Variational Models of Network Formation and Ion Transport: Applications to Perfluorosulfonate Ionomer Membranes

Abstract
: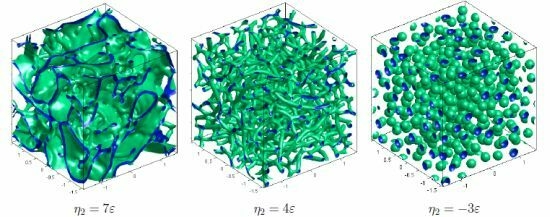
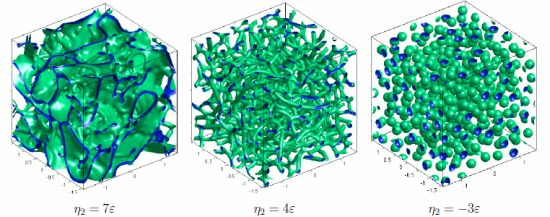{kind=link}
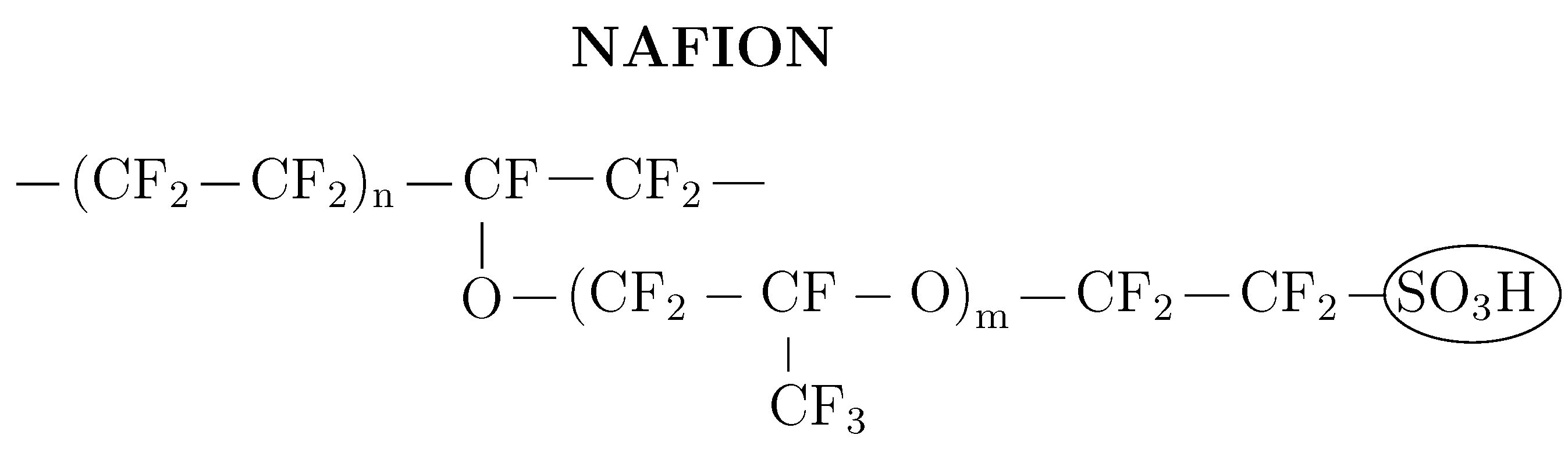{kind=link}
{kind=link}
{kind=link}
{kind=link}
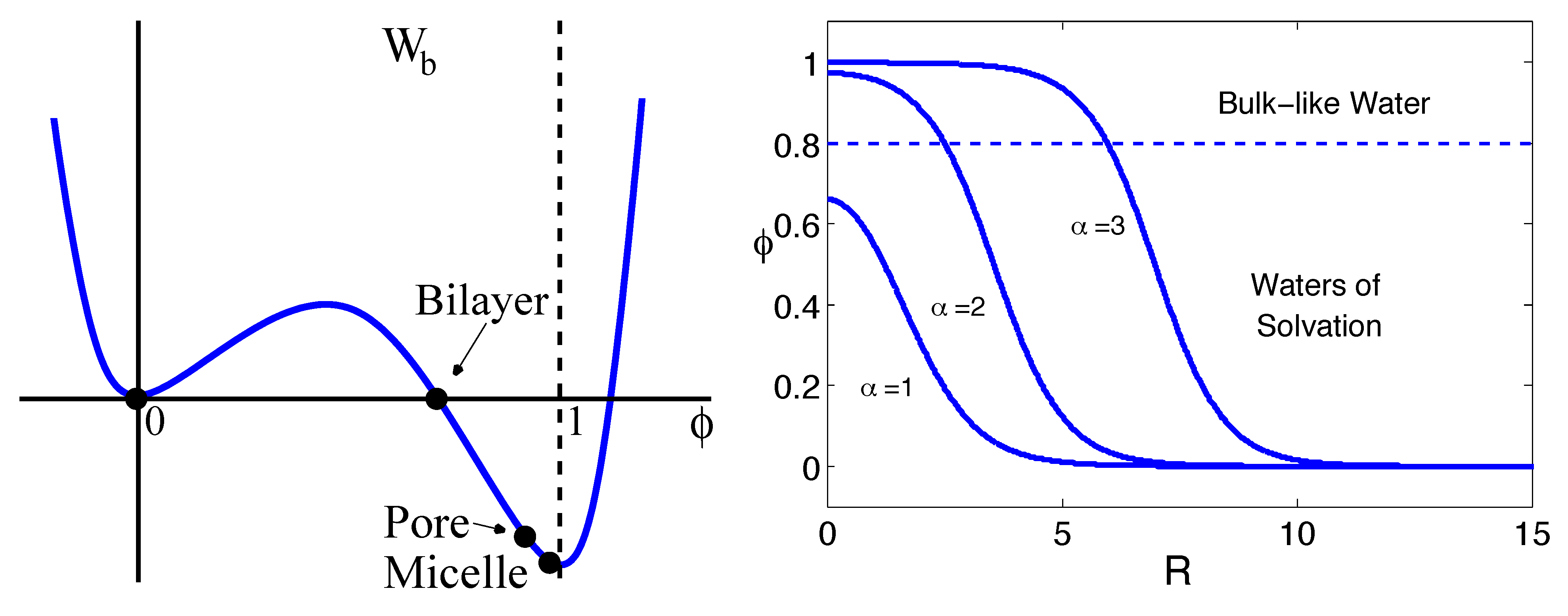{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Network Formation in Binary Mixtures
2.1. Gradient Flows of the FCH Energy
2.2. Interpretation of Energetic Terms and Parameters
2.3. Sharp-Interface Reductions of the FCH Energy
3. Applications of the Binary FCH Energy
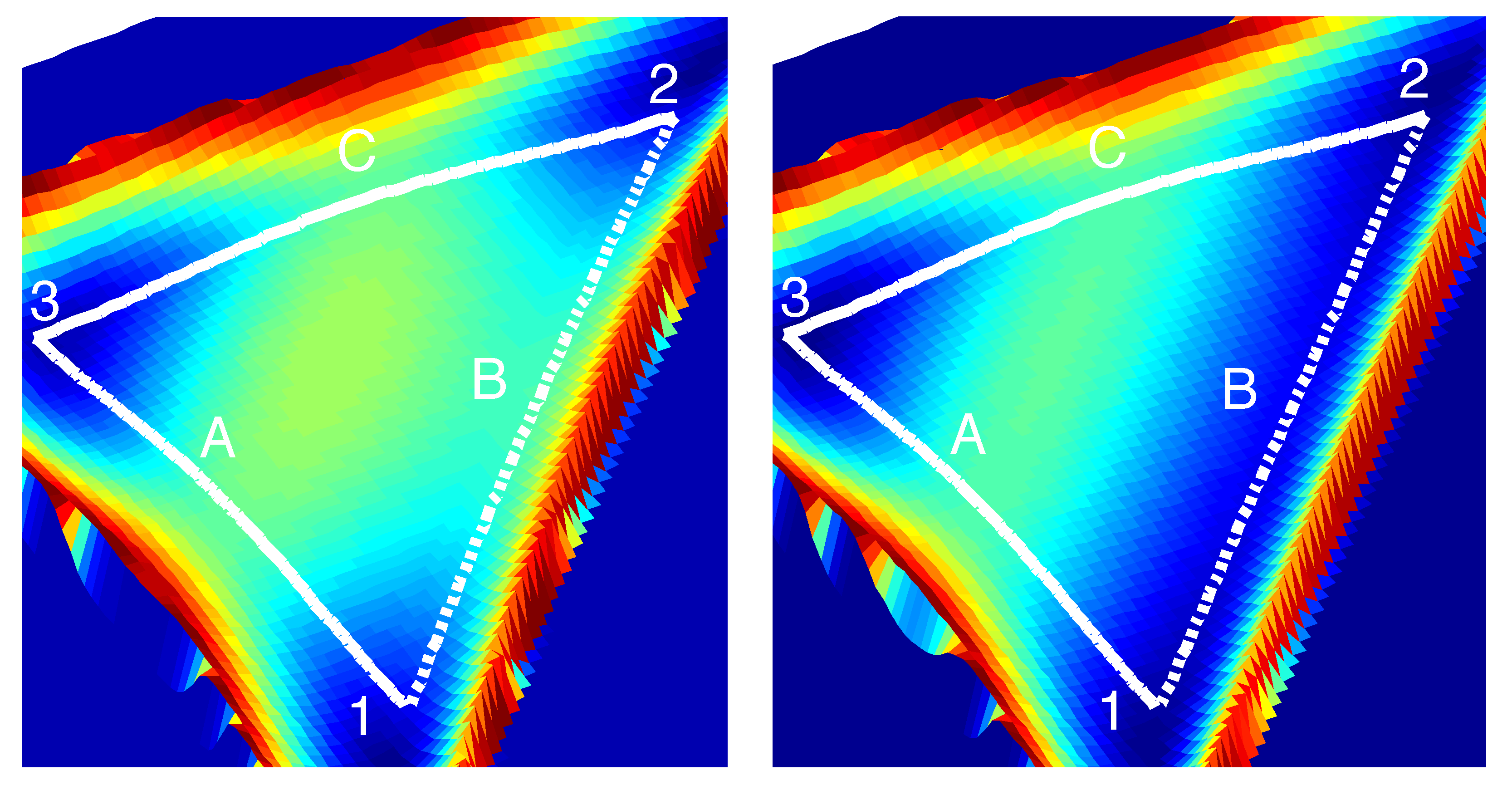
3.1. A Study of Nafion: Comparison to SAXS Data
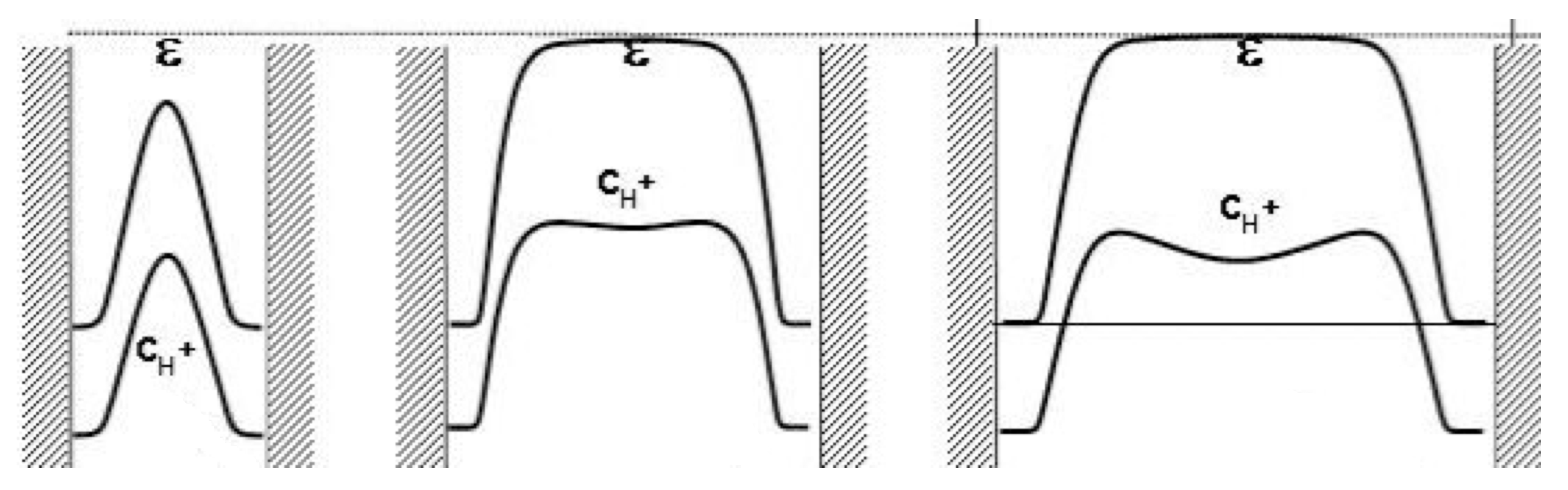
3.2. Extensions to Incorporation of Ionic Conduction
4. Multi-Component Extensions
4.1. Casting and Annealing of PFSAs
5. Conclusions
Acknowledgements
References
- Ameduri, B. From Vinyidene Fluoride (VDF) to the applications of VDF-containing polymers and co-polymers: Recent developments and future trends. Chem. Rev. 2009, 109, 6632–6686. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, E.; Beyer, F. Self-Assembling Nanomembranes through Electrostatic Melt Processing of Copolymer Films; ARL-TR-2800; Army Research Laboratory: Adelphi, MD, USA, 2002. [Google Scholar]
- Wu, D.S.; Paddison, S.J.; Elliott, J.A. Effect of molecular weight on hydrated morphologies of the short-side-chain perfluorosulfonic acid membrane. Macromolecules 2009, 42, 3358–3367. [Google Scholar] [CrossRef]
- Eikerling, M.; Paddison, S.J.; Pratt, L.R.; Zawodzinski, T.A. Defect structure for proton transport in a triflic acid monohydrate solid. Chem. Phys. Lett. 2003, 368, 108–114. [Google Scholar] [CrossRef]
- Promislow, K.; Wetton, B. PEM fuel cells: A mathematical overview. SIAM J. Appl. Math. 2009, 70, 369–409. [Google Scholar] [CrossRef]
