systems (particularly when traditional grafting-to brushes were from existing batches of polymer that could be extensively characterised) that could be adequately compared to the simple scaling theories. Highly controlled polydispersity, however, is not necessarily required in order for a polymer brush to have functionality.
2.4. Atom Transfer Radical Polymerization (ATRP)
“Living” Polymerisation methods are important for the synthesis of well-defined polymeric materials. Living, or “controlled”, polymerizations [25] have distinct advantages over conventional Radical Polymerization. Conventional Radical Polymerization is a simple, inexpensive method of creating polymers, at the cost of control over a polymer's polydispersity (molecular weight distribution) and architecture (structure). It clearly follows that in order to design and build molecular structures a degree of control is required in the synthesis. One such living polymerization method offering this control is Atom Transfer Radical Polymerization. ATRP is a relatively modern technique having only been developed in the 1990s [26,27], but has rapidly expanded in the past few years [28]. Excellent reviews on the mechanism and the developments in ATRP have been published [21,28,29] to which the reader is directed for a more in-depth discussion.
During living radical polymerization, the propagation proceeds with irreversible chain transfer in the absence of termination until all of the monomer is consumed. This gives good control of the polydispersity and therefore allows the pre-determination of molecular weights using stoichiometry, as (at least in the idealised picture of a polymer brush) all the polymer chain lengths should be approximately the same. The monomer sequentially attaches to the radical chain end, transferring the radical across to the “new” terminal carbon. Whilst there is still monomer present, the polymer will continue to grow as the halide moves back and forth between the species; this is the key step, and gives rise to the term “atom transfer”. This chemistry can also be used when an initiator is tethered to a silicon substrate to create a grafted polymer brush. The mechanism is identical, with the only difference being that initiation occurs on a fixed base from which the polymer chain grows [30]. The native silicon oxide surface (between 15–20 Å) is coated with a monolayer of a chlorosilane-terminated to form an ATRP initiator (e.g., 2-bromo-2-methyl-propionic acid 11-trichlorosilanyl-undecyl ester) [22,31], capable of releasing a halogen to create a free radical. The 11-trichlorosilane compound is hydrolysed by trace amounts of water, whereby HCl is liberated. The native oxide layer on the silicon surface is comprised of hydroxyl end-groups, which undergo a condensation reaction with the hydrolysed chlorosilane generating Si-O-Si linkages between the ATRP initiator and the silicon wafer. The silicon wafer is then placed within an ATRP reaction vessel, under normal ATRP conditions, to produce a polymer brush.
Polymerization techniques such as ATRP are under constant development, not only in order to produce better-controlled polymer brushes with tolerance to a wide range of monomer functionalities, but also to make the processes faster, easier, and cheaper. ARGET ATRP [32], for example, greatly relaxes the need to keep oxygen away from ATRP reagents by “mopping up” unwanted oxygen with a reducing agent. Electrochemically mediated ATRP (eATRP) allows the electrochemical control of reaction rates by tuning the oxidation states of the catalysts [33].
3. Temperature Responsive Brushes and PEG Brushes
One of the most commonly studied thermally responsive polymers is poly(N-isopropylacrylamide) (PNIPAM). The huge interest in this polymer is due to the temperature at which it changes conformation is similar to the temperature in a physiological environment. PNIPAM is a water soluble polymer that displays a lower critical solution temperature at around 32 °C in aqueous solution, and so undergoes a coil to globule transition above this critical temperature.
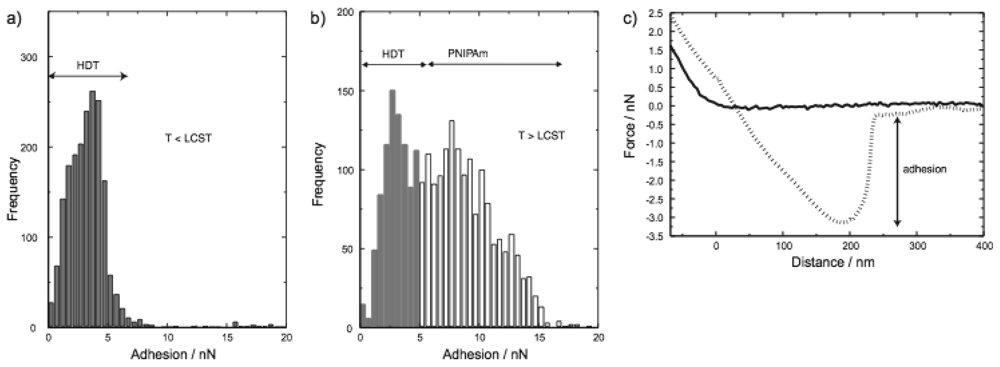
The potential for PNIPAM as a switchable surface was shown by Jones et al. [34] by performing a series of high resolution force microscopy measurements to measure the change in adhesion for a patterned PNIPAM polymer brush. Their work showed that their layers possess a reversible phase transition between hydrophilic and hydrophobic when traversing the LCST temperature. This can be clearly seen in Figure 4(c) where two example retraction curves are shown, one below and one above the LCST, with a large difference in adhesion.
Work by the Zauscher group also looked at the patterning and response of PNIPAM brushes and again used scanning probe techniques (SPM) to measure the response this time as a function of solvent conditions. They could switch the conformation of the brush chains from swollen in sodium chloride to collapsed in a 1:1 mixture of methanol and sodium chloride. Their SPM measurements showed that the swelling of their polymer brushes was confined mostly to the out of plane direction [35,36].
3.1. Poly(ethylene oxide) Brushes
Poly(ethylene oxide) or poly(ethylene glycol) (abbreviated to PEO and PEG respectively and essentially referring to the same polymer) brushes are widely studied due to their resistance to protein adsorption, although the nature of this resistance is poorly understood. Neutron reflectometry in combination with isotopic substitution of hydrogen for deuterium (“contrast variation”) may be used to distinguish the nature of adsorption of proteins in polymer brush layers, by building profiles of the polymer and protein in the direction normal to the grafting surface [37]. However, there are many difficulties in trying to understand the complexities and mechanisms that underlie protein resistance due to the lack of contrast between PEO and a hydrogenous protein. The field of protein resistant surfaces is very active; and whilst these materials cannot be described as responsive they can be combined with other polymers to enable the system to resist non-specific protein absorption that would over a short space of time in a biological environment cover the whole layer reducing the surface functionality of the overall film. For further background reading on this subject please refer to the following papers highlighted on this subject [38–41].
