
Conformational Properties of Active Semiflexible Polymers


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Model of Active Polymer
3. Solution of Equation of Motion
4. Results
4.1. Center-of-Mass Motion
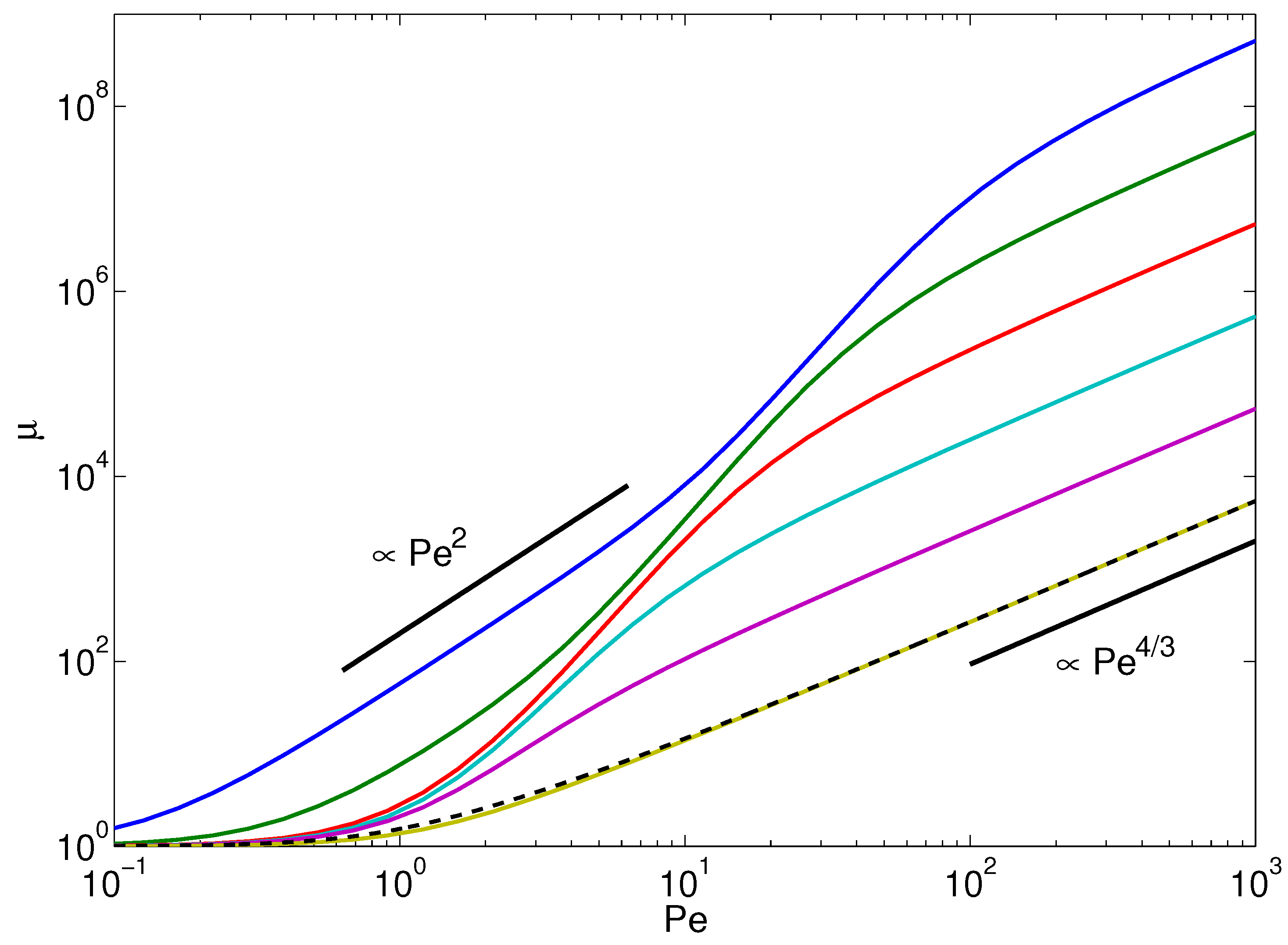
4.2. Lagrangian Multiplier: Stretching Coefficient
- For a passive polymer, implies .
- For and , i.e., ,which yields (cf. Figure 3). Here, there remains a polymer-length dependence for , namely .