- Rubatat, L.; Gebel, G.; Diat, O. Fibriallar structure of Nafion: Matching Fourier and real space studies of corresponding films and solutions. Macromolecules 2004, 37, 7772–7783. [Google Scholar] [CrossRef]
- Lee, K.-M.; Hsu, C.-Y.; Chiu, W.-H.; Tsui, M.-C.; Tung, Y.-L.; Tsai, S.-Y.; Ho, K.-C. Dye-sensitized solar cells with mirco-porous TiO2 electrode and gel polymer electrolytes prepared by in situ cross-link reaction. Sol. Energy Mater. Sol. Cells 2009, 93, 2003–2007. [Google Scholar] [CrossRef]
- Peet, J.; Heeger, A.; Bazan, G. “Plastic” solar cells: Self-assembly of bulk hetrojunction nanomaterials by spontaneous phase separation. Acc. Chem. Res. 2009, 42, 1700–1708. [Google Scholar] [CrossRef]
- Crossland, E.; Kamperman, M.; Nedelcu, M.; Ducati, C.; Wiesner, U.; Smilgies, D.; Toombes, G.; Hillmyer, M.; Ludwigs, S.; Steiner, U.; Snaith, H. A Bicontinuous double gyroid hybrid solar cell. Nano Lett. 2009, 9, 2807–2812. [Google Scholar] [CrossRef]
- Arora, P.; Zhang, Z.M. Battery separators. Chem. Rev. 2004, 104, 4419–4462. [Google Scholar] [CrossRef]
- Ichikawa, T.; Yoshio, M.; Hamaskai, A.; Kagimoto, J.; Ohno, H.; Kato, T. 3D interconnected ionic nano-channels formed in polymer films: Self-organization and polymerization of thermotropic bicontinuous cubic liquid cyrstals. J. Am. Chem. Soc. 2011, 133, 2163–2169. [Google Scholar] [CrossRef]
- Caflisch, R.E. The fluid dynamic limit of the nonlinear Boltzmann equation. Commun. Pure Appl. Math. 1980, 33, 651–666. [Google Scholar] [CrossRef]
- Golse, F. Hydrodynamic Limits. In Proceedings of the European Congress of Mathematics, Stockholm, Sweden, 27 June–2 July 2004; European Mathematics Society: Zürich, Sweden, 2005; pp. 699–717. [Google Scholar]
- Müller, M. Concurrent coupling between a particle simulation and a continuum description. Eur. Phys. J. Spec. Top. 2009, 177, 149–164. [Google Scholar] [CrossRef]