4. Polyelectrolyte Brushes: Response to Salt and pH
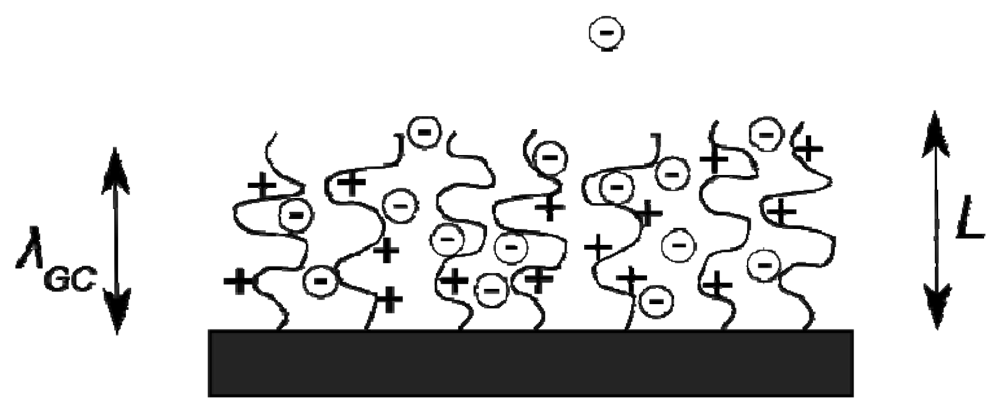
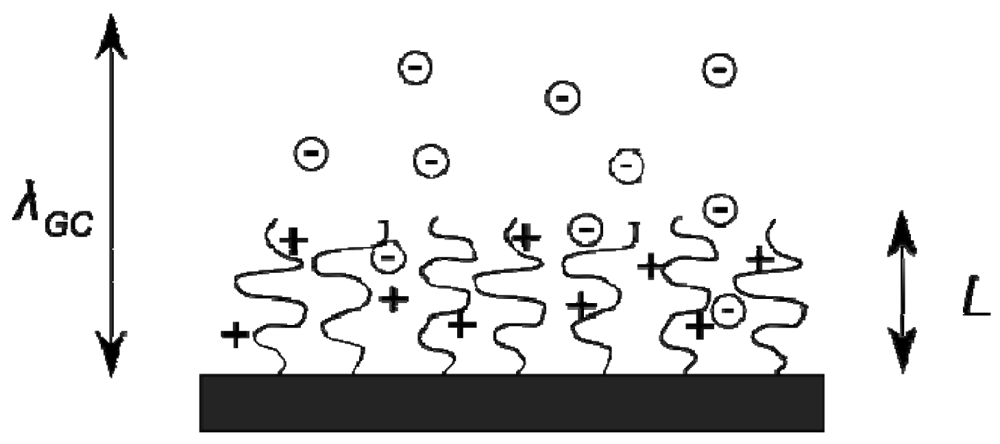
When a polymer brush is formed from strong or weak polyelectrolyte molecules, electrostatic interactions introduce a rich variety of behavioural regimes in addition to the neutral brush properties [18]. At typical values of grafting density (σg = 0.4 nm−2) and degree of polymerisation (N ∼ 250), even a modestly charged polymer brush has a significant surface charge density, and therefore binds a layer of counterions in the vicinity of the surface. The binding of the counterion layer is dependent upon the surface charge density and is characterised by the Gouy-Chapman length λGC [18], which describes the length scale within which the counterions are largely confined to the surface, see Figure 5. A brush of charge fraction α, grafting density σg and degree of polymerisation N has a surface charge density of αNeσg so that
The counterion binding length scale λGC decreases with increasing surface charge density, hence it is dependent upon the charge fraction, degree of polymerisation and grafting density of brushes. When λgc becomes comparable to the height of a charged polymer brush, counterions within the brush begin to exert an osmotic pressure upon the brush segments. The strength of this effect in comparison to the bare electrostatic repulsion between the charged monomers upon the brush is the key factor in the definition of the resulting regime behaviour.
4.1. The Various Regimes of Strong and Weak Polyelectrolyte Brushes
4.1.1. Neutral Brush
In the neutral brush regime the brush swelling is dictated by the equilibrium between the excluded volume of monomers and the entropic elasticity of the chains and the scaling relation is given by Equation 1. A partly charged brush may still behave as though is were completely neutral and the quasi-neutral brush regime occurs where there is a small amount of charge present, but not enough to dominate the excluded volume interactions.
4.1.2. Osmotic Brush
A strongly charged brush binds its counterions with λGC < L so that the entire counterion layer is effectively confined within the brush, as depicted in Figure 6, resulting in an osmotic pressure due to the increased concentration of counterions within the brush compared to the bulk. The situation where this osmotic pressure is so large that the bare electrostatic repulsion between the monomers may be neglected is known as the osmotic brush (OsB) regime. The regime is valid where the osmotic pressure of the counterions is so large that the excluded volume interactions are negligible [12]. In this regime the height of the brush layer is expected to vary as
An important feature of the osmotic brush regime is that the brush height is not dependent upon the grafting density.
For a weak polyelectrolyte brush (in the specific case of a weak polybase brush) the so-called annealed osmotic regime is described [12] by
The internal charge fraction of a brush in the annealed osmotic regime has complex dependencies upon the chemical environment. Firstly, the brush charge fraction is dependent upon pH leading to the unique pH-responsiveness. Also, at low salt concentrations, negatively charged salt ions may exchange with counter-ions, effectively changing pH of the brush and causing additional swelling. Therefore the scaling theory predicts the brush to swell at low salt concentrations before falling over to the SB regime as described below.
4.1.3. Salted Brush
The addition of salt at concentration CS to a brush-solvent system introduces electrostatic screening due to a decrease in the Debye screening length. The decrease in the Debye screening length reduces the range of the electrostatic interactions, causing them to behave more like short-range excluded volume interactions, and for high salt concentrations the height of the brush is expected to scale as
i.e., , where an increase in salt concentration causes a de-swelling, in the so-called salted brush (SB) regime. This scaling theory is valid for both weak and strong polyelectrolyte brushes when the salt concentration outside the brush is greater than the internal concentration of protons. Strong polyelectrolyte brushes are unaffected by smaller concentrations of salt, which have no effect on the concentration of protons inside the brush.
4.1.4. Charged or Pincus Brush
A moderately charged brush, i.e., a brush with λGC > L, will bind its counter-ions in a diffuse cloud that extends far beyond the physical limit of the brush, as depicted in Figure 7 In this case the osmotic contribution due to the counterions is negligible in comparison to the bare electrostatic repulsion of the charged monomers. This narrow theoretical regime is known as the charged brush or Pincus brush (PB) regime.
Simple calculations of the Gouy-Chapman counterion binding length show that the conditions required for the Charged brush regime are incredibly narrow. Using some parameters typical for an ATRP brush (dry thickness 20 nm, N = 250, σg = 0.4 nm−2 and lB = 0.7 nm), a value of α = 0.0003 is required for the Gouy-Chapman length to be comparable to the dry brush thickness (even less if it is to be compared to the swollen thickness). This is to say that even if 0.03% of the monomers were charged, such a brush would trap the entirety of its counterions, and so it would either be quasi-neutral or osmotic in its swelling behaviour. This is further impacted by the fact that due to the variation in degree of charge along weak polyelectrolyte chains there is almost always non-zero charge present upon a weak polyelectrolyte brush even above the bulk pKa of the free polymer [42]. In conclusion, to measure the Charged brush regime is rather a challenging experimental feat [43].
