1. Introduction
Polyolefins are one of the most globally produced and widely used polymeric materials in the marketplace. The most widespread representative of polyolefins is polypropylene (PP) for its higher melting point, lower density, chemical resistance, rigidity, impact strength and low cost. For these properties, the PP can be used in many manufacturing processes such as compression, foaming, thermoforming, blow moulding, injection moulding, rotational moulding and extrusion coating. PP occurs in two basic types (linear, branched) and is usually processed in a melt state. The key problem with processing of conventional linear PP is its low melt strength, which may limit processing window considerably. Thus, it is not surprising that considerable progress has been made to enhance PP melt strength by introduction of long chain branching via different methods such as electron beam radiation [
1,
2,
3,
4,
5,
6], gamma radiation [
1,
7,
8], UV radiation [
9,
10,
11,
12,
13,
14,
15,
16] or utilizing peroxides (usually in presence of multi-functional monomers) [
17,
18,
19,
20,
21,
22,
23]. It has been reported that branched PPs have a predominantly star-like structure [
5,
24,
25,
26,
27,
28], which causes significant extensional strain hardening inducing a so-called “self-healing” effect leading to improved quality of products (for example, wall thickness uniformity or a better foam structure) due to homogeneous deformation in elongational flows [
28,
29,
30,
31,
32], a more pronounced shear thinning behaviour, and a higher elasticity compared to linear polypropylene [
19,
23,
33,
34]. It is important to mention that the “topological stiffness” is typical for so-called semiflexible polymers such as comb, brush, star, H-shaped and multiarm (pom-pom) branched polymers (including highly branched dendronized polymers having a linear backbone with a very high number of attached dendritic side groups as the extreme case) [
35,
36,
37,
38,
39,
40,
41,
42,
43,
44,
45], i.e., polymers, whose behaviour lies between perfectly flexible and perfectly rigid polymer chains and their molecular elasticity has impact on both single-chain and collective behaviour [
46].
During processing of polypropylenes, degradation reactions can start to occur, which can significantly influence their molecular structure, narrow the processing window, as well as reduce basic properties of the final product considerably. The most attention has been focused on conventional linear PPs. In more detail, it has been shown that free-radical-induced degradation of PP, induced by different amounts of peroxide, leads to reduction in average molecular weight, polydispersity and melt viscosity [
47,
48,
49,
50,
51,
52,
53]. Thermo-mechanical and/or thermo-oxidative degradation of PPs was investigated by using the multiple extrusion process [
54,
55,
56], torque rheometer [
57], rheometric mechanical spectrometer [
58], and windy oven [
59]. It has been demonstrated that Newtonian viscosity [
55], recoverable shear compliance [
55], molecular weight [
54], loss and storage moduli [
58] monotonically decrease with the degradation time mainly due to chain scission of macromolecules driven via β-scission reaction, breakdown of peroxy radicals and shear [
54]. It has been found that during thermo-mechanical degradation, the chain scission at low molecular weight (
MW) is random (i.e.,
MW-independent) but it is increasing with
MW at higher
MW, which is in good agreement with mechanical degradation theory of Bueche, which states that “the probability of chain scission is higher for the high
MW chains to yield smaller molecules of about half the original size” [
56]. The situation was found to be even more complex during thermo-oxidative degradation, during which chain scission can be kinetically favoured near the oxygen-centred radicals leading to a heterogeneous type of degradation and causing change from unimodal to bimodal MWD [
59].
It has been shown that rheology can be used as a powerful tool for understanding degradation of different polymers due to its sensitivity to very small changes in the molecular structure of polymers, as well as for understanding their molecular structure via utilization of advanced constitutive equations, which are based on the molecular arguments [
60,
61,
62,
63,
64,
65]. In more detail, multimode eXtended Pom-Pom (XPP) and PTT-XPP models have been used to understand structural changes of PPs during peroxide-induced modification by the reactive extrusion process [
60,
65]. Even if these models have been found to be useful for a branching level quantification, there are some disadvantages connected with their usage. Firstly, model parameters’ identification process is rather complicated due to high mathematical complexity and huge number of adjustable parameters, which usually require their manual adjustment. Secondly, both models are based on the assumption that branched macromolecules take the “H” shape, which can result in poor model predictions, especially for PPs having different, e.g., star-like, structures [
60].
Even if the degradation behaviour of conventional linear PP is well documented in the open literature, only a little or no information is known about structural changes during thermal degradation of branched polypropylenes, especially with respect to branching, which could be used for process stabilization and optimization of final product properties. With the aim to extend the knowledge in this area, thermal stability of branched polypropylene was investigated in this work by using shear and extensional rheology as well as the GPC technique. Specific attention was paid to quantification and interpretation of the measured data by using four different constitutive equations allowing manual-adjustment-free determination of their parameters as well as quantification/characterization of molecular changes during thermal degradation of branched PP.