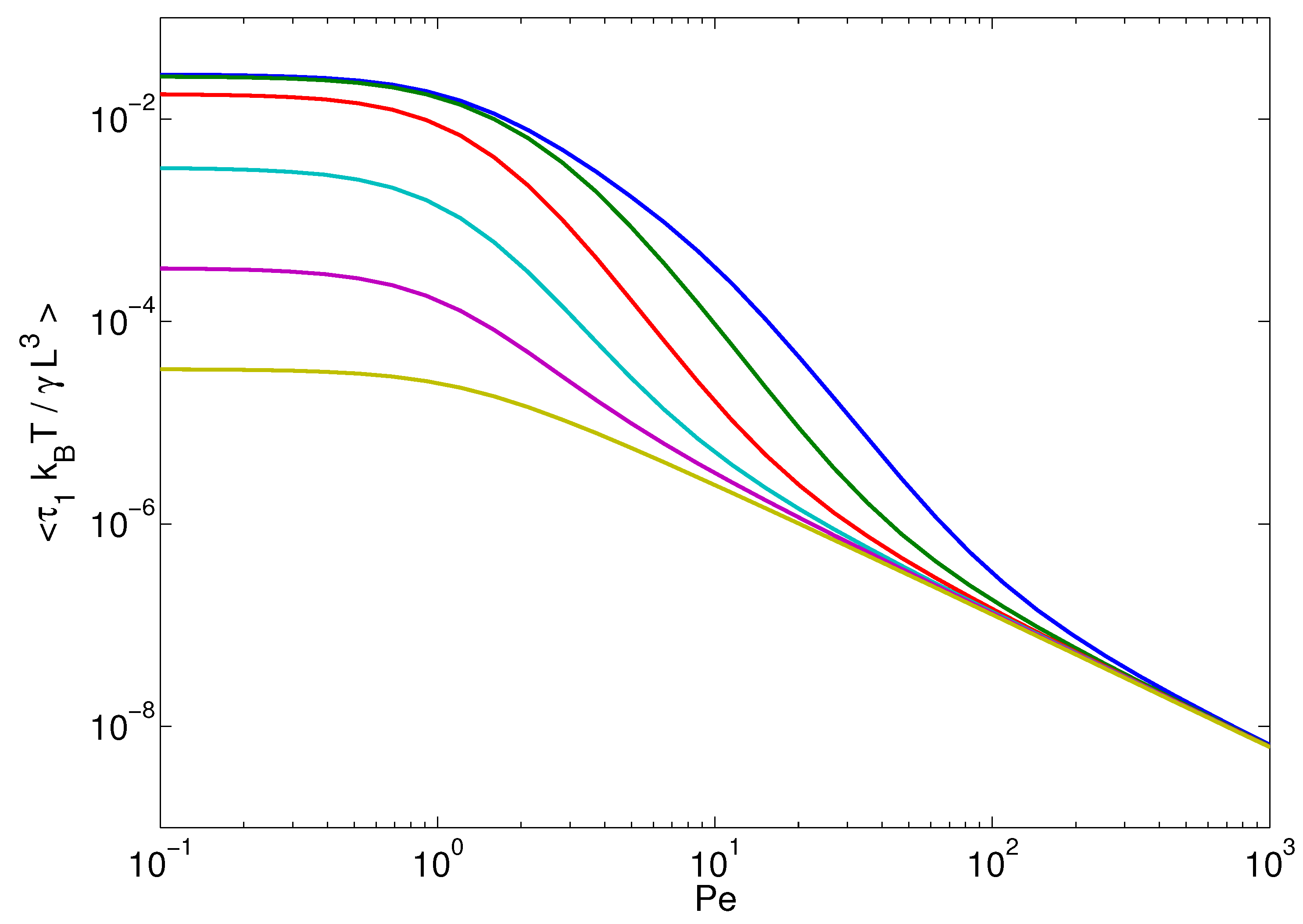
4.3. Relaxation Times
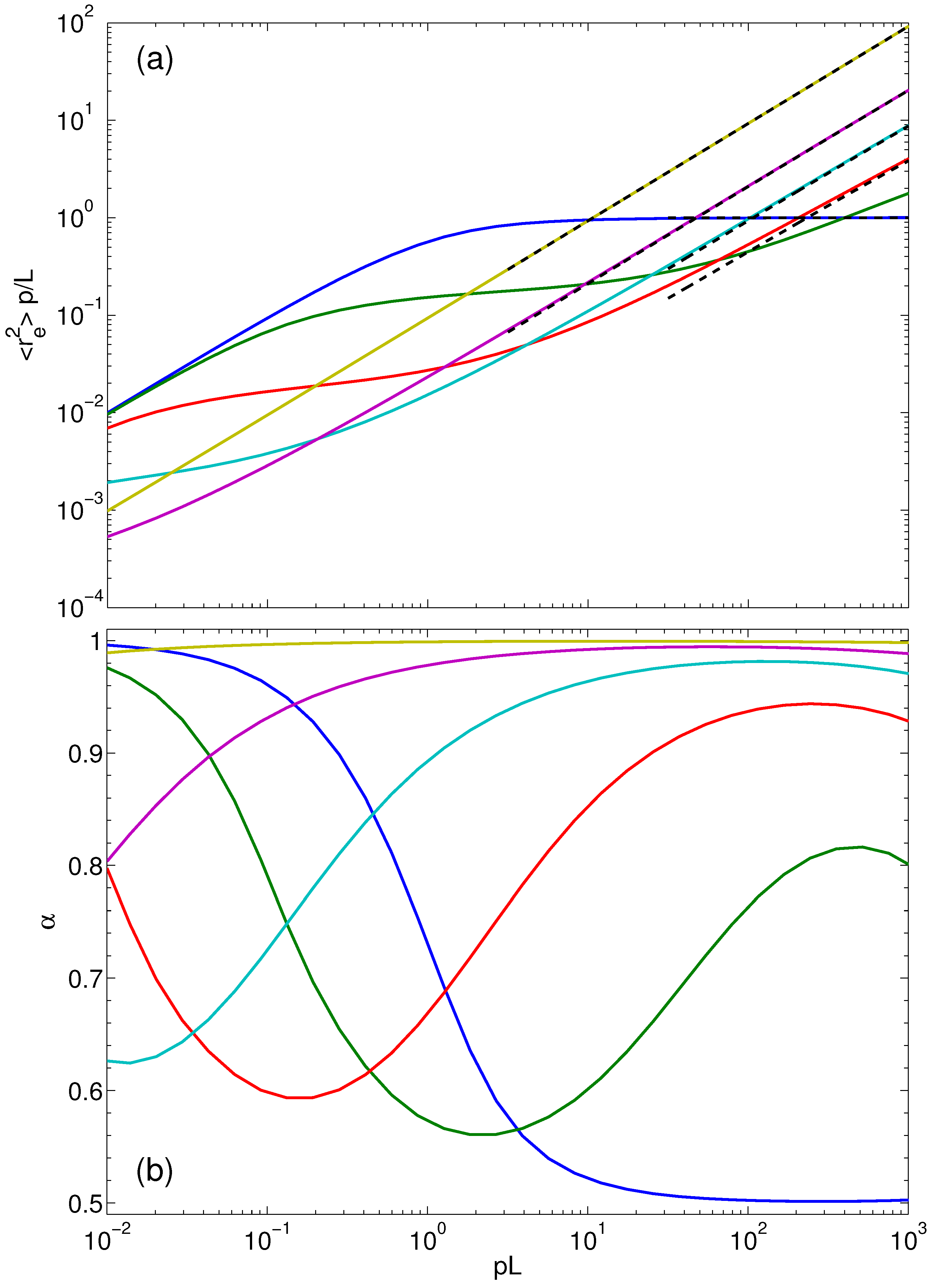
4.4. Mean Square End-to-End Distance
- For finite and , the argument of the hyperbolic tangent function becomes small, and Taylor expansion gives:Insertion of the asymptotic behavior of Equation (33) for the Lagrangian multiplier yields . Hence, the polymers assume nearly stretched conformations independent of the persistence length. This is visible in Figure 6.
- For , such that and , the argument of the hyperbolic tangent function becomes large. By setting the hyperbolic tangent to unity, we obtain:Insertion of the asymptotics of Equation (32) for the stretching coefficient yields . This dependence on the Péclet number is shown in Figure 6 for the polymer with .
5. Summary and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Lauga, E.; Powers, T.R. The hydrodynamics of swimming microorganisms. Rep. Prog. Phys. 2009. [Google Scholar] [CrossRef]