4.2. Polyelectrolyte Brush Experiments
The first theoretical descriptions of the properties of weak polyelectrolyte brushes that laid the foundation for the current understanding emerged in the 1990′s [12], but at this time it was still comparatively difficult to synthesize polyelectrolyte brushes. Early approaches employed block copolymers with hydrophilic charged blocks and hydrophobic anchoring blocks that would segregate to the surface in aqueous solutions to form a charged brush. One of the first examples of chemically grafted brushes in the literature appeared in 1995 [44]. A polystyrene brush was grafted via a chlorosilane linkage to a porous silica surface. The resulting brush was rendered a strong polyelectrolyte (poly(sodium-polystyrene sulfonate), PSSNa) using a sulfonation reaction.
A key work on the determination of the structure of polyelectrolyte brushes was published by Tran (1999) [45], where the PS-PSSNa synthesis route was again used. Ellipsometry was used to determine dry layer thicknesses, allowing the calculation of the grafting density from the known molecular weight of the polymer. Infrared spectroscopy was used to characterise the chemical structure of brush and neutron reflectivity in liquid (water/deuterated water depending on the contrast matching required) was used to characterise density profiles of both monomers and counterions in the brush. It was shown that the brush height was not dependent on grafting density—as is characteristic of the osmotic brush regime, and also that for fixed grafting density the brush height scaled as H ∼ N as expected for the osmotic regime. The counterion profile was shown to closely resemble the monomer density profile. For an electroneutral brush this is confirmation that the counterions are confined inside the brush, and exerting a strong osmotic pressure to counterbalance the conformational elasticity of the chains. The study also considered the addition of salt. For low salt concentrations (lower than the concentration of counterions inside the brush) there was no screening of electrostatic interactions within the brush and it thus stayed in the osmotic regime. However, higher salt concentrations were shown to decrease the brush height as a function of salt concentration to the power −1, which compares favourably to the scaling theory for both weak and strong polyelectrolyte brushes in the salted brush regime. It is interesting to note that at high salt concentrations (5 mol L−1 NaCl) the brush was not collapsed, such that it had expelled solvent and was close to its dry thickness, but was just as stretched as a neutral polymer brush in a good solvent.
Grafting from techniques (e.g., [46]) paved the way for well-characterized densely-grafted polymer brushes. In such techniques, a self assembled monolayer of initiator is laid down on the substrate, from which brushes are grown monomer by monomer, e.g., by anionic polymerisation or atom transfer radical polymerisation, a polymerisation technique capable of producing well-controlled brushes at relatively high grafting density (∼0.4 nm−2). Biesalski and Rühe [47] used ellipsometry to investigate poly(methacrylic acid) PMAA polyacid brushes. A variable-angle null-ellipsometer was used to measure the segment density profile as the pH of the sub-phase was changed. The brushes (dry height of 20 nm) were shown to be highly swollen using ellipsometry (hundreds of nanometres) even at very low pH 1.9 (a weak polyacid would be expected to collapse in acidic conditions). However, the density profile was strongly shifted out to a few micrometers as pH increased. This was understood to correspond to an increase in the charge fraction on the chains, increasing the electrostatic repulsion between monomers. Following on from Tran et al., Biesalski and Rühe [48] again used variable-angle ellipsometry. The thickness of PMAA brushes was studied as a function of pH, exhibiting the expected increase in thickness with increasing pH value.
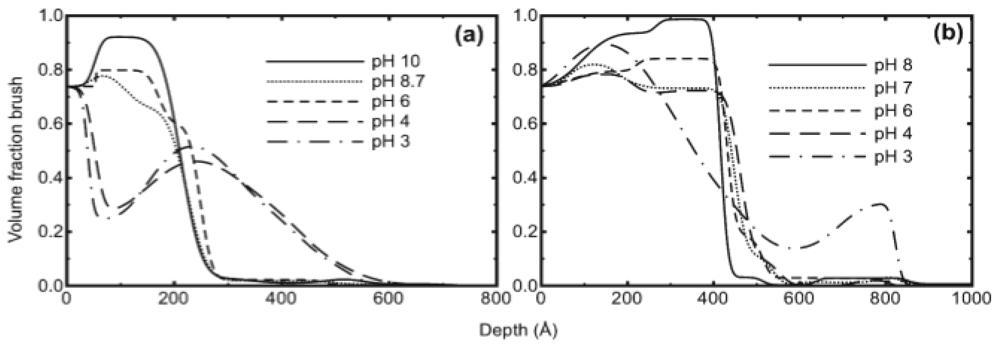
The salt added case was also studied displaying a power law increase of thickness with salt concentration of 1/3 for low salt concentration (and −1/3 at high concentration) as shown in Figure 8. This is a key piece of experimental evidence for the expected behaviour of weak polyelectrolyte brushes, showing the transition between the annealed osmotic brush and salted brush regimes, and strongly supports the validity of the proposed scaling theory. Geoghegan et al. [49] investigated pH-induced swelling of poly[2-(diethylamino)ethyl methacrylate] (PDEAEMA) weak polybase brushes grown using atom transfer radical polymerisation. Neutron reflectivity was used to investigate the polymer volume fraction profile of 13 and 27 nm brushes as a function of pH (Figure 9). Swelling of the brushes was measured as the pH was lowered and the brushes became ionized. A swelling of a factor of 2 between pH 8 and 3 was observed, which is interestingly much less than for the corresponding pH interval from the PMAA studies (e.g., [47]), probably due to the higher grafting density in this study. The form of the polymer volume fraction profiles is intriguing since no aspect of current polymer brush theory supports the notion of a non-monotonic brush profile, but the presence of entanglements could provide an explanation.
A recent study from Sanjuan et al. [50] used both PDMAEMA polybase brushes grown using ATRP, as well as PTMAEMA (poly(2-(trimethylamino)ethyl methacrylate) strong polyelectrolyte brushes produced by quaternisation of PDMAEMA samples. Ellipsometry and neutron reflectivity were carried out to measure the thickness of the brushes, expressed as a swelling ratio. To identify the extremes of the swelling behaviour of the samples, the PDMAEMA brushes were dissolved in methanol so that they essentially behaved as neutral brushes, and the PTMAEMA brushes dissolved in water to approximate the swelling state of a fully charged PDMAEMA brush.
The strong polyelectrolyte PTMAEMA is more swollen than the neutral case due to the presence of electrostatic effects. The degree of swelling in PDMAEMA and PTMAEMA brushes decreased as the grafting density was increased, in accordance with the respective power law exponents of −2/3 and −1 predicted [50] by scaling theory. Measurements on the PTMAEMA strong polybase brushes also illustrated the turnover between osmotic brush regime and salted brush regime as the salt concentration is increased. The thickness scales to the predicted power law exponent of −1, which was reproduced for samples of two different grafting densities. This also confirms the group's previous results for strong polyelectrolytes [45]. These vindications of the scaling approximations are particularly rewarding and difficult to obtain since real experimental systems rarely conform without ambiguity to the precisely defined regime behaviour. In the same study [50], the behaviour of PDMAEMA weak polybase brushes in response to changing pH was also presented, using data from both ellipsometry and neutron reflectivity. The brushes were fully swollen at low pH (at a size similar to that of the strong polyelectrolyte PTMAEMA in water). As the pH was increased the brush height decreased, tending towards the value for PDMAEMA in methanol (neutral polymer behaviour). Polyampholyte brushes, that is brushes with both polyacid and polybase groups appearing upon the same chain, have shown a larger pH response than homopolymer polybase brushes by responding at both extremes of the pH scale instead of just one [51]. Parnell et al. [52] measured the real-time swelling and collapse of a poly(methacrylic acid) weak polyacid brush using atomic force microscopy. Polymer brush material was removed from the silicon surface using a scratch from a scalpel blade, taking care not to damage the substrate. Contact mode AFM was used to measure the step height of the edge of the intact polymer layer as solutions of pH 10.5 and 3 were introduced to the sample environment. Figure 10 clearly shows the change in height of the polymer brush as it becomes swollen at high pH and relatively collapsed at low pH. The actuating timescales of responsive polymer brushes have implications for applications in particular for the area of microfluidics, where channels or pores could be restricted/actuated by the fast switching of responsive brushes to enable sorting of molecules.