- Knox, C.; Voth, G.A. Probing selected morphological models of hydrated nafion using large-scale molecular dynamics simulations. J. Phys. Chem. B 2010, 144, 3205–3218. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.G.; LeBard, D.N.; DeVane, R.; Shinoda, W.; Kohlmeyer, A.; Klein, M.L. Micellization studied by gpu-accelerated coarse-grained molecular dynamics. J. Chem. Theory Comput. 2011, 7, 4135–4145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Promislow, K.; Martin, J.; Wang, H.; Balcom, B. Bi-modal water transport behavior across a simple Nafion membrane. J. Power Sources 2011, 196, 8525–8530. [Google Scholar] [CrossRef]
- Weber, A.Z.; Newman, J. Modeling transport in polymer-electrolyte membranes. Chem. Rev. 2004, 104, 4679–4726. [Google Scholar] [CrossRef]
- Weber, A.Z.; Newman, J. Transport in polymer-electrolyte membranes—II. Mathematical model. J. Electrochem. Soc. 2004, 151, A311–A325. [Google Scholar] [CrossRef]
- Eikerling, E.; Berg, P. Poroelectrostatic theory of water sorption and swelling in polymer electrolyte membranes. Soft Matter 2011, 7, 5976–5990. [Google Scholar] [CrossRef]
- Mauritz, K.A.; Moore, R.B. State of understanding of nafion. Chem. Rev. 2004, 104, 4535–4585. [Google Scholar] [CrossRef]
- Doyle, M.; Rajendran, G. Perfluorinated membranes. In Handbook of Fuel Cells—Fundamentals, Technology and Applications; Vielstich, W., Lamm, A., Gasteiger, H.A., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2003; Volume 3, pp. 351–395. [Google Scholar]
- Hsu, W.; Gierke, T. Ion transport and clustering in Nafion perfluorinated membranes. J. Membr. Sci. 1983, 13, 307–326. [Google Scholar] [CrossRef]
- Cahn, J.W.; Hilliard, J.E. Free energy of a nonuniform system. I. Interfacial free energy. J. Chem. Phys. 1958, 28, 258–267. [Google Scholar] [CrossRef]
- Morgan, F. Geometric Measure Theory: A Beginners Guide; Academic Press: Waltham, MA, USA, 2000. [Google Scholar]