- Ramaswamy, S. The mechanics and statistics of active matter. Annu. Rev. Condens. Matter Phys. 2010, 1, 323–345. [Google Scholar] [CrossRef]
- Vicsek, T.; Zafeiris, A. Collective motion. Phys. Rep. 2012. [Google Scholar] [CrossRef]
- Romanczuk, P.; Bär, M.; Ebeling, W.; Lindner, B.; Schimansky-Geier, L. Active brownian particles. Eur. Phys. J. Spec. Top. 2012, 202, 1–162. [Google Scholar] [CrossRef]
- Marchetti, M.C.; Joanny, J.F.; Ramaswamy, S.; Liverpool, T.B.; Prost, J.; Rao, M.; Simha, R.A. Hydrodynamics of soft active matter. Rev. Mod. Phys. 2013, 85, 1143. [Google Scholar] [CrossRef]
- Elgeti, J.; Winkler, R.G.; Gompper, G. Physics of microswimmers—single particle motion and collective behavior: A review. Rep. Prog. Phys. 2015. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, C.; Di Leonardo, R.; Löwen, H.; Reichhardt, C.; Volpe, G.; Volpe, G. Active Brownian Particles in Complex and Crowded Environments. 2016. Available online: https://arxiv.org/abs/1602.00081 (accessed on 6 June 2016).