The stimuli-responsive behaviour of polymers has been exploited on the micron length scale by grafting responsive polymer films or brushes to microcantilevers. Environmentally induced conformational changes in the polymer layers produce mechanical stresses and hence bending in the cantilevers. This bending is typically monitored using laser and photodiode in an AFM or similar apparatus. Again, the development of versatile surface-initiated radical polymerisation techniques made it possible to covalently graft polymer brush layers to microcantilevers [53], offering a new range of robust responsive coatings. These systems have been used to good effect as chemical sensors [54], and conversely as actuators [55].
Although this review focuses on solid/liquid interfaces, there is in interest brushes formed at the air/water interface by the use of block copolymers with a hydrophobic anchoring block that forms a collapsed film at the interface allowing a hydrophilic block to dangle into the solution. Correct selection of the block lengths allows the creation of relatively densely grafted brushes. Matsuoka and co-workers [56] have performed elegant reflectometry studies of the salt response of weak and strong polyacid brushes at the air/water interface. Furthermore, it is possible to transfer copolymer brushes from the air water interface to a solid substrate such as silicon (with appropriate surface modification) using the Langmuir-Schaeffer technique [57].
In part due to the creativity of polymer chemists, and increasingly biological scientists, there exists a wide family of brush-like systems formed from end-attached polymers for which the scaling theories presented here are not well suited. These could be mixed brushes [58], copolymer brushes [40,59], patterned [60] or confined brushes [61] or layers of end-attached biopolymers such as DNA [62] that have additional complexities not described by the homopolymer scaling theories. Furthermore, the scaling theories are not perfect and even in their limited scope represent approximations of the real situation. A good example of this was demonstrated by Dong and co-workers [42] who showed that the degree of dissociation of weak polyelectrolyte brushes varies with distance from the grafting surface, while it is commonly treated as independent of position [12]. Self-consistent field theory and molecular dynamics present a finer grained picture, but naturally lack the simplicity of scaling theory.
5. Voltage, Electric Field, or Electrochemical Response: Present and Future
The presence of electrical charge in polyelectrolyte brushes offers additional possibilities for control of their structure through the use of electrostatic interactions. In the absence of an externally applied field, electrostatic interactions between charged monomers, surface charges, and counter-ions all play important roles in dictating the structure and swelling state of polyelectrolyte brushes, accounting for the increased swelling of strong polyelectrolyte brushes with respect to their neutral equivalent, and for the pH-response of weak polyelectrolyte brushes. It follows naturally that externally applied electric fields could be used to manipulate charges within polyelectrolyte brushes to realise changes in brush structure. The use of applied fields offers a number of potential advantages over established physical-chemical switching methods. These established methods include changing salt concentration, solvent quality, or in the case of weak polyelectrolytes, changing the pH. The use of an externally applied electric field removes the need to physically or chemically change the solution in contact with the brush, allowing remote control of the brush system to be realised. The use of patterning techniques and pixelation of surface electrodes could bring about spatial control of the applied field and hence brush structure. The ease with which chosen electrical stimuli may be produced with simple circuitry enhances possibility of the integration of electro-responsive polymers into technology.
To date, only a few theoretical and experimental demonstrations of electrically induced changes in the structure of a charged polymer brush have been published. Heine and Wu [63] used self-consistent field theory to calculate changes in the volume fraction profile of end-charged homopolymer and copolymer brushes (Figure 11). When the charge on the surface was of the same magnitude as the end group charge, the chain ends were repelled from the surface causing a stretched brush conformation. Conversely, when the charge on the surface had an opposite sign to that upon the end groups, they were attracted towards the surface causing folding and eventually complete looping of the chains to the surface, resulting in a relatively collapsed volume fraction profile. Tsori and co-workers have predicted interfacial instabilities in such systems [64].
Zhou and Huck [65] demonstrated that AFM microcantilevers grafted on one side with a poly[2-(methacrylolyloxy)ethyl] trimethyl ammonium chloride (PMETAC) strong polyelectrolyte brush layer could be actuated by the application of a voltage between the tip holder and a remote electrode within a liquid cell containing weak salt solutions. This effect has been explained by the surface stresses due to voltage-induced conformational changes within the polyelectrolyte brush layers. A model based upon a modified Poisson-Boltzmann equation coupled to Stoney's equation [65] predicted well the bending of the cantilever as a function of applied voltage. It also predicted changes in the volume fraction profile of the polyelectrolyte brush (see Figure 12), though these were never demonstrated experimentally. Specifically, it was predicted that a strong polycationic brush would be swollen by a positive applied bias (positive voltage at the grafting surface) and de-swollen by a negative applied bias (negative voltage at the grafting surface).
Molecular dynamics simulations have shown highly promising results for the fast, cyclic actuation of polyelectrolyte brushes in applied electric fields. Ouyang [66] has demonstrated that partially charged, strong polyelectrolyte brushes may be swollen by an applied voltage, and that the response frequency of the system can reach several hundred MHz. This kind of reproducible and fast response is exciting for the design of devices such as micro- and nano-fluidics where opening and closing of pores is required on demand [67] (temperature and pH responsive polymers are also key contenders in the control of microfluidic pores, see Section 6). Another area of interest is the design of smart surfaces where spatial and temporal control of the surface brush coating is highly desirable.
We have recently demonstrated that weak polyelectrolyte brushes may be partially or strongly swollen by applied voltages [69]. Poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) weak polybase brushes were grown from silicon substrates. Changes in the profiles of the brushes in response to DC voltages applied between the brush substrate and a parallel electrode some distance away in the surrounding water were measured using ellipsometry and neutron reflectometry [70]. Neutron reflectometry data (Figure 13) showed that positive applied voltages caused moderate swelling, strong swelling, and finally damage of the brush as the magnitude of the voltage was increased from 0.5 to 5 V. In this case positive bias means the positive terminal of the voltage supply was connected to the brush substrate. The range of swelling was shown to be larger than that accessible via pH-induced swelling, and this coupled with the versatility of electrical stimuli over physical-chemical stimuli (e.g., changing the solution pH) makes the future of voltage-induced actuation appear very promising. Migliorini [71] has used self-consistent field theory to study the effect of electric fields upon semi-dilute, strong polyelectrolyte brushes, predicting swelling of polycationic brushes upon application of a positive bias (positive terminal in contact with the brush grafting substrate).