- de Giorgi, E. Some remarks on Γ-Convergence and least squares methods. In Composite Media and Homogenization Theory; Dal Maso, G., Dell-Antonio, G.F., Eds.; Birkhäuser: Basel, Switzerland, 1991; pp. 135–142. [Google Scholar]
- Promislow, K.; Zhang, H. Critical points of functionalized lagrangians. Discret. Contin. Dyn. Syst. Ser. A 2012, in press. [Google Scholar] [CrossRef]
- Canham, P. Minimum energy of bending as a possible explanation of biconcave shape of human red blood cell. J. Theor. Biol. 1970, 26, 61–81. [Google Scholar] [CrossRef]
- Helfrich, W. Elastic properties of lipid bilayers—Theory and possible experiments. Z. Naturforsch. C 1973, 28, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Kamien, R. The geometry of soft materials: A primer. Rev. Mod. Phys. 2002, 74, 953–971. [Google Scholar] [CrossRef]
- Liu, C.; Walkington, N. An eulerian description of fluids containing visco-elastic particles. Arch. Ration. Mech. 2001, 159, 229–252. [Google Scholar] [CrossRef]
- Gavish, N.; Hayrapetyan, G.; Promislow, K.; Yang, L. Curvature driven flow of bi-layer interfaces. Physica D 2011, 240, 675–693. [Google Scholar] [CrossRef]
- Freger, V. Hydration of ionomers and schroeder’s paradox in nafion. J. Phys. Chem. B 2009, 113, 24–36. [Google Scholar] [CrossRef]
- Paddison, S.J. The modeling of molecular structure and ion transport in sulfonic acid based ionomer membranes. J. New. Mat. Electorchem. Syst. 2001, 4, 197–207. [Google Scholar]
- Kreuer, K.D.; Paddison, S.J.; Spohr, E.; Schuster, M. Transport in proton conductors for fuel-cell applications: Simulations, elementary reactions, and phenomenology. Chem. Rev. 2004, 104, 4637–4678. [Google Scholar] [CrossRef]
- Paul, R.; Paddison, S.J. A statistical mechanical model for the calculation of the permittivity of water in hydrated polymer electrolyte membrane pores. J. Chem. Phys. 2001, 115, 7762–7771. [Google Scholar] [CrossRef]