- Marchetti, M.C.; Fily, Y.; Henkes, S.; Patch, A.; Yllanes, D. Minimal model of active colloids highlights the role of mechanical interactions in controlling the emergent behavior of active matter. Curr. Opin. Colloid Interface Sci. 2016, 21, 34–43. [Google Scholar] [CrossRef]
- Zöttl, A.; Stark, H. Emergent behavior in active colloids. J. Phys. Condens. Matter 2016. [Google Scholar] [CrossRef] [PubMed]
- Nédélec, F.J.; Surrey, T.; Maggs, A.C.; Leibler, S. Self-organization of microtubules and motors. Nature 1997, 389, 305–308. [Google Scholar] [PubMed]
- Howard, J. Mechanics of Motor Proteins and the Cytoskeleton; Sinauer Associates: Sunderland, MA, USA, 2001. [Google Scholar]
- Kruse, K.; Joanny, J.F.; Jülicher, F.; Prost, J.; Sekimoto, K. Asters, vortices, and rotating spirals in active gels of polar filaments. Phys. Rev. Lett. 2004. [Google Scholar] [CrossRef] [PubMed]
- Bausch, A.R.; Kroy, K. A bottom-up approach to cell mechanics. Nat. Phys. 2006, 2, 231–238. [Google Scholar] [CrossRef]
- Jülicher, F.; Kruse, K.; Prost, J.; Joanny, J.F. Active behavior of the cytoskeleton. Phys. Rep. 2007, 449, 3–28. [Google Scholar] [CrossRef]
- Harada, Y.; Noguchi, A.; Kishino, A.; Yanagida, T. Sliding movement of single actin filaments on one-headed myosin filaments. Nature 1987, 326, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Schaller, V.; Weber, C.; Semmrich, C.; Frey, E.; Bausch, A.R. Polar patterns of driven filaments. Nature 2010, 467, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Prost, J.; Jülicher, F.; Joanny, J.F. Active gel physics. Nat. Phys. 2015, 11, 111–117. [Google Scholar] [CrossRef]
- Berg, H.C. E. Coli in Motion; Biological and Medical Physics Series; Springer: New York, NY, USA, 2004. [Google Scholar]
- Scharf, B. Real-time imaging of fluorescent flagellar filaments of rhizobium lupini H13-3: Flagellar rotation and ph-induced polymorphic transitions. J. Bacteriol. 2002, 184, 5979–5986. [Google Scholar] [CrossRef] [PubMed]
- Copeland, M.F.; Weibel, D.B. Bacterial swarming: A model system for studying dynamic self-assembly. Soft Matter 2009, 5, 1174–1187. [Google Scholar] [CrossRef] [PubMed]
- Kearns, D.B. A field guide to bacterial swarming motility. Nat. Rev. Microbiol. 2010, 8, 634–644. [Google Scholar] [CrossRef] [PubMed]
- Cordoba, A.; Schieber, J.D.; Indei, T. A single-chain model for active gels I: active dumbbell model. RSC Adv. 2014, 4, 17935–17949. [Google Scholar] [CrossRef]
- Sumino, Y.; Nagai, K.H.; Shitaka, Y.; Tanaka, D.; Yoshikawa, K.; Chate, H.; Oiwa, K. Large-scale vortex lattice emerging from collectively moving microtubules. Nature 2012, 483, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Howse, J.R.; Jones, R.A.L.; Ryan, A.J.; Gough, T.; Vafabakhsh, R.; Golestanian, R. Self-motile colloidal particles: From directed propulsion to random walk. Phys. Rev. Lett. 2007. [Google Scholar] [CrossRef] [PubMed]
- Volpe, G.; Buttinoni, I.; Vogt, D.; Kümmerer, H.J.; Bechinger, C. Microswimmers in patterned environments. Soft Matter 2011, 7, 8810–8815. [Google Scholar] [CrossRef]
- Buttinoni, I.; Bialké, J.; Kümmel, F.; Löwen, H.; Bechinger, C.; Speck, T. Dynamical clustering and phase separation in suspensions of self-propelled colloidal particles. Phys. Rev. Lett. 2013. [Google Scholar] [CrossRef] [PubMed]
- Ten Hagen, B.; Kümmel, F.; Wittkowski, R.; Takagi, D.; Löwen, H.; Bechinger, C. Gravitaxis of asymmetric self-propelled colloidal particles. Nat. Commun. 2014. [Google Scholar] [CrossRef] [PubMed]