The study of polyelectrolyte brushes with standard electrochemical methods has received increasing attention, although these studies typically lack information about the effect of bias on brush structure. Brushes may readily be grafted onto electrochemical electrodes and then their conformation or oxidation state changed using electrochemical stimuli or measured using typical electrochemical means, e.g., cyclic voltammetry [72]. For example, indium tin oxide (ITO) electrodes modified with brushes of poly(4-vinyl pyridine) were shown to be reversibly inactivated to certain redox species by the application of a voltage [73]. Electrochemical switching of polyelectrolyte brush wetting properties [74,75] have been demonstrated, as well as reversible “locking”, which involves decoupling the response of a brush from the ionic strength of the surrounding solution [76]. These examples of electrochemically “smart” surfaces indicate progress towards a class of versatile and intelligent switchable surfaces and sensors based on polyelectrolyte brushes.
6. Applications and Devices
The development of truly functional responsive polymer devices is a natural consequence of the huge progress in surface chemistry and architecture of responsive polymer brushes that we have tried to summarise in this review. It is also of great benefit commercially and technologically that very thin polymer films can have such a marked effect on the overall properties and functionality of potential real world devices. This is mostly due to the important part that interfaces play in their interaction with molecules and solutes. We have tried to include a cross section of possible devices or systems that would lend themselves to being utilized as real world devices. We have not discussed applications of polymer brushes where the responsive nature is not exploited (e.g., anti-fouling coatings [77]).
La Spina et al. [78] demonstrated switchable adhesion between a weak polyacid gel and a weak polybase brush grafted from a silicon wafer (see Figure 14 for the experimental arrangement), mediated by the environmental pH value. In the correct conditions, between 3 and 7 pH the gel and brush were attached with a work of adhesion on the order of 200 mN/m, as measured using a modified Johnson-Kendall-Roberts (JKR) experiment, whilst at pH 2 or below the brush and gel could be separated without damage. This experiment shows an example of the modification of a surface with a nanoscale responsive polymer brush layer in order to create a macroscopic physical adhesion effect, in addition to presenting several interesting questions relating to the polymer physics of the adhesion. Recently, studies of switchable adhesion properties of temperature-responsive polymer brushes have been demonstrated using atomic force microscopy [79], while adhesion between oppositely charged planar polyelectrolyte brushes has been demonstrated [80].
A very novel experiment utilizing grafted polyelectrolyte brushes was carried out by Ito et al. [81]. Their study attached Poly(methacrylic acid) brushes onto a polycarbonate membrane with pore sizes of 200 nm. They compared the water flow with and without the attachment of the brush and showed that for the ungrafted membrane the water permeation rate was independent of pH whilst the grafted brush exhibited a strong pH dependence (see Figure 15). At a pH close to the pKa of Poly(methacrylic acid) (pKa of 4) the flow reduced dramatically as the polymer brush collapsed and consequently reduced the size of the grafted pore and the water flow rate. This kind of filtering could be used in series to remove ions, as salts will also cause a collapse of a polymer brush. This could be used in series with other filters to purify water for drinking, also some polymer brushes are not prone to the type of protein adsorption, which over time renders water filters useful and necessitates their replacement (at huge cost).
Huber and co-workers demonstrated the controlled holding and release of proteins by utilizing the temperature response of PNIPAM in a custom made microfluidic device [82]. At room temperature, while the polymer was swollen, there was negligible adsorption of globular protein, whereas above the LCST when the polymer brush collapses allowing extensive adsorption. They showed excellent control over the PNIPAM brush layer by using highly integrated micro-hotplates that can be spatially addressed with fine spatial control and also rapidly addressed. They were able to heat above the LCST in less than 1 ms whilst cooling took longer as it was not actively cooled. Figure 16 shows the patterned electrode used (Figure 16(a)) to heat the PNIPAM layers along with a sequence (Figure 16(b)) showing the release of protein after the layer was taken below the LCST.
Many applications of polymer brushes in the future may rely on the combination of patterning techniques with polymer brush synthesis to achieve spatial control over brush structure, as demonstrated in the last example. One aim of this technique would be to realize transport of nano- and micro-scale cargoes across surfaces [83].
7. Conclusions
The name “polymer brush” can really refer to many things, though these varied systems all have the defining feature of having polymer chains grafted at one end to a substrate at sufficient density to overlap and stretch away from the surface. By no means an exhaustive survey shows that polymer brush chains can be simple homopolymers, block copolymers or more complex grafted polymers; they may be neutral, strong or weak polyelectrolyes or even polyampholytes; polymer brushes may be mixed, binary, patterned, or confined.
Because of the interactions between neighbouring brush chains and between the brush and the local environment, polymer brushes are responsive, exhibiting changes in structure and properties in response to external stimuli. Polymer brush research has moved on from the early days of perfecting brush formation, through increasing complexities of polymer synthesis, to today's intricately designed and synthesized electrochemically responsive brushes, for example. In parallel, our understanding of polymer brushes has progressed from describing the behaviour of simple end-grafted homopolymers to complex theory and simulation to predict the structure and behaviour of brushes in response to new and varied stimuli.
Polymer brush research requires, and shows, a delicate balance between continuous changing of the chemistry of brushes in order to achieve specific properties and the dedicated study of single systems in sufficient detail in order bring forward a full understanding. The development of surface-initiated polymerization has allowed us to create polymer brushes with a level of control previously confined to the imagination. This increase in control and ease of brush polymerisation allows us to cover large surfaces with well-defined polymer brushes in an industrial context, while at the opposite length scale, we create complex patterned brush surfaces for micro- and nano-technology. The development of non-invasive stimuli for switching polymer brushes, such as voltage-induced swelling, could lead to a new generation of highly functional devices that will allow surface properties to be remotely manipulated.


















Acknowledgments
AJP was funded by the EPSRC Soft Nanotechnology grant EP/E046215/1. MPW is funded by the Cooperative Research Centre for Polymers ( www.crcp.com.au).