- Paul, R.; Paddison, S.J. Structure and dielectric saturation of water in hydrated polymer electrolyte membranes: Inclusion of the internal field energy. J. Phys. Chem. B 2004, 108, 13231–13241. [Google Scholar] [CrossRef]
- Sumeet, J.; Bates, F. Consequences of nonergodicity in aqueous binary PEO-PB micellar dispersions. Macromolecules 2004, 37, 1511–1523. [Google Scholar]
- Elliott, J.A.; Hanna, S.; Elliott, A.M.S.; Cooley, G.E. Interpretation of the small-angle X-ray scattering from swollen and oriented perfluorinated ionomer membranes. Macromolecules 2000, 33, 4161–4171. [Google Scholar] [CrossRef]
- Kim, H.-H.; Glinka, C.J.; Grot, S.A.; Grot, W.G. SANS study of the effects of water vapor sorption in the nanoscale structure of perfluorinated sulfonic acid (Nafion) membranes. Macromolecules 2006, 39, 4775–4787. [Google Scholar] [CrossRef]
- Phillips, K.A.; Moore, R.B. Mechanical and transport property modifications of perfluorosulfonate ionomer membranes prepared with mixed organic and inorganic counterions. J. Polym. Sci. Part B Polym. Phys. 2006, 44, 2267. [Google Scholar] [CrossRef]
- Starkweather, H.W. Crystallinity in perfluorosulfonic acid ionomers and related polymers. Macromolecules 1982, 15, 320–323. [Google Scholar] [CrossRef]
- Krivandin, A.V.; Solov’ena, A.B.; Glagolev, N.N.; Shatalova, O.V.; Kotova, S.L. Structure alterations of perfluorinated sulfocationic membranes under the actoni of ethylene glycol (SAXS and WAXS) studies. Polymer 2003, 44, 5789–5796. [Google Scholar] [CrossRef]
- Schmidt-Rohr, K.; Chen, Q. Parallel cylindrical water nanochannels in Nafion fuel-cell membranes. Nat. Mater. 2008, 7, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Rubatat, L.; Rollet, A.L.; Gebel, G.; Diat, O. Evidence of elongated polymeric aggregates in Nafion. Macromolecules 2002, 35, 4050–4055. [Google Scholar] [CrossRef]