2. Experimental
In this work, branched PP Daploy WB180HMS (Borealis Polyolefine, Linz, Austria) has been used. In the first step, the polymer was thermally degraded. For such a purpose, polymer was added into a Rosand RH7-2 twin bore capillary rheometer (Rosand Precision, Stourbridge, England), melted and kept at given times (1, 3, 5 and 7 h) at 240 °C. Obtained thermally degraded polymers together with the virgin polymer were consequently pressed to plates from which the final testing samples were prepared. Linear viscoelastic properties were measured on Advanced Rheometric Expansion System (ARES 2000 model, Rheometric Scientific, Piscataway, NJ, USA) at 170 °C, 180 °C and 190 °C in parallel plates mode. In order to capture Newtonian plateau, shear creep measurements were performed at the reference temperature equal to 180 °C. Transient uniaxial extensional viscosity, η
E, was determined at 170 °C, 180 °C and 190 °C on Sentmanat Extensional Rheometer (SER-HV-A01 model, Xpansion Instruments, Tallmadge, OH, USA [
66,
67,
68]) attached to ARES 2000 rotational rheometer and the obtained measured data were shifted to the reference temperature. It is important to mentioned that time-resolved mechanical spectroscopy was used to ensure that there is virtually no melt degradation during rheological characterization of tested polymer samples for the given temperature range (170 °C–190 °C). Finally, high temperature Gel Permeation Chromatography (GPC) was used to determine number average molecular weight,
Mn, weight average molecular weight,
Mw, Z-average molecular weight,
Mz, and
Z+1-average molecular weight,
Mz+1, for all tested samples.
4. Results and Discussion
The generalized Maxwell model was employed to fit the measured frequency-dependent loss and storage moduli at reference temperature 180 °C for virgin and degraded PPs to generate discrete relaxation spectra (in terms of finite number of λ
k and
Gk pairs), which are provided in
Table 1.
Average relaxation time,
, for each sample was determined according to [
80] as follows:
Basic rheological measurements in the small-amplitude oscillatory shear flow together with the average relaxation time are summarized in
Figure 1,
Figure 2,
Figure 3,
Figure 4 and
Figure 5, from which the following conclusions can be formulated.
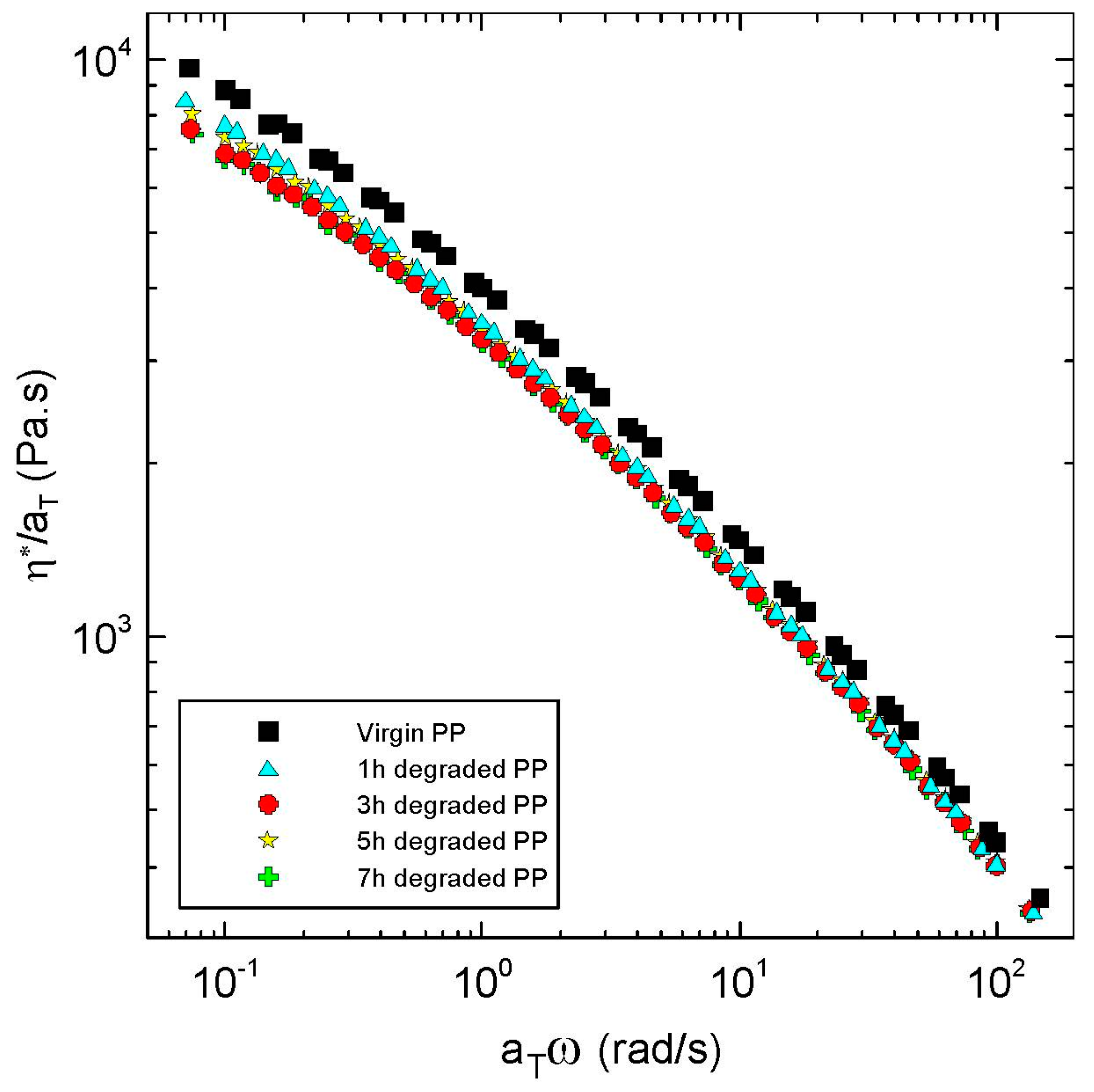
Firstly, it can be considered that the tested polypropylene is thermorheologically simple because all measured points coincide into one single curve in the van Gurp-Palmen plot, which is provided in
Figure 1. This is typical for the star-like branched polypropylenes but not for the polyethylenes having typically tree-like branching and thermorheologically complex behavior [
27]. Thermorheological simplicity of the tested polymer sample justifies utilization of the time-temperature superposition principle to generate master curves in this work.
Secondly, complex viscosity (
Figure 2), shear elasticity captured through Tanδ here (