- Winkler, R.G. Dynamics of flexible active Brownian dumbbells in the absence and the presence of shear flow. Soft Matter 2016, 12, 3737–3749. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Karrila, S.J. Microhydrodynamics: Principles and Selected Applications; Butterworth-Heinemann: Boston, MA, USA, 1991. [Google Scholar]
- Drescher, K.; Dunkel, J.; Cisneros, L.H.; Ganguly, S.; Goldstein, R.E. Fluid dynamics and noise in bacterial cell-cell and cell-surface scattering. Proc. Natl. Acad. Sci. USA 2011, 108, 10940–10945. [Google Scholar] [CrossRef] [PubMed]
- Drescher, K.; Goldstein, R.E.; Michel, N.; Polin, M.; Tuval, I. Direct measurement of the flow field around swimming microorganisms. Phys. Rev. Lett. 2010. [Google Scholar] [CrossRef] [PubMed]
- Guasto, J.S.; Johnson, K.A.; Gollub, J.P. Oscillatory Flows Induced by Microorganisms Swimming in Two Dimensions. Phys. Rev. Lett. 2010. [Google Scholar] [CrossRef] [PubMed]
- Watari, N.; Larson, R.G. The hydrodynamics of a run-and-tumble bacterium propelled by polymorphic helical flagella. Biophys. J. 2010, 98, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Yang, M.; Gompper, G.; Winkler, R.G. Modelling the mechanics and hydrodynamics of swimming E. coli. Soft Matter 2015, 11, 7867–7876. [Google Scholar] [CrossRef] [PubMed]
- Ghose, S.; Adhikari, R. Irreducible representations of oscillatory and swirling flows in active soft matter. Phys. Rev. Lett. 2014. [Google Scholar] [CrossRef] [PubMed]
- Klindt, G.S.; Friedrich, B.M. Flagellar swimmers oscillate between pusher- and puller-type swimming. Phys. Rev. E 2015. [Google Scholar] [CrossRef] [PubMed]
- Peruani, F.; Schimansky-Geier, L.; Bär, M. Cluster dynamics and cluster size distributions in systems of self-propelled particles. Eur. Phys. J. Spec. Top. 2010, 191, 173–185. [Google Scholar] [CrossRef]
- Fily, Y.; Marchetti, M.C. Athermal phase separation of self-propelled particles with no alignment. Phys. Rev. Lett. 2012. [Google Scholar] [CrossRef] [PubMed]
- Bialké, J.; Speck, T.; Löwen, H. Crystallization in a dense suspension of self-propelled particles. Phys. Rev. Lett. 2012. [Google Scholar] [CrossRef] [PubMed]
- Redner, G.S.; Hagan, M.F.; Baskaran, A. Structure and dynamics of a phase-separating active colloidal fluid. Phys. Rev. Lett. 2013. [Google Scholar] [CrossRef] [PubMed]
- Wysocki, A.; Winkler, R.G.; Gompper, G. Cooperative motion of active Brownian spheres in three-dimensional dense suspensions. EPL 2014. [Google Scholar] [CrossRef]
- Ten Hagen, B.; Wittkowski, R.; Takagi, D.; Kümmel, F.; Bechinger, C.; Löwen, H. Can the self-propulsion of anisotropic microswimmers be described by using forces and torques? J. Phys. 2015. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Ripoll, M. A self-propelled thermophoretic microgear. Soft Matter 2014, 10, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Solon, A.P.; Stenhammar, J.; Wittkowski, R.; Kardar, M.; Kafri, Y.; Cates, M.E.; Tailleur, J. Pressure and phase equilibria in interacting active brownian spheres. Phys. Rev. Lett. 2015. [Google Scholar] [CrossRef] [PubMed]
- Solon, A.P.; Fily, Y.; Baskaran, A.; Cates, M.E.; Kafri, Y.; Kardar, M.; Tailleur, J. Pressure is not a state function for generic active fluids. Nat. Phys. 2015, 11, 673–678. [Google Scholar] [CrossRef]
- Takatori, S.C.; Yan, W.; Brady, J.F. Swim pressure: Stress generation in active matter. Phys. Rev. Lett. 2014. [Google Scholar] [CrossRef] [PubMed]
- Maggi, C.; Marconi, U.M.B.; Gnan, N.; Di Leonardo, R. Multidimensional stationary probability distribution for interacting active particles. Sci. Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Ginot, F.; Theurkauff, I.; Levis, D.; Ybert, C.; Bocquet, L.; Berthier, L.; Cottin-Bizonne, C. Nonequilibrium equation of state in suspensions of active colloids. Phys. Rev. X 2015. [Google Scholar] [CrossRef]