- Paddison, S.J.; Promislow, K.S. Device and Materials Modeling of Polymer Electrolyte Membrane Fuel Cells; Springer: New York, NY, USA, 2009. [Google Scholar]
- Wu, D.S.; Paddison, S.J.; Elliott, J.A. A comparative study of hydrated morphologies of perfluorosulfonic acid fuel cell membranes with mescopic simulations. Energy Environ. Sci. 2008, 1, 284–293. [Google Scholar] [CrossRef]
- Litt, M.H. A reevaluation of nafion morphology. ACS Polym. Prepr. 1997, 38, 80–81. [Google Scholar]
- Whang, G.S.; Kaviany, M.; Gostick, J.T.; Kientiz, B.; Weber, A.Z.; Kim, M.W. Role of water states on water uptake and proton transport in Nafion using molecular simulations and bimodal network. Polymer 2011, 52, 2584–2593. [Google Scholar]
- Nemet-Nasser, S. Micromechanics of actuation of ionic polymer-metal composites. J. Appl. Phys. 2002, 92, 2899–2915. [Google Scholar] [CrossRef]
- Zawodzinski, T.A.; Derouin, C.; Radzinski, S.; Sherman, R.S.; Smith, V.T.; Springer, T.E.; Gottesfeld, S. Water uptake by and transport through Nafion 117 membranes. J. Electrochem. Soc. 1993, 140, 1041–1047. [Google Scholar] [CrossRef]
- Dogonadzem, R.R.; Kornyshev, A.A. Polar solvent structure in the theory of ionic solvation. J. Chem. Soc. Farad. Trans. 1974, 70, 1121–1132. [Google Scholar] [CrossRef]
- Kornyshev, A.A. Nonlocal screening of ions in a structured polar liquid–new aspects of solvent description in electrolyte theory. Electrochim. Acta 1981, 26, 1–20. [Google Scholar] [CrossRef]
- Kornyshev, A.A. Nonlocal electrostatics in solvations. In The Chemical Physics of Solvation, Part A; Dogonadze, R.R., Kalman, E., Kornyshev, A.A., Ulstrup, J., Eds.; Elsevier: Amsterdam, The Netherlands, 1985; pp. 77–118. [Google Scholar]
- Rubinstein, A.; Sherman, S. Influence of the solvent structure on the electrostatic interactions in proteins. Biophys. J. 2004, 87, 1544–1557. [Google Scholar] [CrossRef]