Figure 3), Newtonian viscosity (
Figure 4) and average relaxation time (
Figure 5) firstly decrease with the degradation time, then surprisingly increase up to the local maximum at five hours and finally decrease again. This indicates simultaneous occurrence of the chain scission and recombination reactions during thermal degradation of the tested polypropylene. This rheological observation is in very good correspondence with the polydispersity index
Mw/
Mn,
Mz and
Mz+1 molecular weight averages that show exactly the same non-monotonic behaviour during thermal degradation as shown in
Figure 6.
Transient extensional viscosity data for virgin as well as thermally degraded branched PPs evaluated at different extensional strain rates (in the range 0.001–14.3 s
−1) are provided in
Figure 7. Here LVE represents Linear Viscoelastic Envelope determined from discontinuous relaxation spectrum. The “steady-state” uniaxial extensional viscosity data were determined from the peaks appearing on the transient viscosity curves for corresponding extensional strain rates. Obtained “steady-state” extensional viscosity data, normalized by the 3 times Newtonian viscosity (measure of the extensional strain hardening and chain branching [
27,
81]), are provided in
Figure 8 as the function of the extensional strain rate. Interestingly, the level of the branching firstly increases within first three hours of thermal degradation and then decreases as deduced from the measure of the extensional strain hardening provided in
Figure 8. This indicates presence of the recombination reactions between polypropylene chains enhancing the branching level, especially within the first 3 h of thermal degradation.
The complex flow behavior observed in the shear and extensional flows as well as GPC data suggests that during the thermal degradation of branched polypropylene melt, the scission of the longest and branched chains is more dominant than recombination reactions, which leads to production of short branched chains from which some of them recombines together and creates medium length chains with enhanced branching level. Continuous reduction of the longest chains in the polymer melt can explain reduction in Newtonian viscosity and shear elasticity whereas increased amount of medium length chains with enhanced level of branching can explain increase in extensional strain hardening at low degradation times. At higher degradation times, it seems that the process is repeated i.e., the chain scission starts to be more dominant than recombination reactions for medium length chains. Consequent reduction of middle length chains and presence of high amount of short chains with enhanced branching level can explain reduction of extensional strain hardening at long degradation times. This hypothesis is supported by the shift of the maximum steady-state uniaxial extensional viscosity to the higher extensional strain rates range for increased degradation time, as visible in
Figure 8.