- Bertin, E. An equation of state for active matter. Physics 2015, 8, 44. [Google Scholar] [CrossRef]
- Speck, T.; Menzel, A.M.; Bialké, J.; Löwen, H. Dynamical mean-field theory and weakly non-linear analysis for the phase separation of active Brownian particles. J. Chem. Phys. 2015. [Google Scholar] [CrossRef] [PubMed]
- Winkler, R.G.; Wysocki, A.; Gompper, G. Virial pressure in systems of spherical active Brownian particles. Soft Matter 2015, 11, 6680–6691. [Google Scholar] [CrossRef] [PubMed]
- Liverpool, T.B.; Maggs, A.C.; Ajdari, A. Viscoelasticity of solutions of motile polymers. Phys. Rev. Lett. 2001, 86, 4171–4174. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, D.; Thakur, S.; Tao, Y.G.; Kapral, R. Ring closure dynamics for a chemically active polymer. Soft Matter 2014, 10, 9577–9584. [Google Scholar] [CrossRef] [PubMed]
- Chelakkot, R.; Gopinath, A.; Mahadevan, L.; Hagan, M.F. Flagellar dynamics of a connected chain of active, polar, Brownian particles. J. R. Soc. Interface 2013. [Google Scholar] [CrossRef] [PubMed]
- Loi, D.; Mossa, S.; Cugliandolo, L.F. Non-conservative forces and effective temperatures in active polymers. Soft Matter 2011, 7, 10193–10209. [Google Scholar] [CrossRef]
- Ghosh, A.; Gov, N.S. Dynamics of active semiflexible polymers. Biophys. J. 2014, 107, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Isele-Holder, R.E.; Elgeti, J.; Gompper, G. Self-propelled worm-like filaments: Spontaneous spiral formation, structure, and dynamics. Soft Matter 2015, 11, 7181–7190. [Google Scholar] [CrossRef] [PubMed]
- Isele-Holder, R.E.; Jäger, J.; Saggiorato, G.; Elgeti, J.; Gompper, G. Dynamics of self-propelled filaments pushing a load. Soft Matter 2016. [Google Scholar] [CrossRef]
- Laskar, A.; Singh, R.; Ghose, S.; Jayaraman, G.; Kumar, P.B.S.; Adhikari, R. Hydrodynamic instabilities provide a generic route to spontaneous biomimetic oscillations in chemomechanically active filaments. Sci. Rep. 2013. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, G.; Ramachandran, S.; Ghose, S.; Laskar, A.; Bhamla, M.S.; Kumar, P.B.S.; Adhikari, R. Autonomous motility of active filaments due to spontaneous flow-symmetry breaking. Phys. Rev. Lett. 2012. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hou, Z. Motion transition of active filaments: Rotation without hydrodynamic interactions. Soft Matter 2014, 10, 1012–1017. [Google Scholar] [CrossRef] [PubMed]
- Babel, S.; Löwen, H.; Menzel, A.M. Dynamics of a linear magnetic “microswimmer molecule”. EPL 2016. [Google Scholar] [CrossRef]
- Kaiser, A.; Löwen, H. Unusual swelling of a polymer in a bacterial bath. J. Chem. Phys. 2014. [Google Scholar] [CrossRef] [PubMed]
- Valeriani, C.; Li, M.; Novosel, J.; Arlt, J.; Marenduzzo, D. Colloids in a bacterial bath: Simulations and experiments. Soft Matter 2011, 7, 5228–5238. [Google Scholar] [CrossRef]
- Suma, A.; Gonnella, G.; Marenduzzo, D.; Orlandini, E. Motility-induced phase separation in an active dumbbell fluid. EPL 2014. [Google Scholar] [CrossRef]
- Cugliandolo, L.F.; Gonnella, G.; Suma, A. Rotational and translational diffusion in an interacting active dumbbell system. Phys. Rev. E 2015. [Google Scholar] [CrossRef] [PubMed]
- Küchler, N.; Löwen, H.; Menzel, A.M. Getting drowned in a swirl: Deformable bead-spring model microswimmers in external flow fields. Phys. Rev. E 2016. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, A.; Babel, S.; ten Hagen, B.; von Ferber, C.; Löwen, H. How does a flexible chain of active particles swell? J. Chem. Phys. 2015. [Google Scholar] [CrossRef] [PubMed]