- Bazant, M.Z.; Kilic, M.S.; Storey, B.D.; Ajdari, A. Towards an understanding of induced-charge electrokinetics at large applied voltages in concentrated solutions. Adv. Colloid Interface Sci. 2009, 106, 104119:1–104119:83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Hassanali, A.A.; Shin, Y.K.; Knight, C.; Singer, S.J. The water-amorphous silica interface: Analysis of the Stern layer and surface conduction. J. Chem. Phys. 2011, 134, 024705:1–024705:13. [Google Scholar] [CrossRef]
- Promislow, K.; Ryham, R. The Conductive Polymer Membrane Equations. preprint.
- Moore, R.B.; Martin, C.R. Procedure for preparing solution-cast perfluorosulfonate ionomer films and membranes. Anal. Chem. 1986, 58, 2569–2570. [Google Scholar] [CrossRef]
- Moore, R.B.; Martin, C.R. Chemical and morphological properties of solution-cast perfluorosulfonate ionomers. Macromolecules 1988, 21, 1334–1339. [Google Scholar] [CrossRef]
- Maragliano, L.; Vanden-Eijnden, E. A temperature accelerated method for sampling free energy and determining reaction pathways in rare event simulations. Chem. Phys. Lett. 2006, 426, 168–175. [Google Scholar] [CrossRef]









© 2012 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gavish, N.; Jones, J.; Xu, Z.; Christlieb, A.; Promislow, K. Variational Models of Network Formation and Ion Transport: Applications to Perfluorosulfonate Ionomer Membranes. Polymers 2012, 4, 630-655. https://doi.org/10.3390/polym4010630
Gavish N, Jones J, Xu Z, Christlieb A, Promislow K. Variational Models of Network Formation and Ion Transport: Applications to Perfluorosulfonate Ionomer Membranes. Polymers. 2012; 4(1):630-655. https://doi.org/10.3390/polym4010630
Chicago/Turabian StyleGavish, Nir, Jaylan Jones, Zhengfu Xu, Andrew Christlieb, and Keith Promislow. 2012. "Variational Models of Network Formation and Ion Transport: Applications to Perfluorosulfonate Ionomer Membranes" Polymers 4, no. 1: 630-655. https://doi.org/10.3390/polym4010630