References
- Bünsow, J.; Kelby, T.S.; Huck, W.T.S. Polymer brushes: Routes toward mechanosensitive surfaces. Acc. Chem. Res. 2009, 43, 466–474. [Google Scholar]
- Jones, R.A.L. Challenges in soft nanotechnology. Faraday Discuss. 2009, 143, 9–14. [Google Scholar]
- Ryan, A.J.; Crook, C.J.; Howse, J.R.; Topham, P.; Jones, R.A.L.; Geoghegan, M.; Parnell, A.J.; Ruiz-Perez, L.; Martin, S.J.; Cadby, A.; Menelle, A.; Webster, J.R.P.; Gleeson, A.J.; Bras, W. Responsive brushes and gels as components of soft nanotechnology. Faraday Discuss. 2005, 128, 55–74. [Google Scholar]
- Cohen Stuart, M.A.; Huck, W.T.S.; Genzer, J.; Muller, M.; Ober, C.; Stamm, M.; Sukhorukov, G.B.; Szleifer, I.; Tsukruk, V.V.; Urban, M.; Winnik, F.; Zauscher, S.; Luzinov, I.; Minko, S. Emerging applications of stimuli-responsive polymer materials. Nat. Mater. 2010, 9, 101–113. [Google Scholar]
- Tokarev, I.; Motornov, M.; Minko, S. Molecular-engineered stimuli-responsive thin polymer film: A platform for the development of integrated multifunctional intelligent materials. J. Mater. Chem. 2009, 19, 6932–6948. [Google Scholar]
- Ducker, R.; Garcia, A.; Zhang, J.; Chen, T.S.Z. Polymeric and biomacromolecular brush nanostructures: Progress in synthesis, pat-terning and characterization. Soft Matter 2008, 4, 1774–1786. [Google Scholar]
- Ballauff, M.; Borisov, O. Polyelectrolyte brushes. Curr. Opin. Colloid Interface Sci. 2006, 11, 316–323. [Google Scholar]
- Advincula, R.C.; Brittain, W.J.; Caster, K.C.; Rühe, J. Polymer Brushes: Synthesis, Characterization, Applications; Wiley: Hoboken, NJ, USA, 2004. [Google Scholar]
- Minko, S. Responsice polymer brushes. J. Macromol. Sci. Part C Polym. Rev. 2006, 46, 397–420. [Google Scholar]
- Toomey, R.; Tirell, M. Functional polymer brushes in aqueous media from self-assembled and surface-initiated polymers. Annu. Rev. Phys. Chem. 2008, 59, 493–517. [Google Scholar]
- Rühe, J.; Ballauff, M.; Biesalski, M.; Dziezok, P.; Groehn, F.; Johannsmann, D.; Houbenov, N.; Hugenberg, N.; Konradi, R.; Minko, S.; Motornov, M.; Netz, R.R.; Schmidt, M.; Seidel, C.; Stamm, M.; Usov, T.S.D.; Zhang, H. Polyelectrolyte brushes. Adv. Polym. Sci. 2004, 165, 79–150. [Google Scholar]
- Zhulina, B.; Birshtein, T.M.; Borisov, V. Theory of ionizable polymer brushes. Macromolecules 1995, 28, 1491–1499. [Google Scholar]
- Huang, H.Q.; Rankin, S.E.; Penn, L.S.; Quirk, R.P.; Cheong, T.H. Transition from mushroom to brush during formation of a tethered layer. Langmuir 2004, 20, 5770–5775. [Google Scholar]
- de Gennes, P.G. Conformations of polymers attached to an interface. Macromolecules 1980, 13, 1069–1075. [Google Scholar]
- Jones, R.A.L.; Richards, R.W. Polymers at Surfaces and Interfaces; Cambridge University Press: Cambridge, UK, 1999. [Google Scholar]
- Ciferri, A. Supramolecular Polymers, 1st ed.; Marcel Dekker: New York, NY, USA, 2000. [Google Scholar]
- Zhao, B.; Brittain, W.J. Polymer brushes: Surface-immobilized macromolecules. Prog. Polym. Sci. 2000, 25, 677–710. [Google Scholar]
- Boyes, S.G.; Granville, A.M.; Baum, M.; Akgun, B.; Mirous, B.K.; Brittain, W.J. Polymer brushes surface immobilized polymers. Surf. Sci. 2004, 570, 1–12. [Google Scholar]
- Taylor, W.; Jones, R.A.L. Producing high-density high-molecular-weight polymer brushes by a “grafting to” method from a concentrated homopolymer solution. Langmuir 2010, 26, 13954–13958. [Google Scholar]
- Advincula, R.; Zhou, Q.G.; Park, M.; Wang, S.G.; Mays, J.; Sakellariou, G.; Pispas, S.; Hadjichristidis, N. Polymer brushes by living anionic surface initiated polymerization on flat silicon (siox) and gold surfaces: Homopolymers and block copolymers. Langmuir 2002, 18, 8672–8684. [Google Scholar]
- Matyjaszewski, K.; Miller, P.J.; Shukla, N.; Immaraporn, B.; Gelman, A.; Luokala, B.B.; Siclovan, T.M.; Kickelbick, G.; Vallant, T.; Hoffmann, H.; Pakula, T. Polymers at interfaces: Using atom transfer radical polymerization in the controlled growth of homopolymers and block copolymers from silicon surfaces in the absence of untethered sacrificial initiator. Macromolecules 1999, 32, 8716–8724. [Google Scholar]
- Beers, K.L.; Gaynor, S.G.; Matyjaszewski, K.; Sheiko, S.S.; Möller, M. The synthesis of densely grafted copolymers by atom transfer radical polymerization. Macromolecules 1999, 31, 9413–9415. [Google Scholar]
- Balastre, M.; Li, F.; Schorr, P.; Yang, J.C.; Mays, J.W.; Tirrell, M.V. A study of polyelectrolyte brushes formed from adsorption of amphiphilic diblock copolymers using the surface forces apparatus. Macromolecules 2002, 35, 9480–9486. [Google Scholar]
- Himmelhaus, M.; Bastuck, T.; Tokumitsu, S.; Grunze, M.; Livadaru, L.; Kreuzer, H.J. Growth of a dense polymer brush layer from solution. Europhys. Lett. 2003, 64, 378–384. [Google Scholar]
- Kamigaito, M.; Ando, T.; Sawamoto, M. Metal-catalyzed living radical polymerization. Chem. Rev. 2001, 101, 3689–3745. [Google Scholar]
- Wang, J.S.; Matyjaszewski, K. Controlled living radical polymerization atom-transfer radical polymerization in the presence of transition-metal complexes. J. Am. Chem. Soc. 1995, 117, 5614–5615. [Google Scholar]
- Sawamoto, M.; Kato, M.; Kamigaito, M.; Higashimura, T. Polymerization of methyl methacrylate with the carbon tetrachlorideldichlorotris-(triphenylphosphine)ruthedum(ii)/methylaluminum bis(2,6-di-tert-butylphenoxide) initiating system: Possibility of living radical polymerization. Macromolecules 1995, 28, 1721–1723. [Google Scholar]
- Matyjaszewski, K.; Spanswick, J. Controlled/living radical polymerization. Mater. Today 2005, 8, 26–33. [Google Scholar]
- Matyjaszewski, K. Atom Transfer Free Radical Polymerization. In Encyclopedia of Materials: Science and Technology; Elsevier: Amsterdam, The Netherlands, 2001; pp. 355–365. [Google Scholar]