- Harder, J.; Valeriani, C.; Cacciuto, A. Activity-induced collapse and reexpansion of rigid polymers. Phys. Rev. E 2014. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Cherstvy, A.G.; Kim, W.K.; Metzler, R. Facilitation of polymer looping and giant polymer diffusivity in crowded solutions of active particles. New J. Phys. 2015. [Google Scholar] [CrossRef]
- Samanta, N.; Chakrabarti, R. Chain reconfiguration in active noise. J. Phys. A Math. Theor. 2016. [Google Scholar] [CrossRef]
- Laskar, A.; Adhikari, R. Brownian microhydrodynamics of active filaments. Soft Matter 2015, 11, 9073–9085. [Google Scholar] [CrossRef] [PubMed]
- Dua, A.; Cherayil, B.J. Chain dynamics in steady shear flow. J. Chem. Phys. 2000. [Google Scholar] [CrossRef]
- Prabhakar, R.; Prakash, J.R. Gaussian approximation for finitely extensible bead-spring chains with hydrodynamic interactions. J. Rheol. 2006, 50, 561–593. [Google Scholar] [CrossRef]
- Dua, A.; Cherayil, B.J. Effect of stiffness on the flow behavior of polymers. J. Chem. Phys. 2000. [Google Scholar] [CrossRef]
- Winkler, R.G.; Keller, S.; Rädler, J.O. Intramolecular dynamics of linear macromolecules by fluorescence correlation spectroscopy. Phys. Rev. E 2006. [Google Scholar] [CrossRef] [PubMed]
- Munk, T.; Hallatschek, O.; Wiggins, C.H.; Frey, E. Dynamics of semiflexible polymers in a flow field. Phys. Rev. E 2006. [Google Scholar] [CrossRef] [PubMed]
- Winkler, R.G. Conformational and rheological properties of semiflexible polymers in shear flow. J. Chem. Phys. 2010. [Google Scholar] [CrossRef] [PubMed]
- Bird, R.B.; Curtiss, C.F.; Armstrong, R.C.; Hassager, O. Dynamics of Polymer Liquids; John Wiley & Sons: New York, NY, USA, 1987. [Google Scholar]
- Winkler, R.G.; Reineker, P. Finite size distribution and partition functions of gaussian chains: Maximum entropy approach. Macromolecules 1992, 25, 6891–6896. [Google Scholar] [CrossRef]
- Marko, J.F.; Siggia, E.D. Stretching DNA. Macromolecules 1995, 28, 8759–8770. [Google Scholar] [CrossRef]
- Winkler, R.G. Deformation of semiflexible chains. J. Chem. Phys. 2003. [Google Scholar] [CrossRef]
- Winkler, R.G. Equivalence of statistical ensembles in stretching single flexible polymers. Soft Matter 2010, 6, 6183–6191. [Google Scholar] [CrossRef]
- Kierfeld, J.; Niamploy, O.; Sa-yakanit, V.; Lipowsky, R. Stretching of semiflexible polymers with elastic bonds. Eur. Phys. J. E 2004, 14, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Salomo, M.; Kegel, K.; Gutsche, C.; Struhalla, M.; Reinmuth, J.; Skokow, W.; Hahn, U.; Kremer, F. The elastic properties of single double-stranded DNA chains of different lengths as measured with optical tweezers. Colloid Polym. Sci. 2006, 284, 1325–1331. [Google Scholar] [CrossRef] [PubMed]
- Blundell, J.R.; Terentjev, E.M. Stretching semiflexible filaments and their networks. Macromolecules 2009, 42, 5388–5394. [Google Scholar] [CrossRef]
- Lamura, A.; Winkler, R.G. Semiflexible polymers under external fields confined to two dimensions. J. Chem. Phys. 2012. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.P.; Binder, K. Stretching semiflexible polymer chains: Evidence for the importance of excluded volume effects from Monte Carlo simulation. J. Chem. Phys. 2012. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, R.; Underhill, P.T. Models of flexible polymers in good solvents: Relaxation and coil–stretch transition. Soft Matter 2012, 8, 6991–7003. [Google Scholar] [CrossRef]