- Edmondson, S.; Osborne, V.L.; Huck, W.T. Polymer brushes via surface-initiated polymerizations. Chem. Soc. Rev. 2004, 33, 14–22. [Google Scholar]
- Parnell, A.J.; Martin, S.J.; Dang, C.C.; Geoghegan, M.; Jones, R.A.L.; Crook, C.J.; Howse, J.R.; Ryan, A.J. Synthesis, characterization and swelling behaviour of poly(methacrylic acid) brushes synthesized using atom transfer radical polymerization. Polymer 2009, 50, 1005–1014. [Google Scholar]
- Matyjaszewski, K.; Hongchen, D.; Jakubowski, W.; Pietrasik, J.; Kusumo, A. Grafting from surfaces for “everyone”: ARGET ATRP in the presence of air. Langmuir 2007, 23, 4528–4531. [Google Scholar]
- Magenau, A.J.D.; Strandwitz, N.C.; Gennaro, A.; Matyjaszewski, K. Electrochemically mediated atom transfer radical polymerization. Science 2011, 332, 81–84. [Google Scholar]
- Jones, D.M.; Smith, J.R.; Huck, W.T.S.; Alexander, C. Variable adhesion of micropatterned thermoresponsive polymer brushes: AFM investigations of poly(N-isopropylacrylamide) brushes prepared by surface-initiated polymerizations. Adv. Mater. 2002, 14, 1130–1134. [Google Scholar]
- Kaholek, M.; Lee, W.-K.; Ahn, S.-J.; Ma, H.; Caster, K.C.; LaMattina, B.; Zauscher, S. Stimulus-responsive poly(N-isopropylacrylamide) brushes and nanopatterns prepared by surface-initiated polymerization. Chem. Mater. 2004, 16, 3688–3696. [Google Scholar]
- Kaholek, M.; Lee, W.-K.; LaMattina, B.; Caster, K.C.; Zauscher, S. Fabrication of stimulus-responsive nanopatterned polymer brushes by scanning-probe lithography. Nano Lett. 2004, 4, 373–376. [Google Scholar]
- Halperin, A.; Fragneto, G.; Schollier, A.; Sferrazza, M. Primary versus ternary adsorption of proteins onto PEG brushes. Langmuir 2007, 23, 10603–10617. [Google Scholar]
- Prime, K.L.; Whitesides, G.M. Adsorption of proteins onto surfaces containing end-attached oligo(ethylene oxide): A model system using self-assembled monolayers. J. Am. Chem. Soc. 1993, 115, 10714–10721. [Google Scholar]
- Ma, H.; Hyun, J.; Stiller, P.; Chilkoti, A. “Non-Fouling” oligo(ethylene glycol)-functionalized polymer brushes synthesized by surface-initiated atom transfer radical polymerization. Adv. Mater. 2004, 16, 338–341. [Google Scholar]
- Ye, M.A.; Zhang, D.; Han, L.; Tejada, J.; Ortiz, C. Synthesis, Preparation, and conformation of stimulus-responsive end-grafted poly(methacrylic acid-gethyleneglycol) layers. Soft Matter. 2006, 2, 243–256. [Google Scholar]
- Skoda, M.W.A.; Schreiber, F.; Jacobs, R.M.J.; Webster, J.R.P.; Wolff, M.; Dahint, R.; Schwendel, D.; Grunze, M. Protein density profile at the interface of water with oligo(ethyleneglycol) self-assembled monolayers. Langmuir 2009, 25, 4056–4064. [Google Scholar]
- Dong, R.R.; Lindau, M.; Ober, C.K. Dissociation behavior of weak polyelectrolyte brushes on a planar surface. Langmuir 2009, 25, 4774–4779. [Google Scholar]
- Israels, R.; Leermakers, F.A.M.; Fleer, G.J.; Zhulina, E.B. Charged polymeric brushes—Structure and scaling relations. Macromolecules 1994, 27, 3249–3261. [Google Scholar]
- Mir, Y.; Auroy, P.; Auvray, L. Density profile of polyelectrolyte brushes. Phys. Rev. Lett. 1995, 75, 2863–2866. [Google Scholar]
- Tran, Y.; Auroy, P.; Lee, L.T. Determination of the structure of polyelectrolyte brushes. Macromolecules 1999, 32, 8952–8964. [Google Scholar]
- Prucker, O.; Rühe, J. Synthesis of poly(styrene) monolayers attached to high surface area silica gels through self-assembled monolayers of azo initiators. Macromolecules 1998, 31, 592–601. [Google Scholar]
- Biesalski, M.; Rühe, J.; Johannsmann, D. Segment density profiles of polyelectrolyte brushes determined by Fourier transform ellipsometry. J. Chem. Phys. 1999, 111, 7029–7037. [Google Scholar]
- Biesalski, M.; Johannsmann, D.; Rühe, J. Synthesis and swelling behavior of a weak polyacid brush. J. Chem. Phys. 2002, 117, 4988–4994. [Google Scholar]
- Geoghegan, M.; Ruiz-Perez, L.; Dang, C.C.; Parnell, A.J.; Martin, S.J.; Howse, J.R.; Jones, R.A.L.; Golestanian, R.; Topham, P.D.; Crook, C.J.; Ryan, A.J.; Sivia, D.S.; Webster, J.R.P.; Menelle, A. The pH-induced swelling and collapse of a polybase brush synthesized by atom transfer radical polymerization. Soft Matter. 2006, 2, 1076–1080. [Google Scholar]
- Sanjuan, S.; Perrin, P.; Pantoustier, N.; Tran, Y. Synthesis and swelling behavior of pH-responsive polybase brushes. Langmuir 2007, 23, 5769–5778. [Google Scholar]
- Sanjuan, S.; Tran, Y. Stimuli-responsive interfaces using random polyampholyte brushes. Macromolecules 2008, 41, 8721–8728. [Google Scholar]
- Parnell, A.J.; Martin, S.J.; Jones, R.A.L.; Vasilev, C.; Crook, C.J.; Ryan, A.J. Direct visualization of the real time swelling and collapse of a poly(methacrylic acid) brush using atomic force microscopy. Soft Matter. 2009, 5, 296–299. [Google Scholar]
- Bumbu, G.G.; Kircher, G.; Wolkenhauer, M.; Berger, R.; Gutmann, J.S. Synthesis and characterization of polymer brushes on micromechanical cantilevers. Macromol. Chem. Phys. 2004, 205, 1713–1720. [Google Scholar]
- Abu-Lail, N.I.; Kaholek, M.; LaMattina, B.; Clark, R.L.; Zauscher, S. Micro-cantilevers with end-grafted stimulus-responsive polymer brushes for actuation and sensing. Sens. Actuat. B Chem. 2006, 114, 371–378. [Google Scholar]
- Zhou, F.; Shu, W.; Welland, M.E.; Huck, W.T.S. Highly reversible and multi-stage cantilever actuation driven by polyelectrolyte brushes. J. Am. Chem. Soc. 2006, 128, 5326–5327. [Google Scholar]
- Matsuoka, H.; Suetomi, Y.; Kaewsaiha, P.; Matsumoto, K. Nanostructure of a poly(acrylic acid) brush and its transition in the amphiphilic diblock copolymer monolayer on the water surface. Langmuir 2009, 25, 13752–13762. [Google Scholar]
- Currie, E.P.K.; Sieval, A.B.; Fleer, G.J.; Stuart, M.A.C. Polyacrylic acid brushes: Surface pressure and salt-induced swelling. Langmuir 2000, 16, 8234–8333. [Google Scholar]