- Manca, F.; Giordano, S.; Palla, P.L.; Cleri, F.; Colombo, L. Theory and Monte Carlo simulations for the stretching of flexible and semiflexible single polymer chains under external fields. J. Chem. Phys. 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manca, F.; Giordano, S.; Palla, P.L.; Cleri, F.; Colombo, L. Response to “Comment on ’Elasticity of flexible and semiflexible polymers with extensible bonds in the Gibbs and Helmholtz ensembles”’. J. Chem. Phys. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliafar, S.; Vezenov, D.; Jagota, A. In-plane force–extension response of a polymer confined to a surface. Eur. Polym. J. 2014, 51, 151–158. [Google Scholar] [CrossRef]
- Alexeev, A.V.; Maltseva, D.V.; Ivanov, V.A.; Klushin, L.I.; Skvortsov, A.M. Force-extension curves for broken-rod macromolecules: Dramatic effects of different probing methods for two and three rods. J. Chem. Phys. 2015. [Google Scholar] [CrossRef] [PubMed]
- Winkler, R.G.; Reineker, P.; Harnau, L. Models and equilibrium properties of stiff molecular chains. J. Chem. Phys. 1994. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Clarendon Press: Oxford, UK, 1986. [Google Scholar]
- Bawendi, M.G.; Freed, K.F. A Wiener integral model for stiff polymer chains. J. Chem. Phys. 1985. [Google Scholar] [CrossRef]
- Battacharjee, S.M.; Muthukumar, M. Statistical mechanics of solutions of semiflexible chains: A path integral formulation. J. Chem. Phys. 1987. [Google Scholar] [CrossRef]
- Langowski, J.B.; Noolandi, J.; Nickel, B. Stiff chain model—Functional integral approach. J. Chem. Phys. 1991, 95, 1266. [Google Scholar] [CrossRef]
- Ha, B.Y.; Thirumalai, D. A mean-field model for semiflexible chains. J. Chem. Phys. 1995, 103, 9408–9412. [Google Scholar] [CrossRef]
- Harnau, L.; Winkler, R.G.; Reineker, P. Dynamic properties of molecular chains with variable stiffness. J. Chem. Phys. 1995, 102, 7750. [Google Scholar] [CrossRef]
- Winkler, R.G.; Harnau, L.; Reineker, P. Distribution functions and dynamical properties of stiff macromolecules. Macromol. Theory Simul. 1997, 6, 1007–1035. [Google Scholar] [CrossRef]
- Winkler, R.G. Semiflexible polymers in shear flow. Phys. Rev. Lett. 2006. [Google Scholar] [CrossRef] [PubMed]
- Winkler, R.G. Diffusion and segmental dynamics of rod-like molecules by fluorescence correlation spectroscopy. J. Chem. Phys. 2007. [Google Scholar] [CrossRef] [PubMed]
- Öttinger, H.C. Stochastic Processes in Polymeric Fluids; Springer: Berlin, Germany, 1996. [Google Scholar]
- Kratky, O.; Porod, G. Röntgenuntersuchung gelöster Fadenmoleküle. Recl. Trav. Chim. PaysBas 1949. [Google Scholar] [CrossRef]
- Aragón, S.R.; Pecora, R. Dynamics of wormlike chains. Macromolecules 1985, 18, 1868. [Google Scholar] [CrossRef]
- Flory, P.J. Statistical Mechanics of Polymer Chains; John Wiley & Sons: New York, NY, USA, 1989. [Google Scholar]
- Rubinstein, M.; Colby, R.C. Polymer Physics; Oxford University Press: Oxford, UK, 2003. [Google Scholar]
- The Equation (10) of Reference 28 contains an error. The factor 2 in front of should be replaced by unity.
- Stenhammar, J.; Marenduzzo, D.; Allen, R.J.; Cates, M.E. Phase behaviour of active Brownian particles: The role of dimensionality. Soft Matter 2014, 10, 1489–1499. [Google Scholar] [CrossRef] [PubMed]







© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eisenstecken, T.; Gompper, G.; Winkler, R.G. Conformational Properties of Active Semiflexible Polymers. Polymers 2016, 8, 304. https://doi.org/10.3390/polym8080304
Eisenstecken T, Gompper G, Winkler RG. Conformational Properties of Active Semiflexible Polymers. Polymers. 2016; 8(8):304. https://doi.org/10.3390/polym8080304
Chicago/Turabian StyleEisenstecken, Thomas, Gerhard Gompper, and Roland G. Winkler. 2016. "Conformational Properties of Active Semiflexible Polymers" Polymers 8, no. 8: 304. https://doi.org/10.3390/polym8080304