- LeMieux, M.C.; Julthongpiput, D.; Bergman, K.N.; Cuong, P.D.; Ahn, H.S.; Lin, Y.H.; Tsukruk, V.V. Ultrathin binary grafted polymer layers with switchable morphology. Langmuir 2004, 20, 10046–10054. [Google Scholar]
- Yu, K.; Wang, H.; Xue, L.; Han, Y. Stimuli-responsive polyelectrolyte block copolymer brushes synthesized from the Si wafer via atom-transfer radical polymerization. Langmuir 2007, 23, 1443–1452. [Google Scholar]
- Jonas, A.M.; Hu, Z.; Glinel, K.; Huck, W.T.S. Chain entropy and wetting energy control the shape of nanopatterned polymer brushes. Macromolecules 2008, 41, 6859–6863. [Google Scholar]
- Jonas, A.M.; Hu, Z.; Glinel, K.; Huck, W.T.S. Effect of nanoconfinement on the collapse transition of responsive polymer brushes. Nano Lett. 2008, 8, 3819–3824. [Google Scholar]
- Kjallman, T.H.M.; Nelson, A.; James, M.; Dura, J.A.; Travas-Sejdic, J.; McGillivray, D.J. A neutron reflectivity study of the interfacial and thermal behaviour of surface-attached hairpin DNA. Soft Matter 2011, 7, 5020–5029. [Google Scholar]
- Heine, D.; Wu, D.T. A switchable polymer layer: Chain folding in end-charged polymer brushes. J. Chem. Phys. 2001, 114, 5313–5321. [Google Scholar]
- Tsori, Y.; Andelman, D.; Joanny, J.-F. Interfacial instability of charged-end group polymer brushes. Europhys. Lett. 2008, 82, 46001:1–46001:6. [Google Scholar]
- Zhou, F.; Biesheuvel, P.M.; Choi, E.-Y.; Shu, W.; Poetes, R.; Steiner, U.; Huck, W.T.S. Polyelectrolyte brush amplified electroactuation of microcantilevers. Nano Lett. 2008, 8, 725–730. [Google Scholar]
- Ouyang, H.; Xia, Z.; Zhe, J. Static and dynamic responses of polyelectrolyte brushes under external electric field. Nanotechnology 2009. [Google Scholar] [CrossRef]
- Ouyang, H.; Xia, Z.; Zhe, J. Voltage-controlled flow regulating in nanofluidic channels with charged polymer brushes. Microfluid. Nanofluid. 2010, 9, 915–922. [Google Scholar]
- Heine, D.; Wu, D.T. A switchable polymer layer: Chain folding in end-charged polymer brushes. J. Chem. Phys. 2001, 114, 5313–5321. [Google Scholar]
- Weir, M.P.; Heriot, S.Y.; Martin, S.J.; Parnell, A.J.; Holt, S.A.; Webster, J.R.P.; Jones, R.A.L. Voltage-induced swelling and deswelling of weak polybase brushes. Langmuir 2011, 27, 11000–11007. [Google Scholar]
- Webster, J.; Holt, S.; Dalgliesh, R. INTER the chemical interfaces reflectometer on target station 2 at ISIS. Physica B 2006, 385–386, 1164–1166. [Google Scholar]
- Migliorini, G. On the corrections to strong-stretching theory for end-confined, charged polymers in a uniform electric field. Macromolecules 2010, 43, 9168–9180. [Google Scholar]
- Choi, E.-Y.; Azzaroni, O.; Cheng, N.; Zhou, F.; Kelby, T.; Huck, W.T.S. Electrochemical characteristics of polyelectrolyte brushes with electroactive counterions. Langmuir 2007, 23, 10389–10394. [Google Scholar]
- Tam, T.K.; Pita, M.; Trotsenko, O.; Motornov, M.; Tokarev, I.; Halamek, J.; Minko, S.; Katz, E. Reversible “closing” of an electrode interface functionalized with a polymer brush by an electrochemical signal. Langmuir 2010, 26, 4506–4513. [Google Scholar]
- Azzaroni, O.; Brown, A.A.; Huck, W.T.S. Tunable wettability by clicking counterions into polyelectrolyte brushes. Adv. Mater. 2007, 19, 151–154. [Google Scholar]
- Spruijt, E.; Choi, E.-Y.; Huck, W.T.S. Reversible electrochemical switching of polyelectrolyte brush surface energy using electroactive counterions. Langmuir 2008, 24, 11253–11260. [Google Scholar]
- Moya, S.; Azzaroni, O.; Farhan, T.; Osborne, V.L.; Huck, W.T.S. Locking and unlocking of polyelectrolyte brushes: Toward the fabrication of chemically controlled nanoactuators. Angew. Chem. Int. Ed. 2005, 44, 4578–4581. [Google Scholar]
- Glinel, K.; Jonas, A.M.; Jouenne, T.; Leprince, J.; Galas, L.; Huck, W.T.S. Antibacterial and antifouling polymer brushes incorporating antimicrobial peptide. Bioconjugate Chem. 2009, 20, 71–77. [Google Scholar]
- La Spina, R.; Tomlinson, M.R.; Ruiz-Pérez, L.; Chiche, A.; Langridge, S.; Geoghegan, M. Controlling network/brush interactions to achieve switchable adhesion. Angew. Chem. Int. Ed. 2007, 46, 6460–6463. [Google Scholar]
- Svetushkina, E.; Puretskiy, N.; Ionov, L.; Stamm, M.; Synytska, A. A comparative study on switchable adhesion between thermoresponsive polymer brushes on flat and rough surfaces. Soft Matter. 2011, 7, 5691–5696. [Google Scholar]
- Kobayashi, M.; Terada, M.; Takahara, A. Reversible adhesive-free nanoscale adhesion utilizing oppositely charged polyelectrolyte brushes. Soft Matter 2011. [Google Scholar] [CrossRef]
- Ito, Y.; Park, Y.S.; Imanishi, Y. Visualization of critical pH-controlled gating of a porous membrane grafted with polyelectrolyte brushes. J. Am. Chem. Soc. 1997, 119, 2739–2740. [Google Scholar]
- Huber, D.L.; Manginell, R.P.; Samara, M.A.; Kim, B.I.; Bunker, B.C. Programmed adsorption and release of proteins in a microfluidic device. Science 2003, 301, 352–354. [Google Scholar]
- Prokhorova, S.A.; Kopyshev, A.; Ramakrishnan, A.; Zhang, H.; Rühe, J. Can polymer brushes induce motion of nano-objects? Nanotechnology 2003, 14, 1098–1108. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Weir, M.P.; Parnell, A.J. Water Soluble Responsive Polymer Brushes. Polymers 2011, 3, 2107-2132. https://doi.org/10.3390/polym3042107
Weir MP, Parnell AJ. Water Soluble Responsive Polymer Brushes. Polymers. 2011; 3(4):2107-2132. https://doi.org/10.3390/polym3042107
Chicago/Turabian StyleWeir, Michael P., and Andrew J. Parnell. 2011. "Water Soluble Responsive Polymer Brushes" Polymers 3, no. 4: 2107-2132. https://doi.org/10.3390/polym3042107
APA StyleWeir, M. P., & Parnell, A. J. (2011). Water Soluble Responsive Polymer Brushes. Polymers, 3(4), 2107-2132. https://doi.org/10.3390/polym3042107