In the next step, considering validity of the Cox-Merz rule, deformation rate-dependent shear and extensional viscosities were fitted by the Generalized Newtonian law (Equations (1)–(3)) modified White Metzner model (Equations (4)–(5)), Yao model (Equations (6)–(12)) and Extended Yao model (Equations (13)–(15)). All model parameters are summarized in
Table 2,
Table 3,
Table 4 and
Table 5. Fitting error for each model was evaluated via the Average Root Mean Squared Error (ARMSE) defined as
where δ is the number of measured points,
and
represent measured and predicted points and indexes
S and
E stand for Shear and Extensional viscosities (see
Table 6).
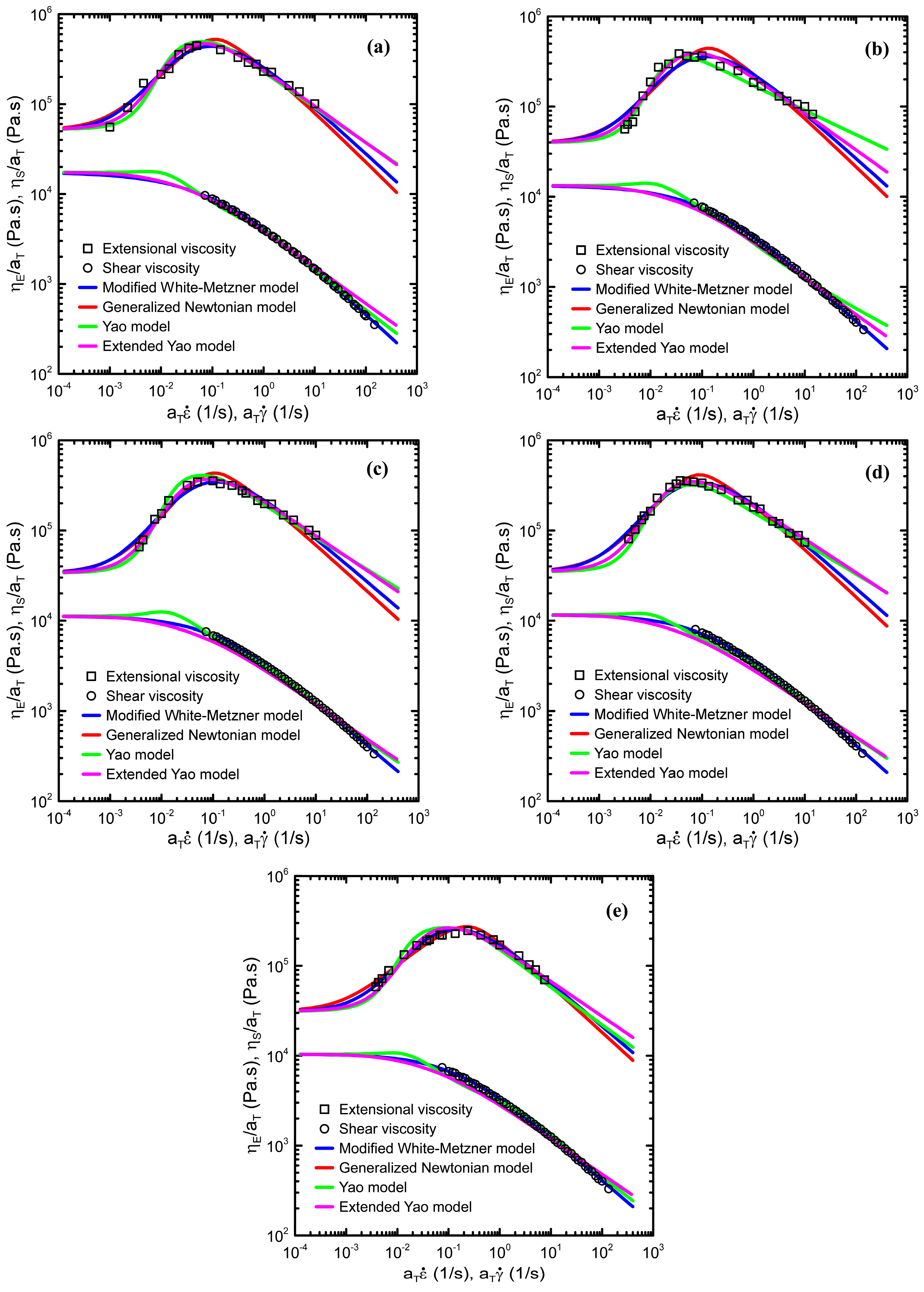
Comparison between model fitting lines and the measured data are provided in
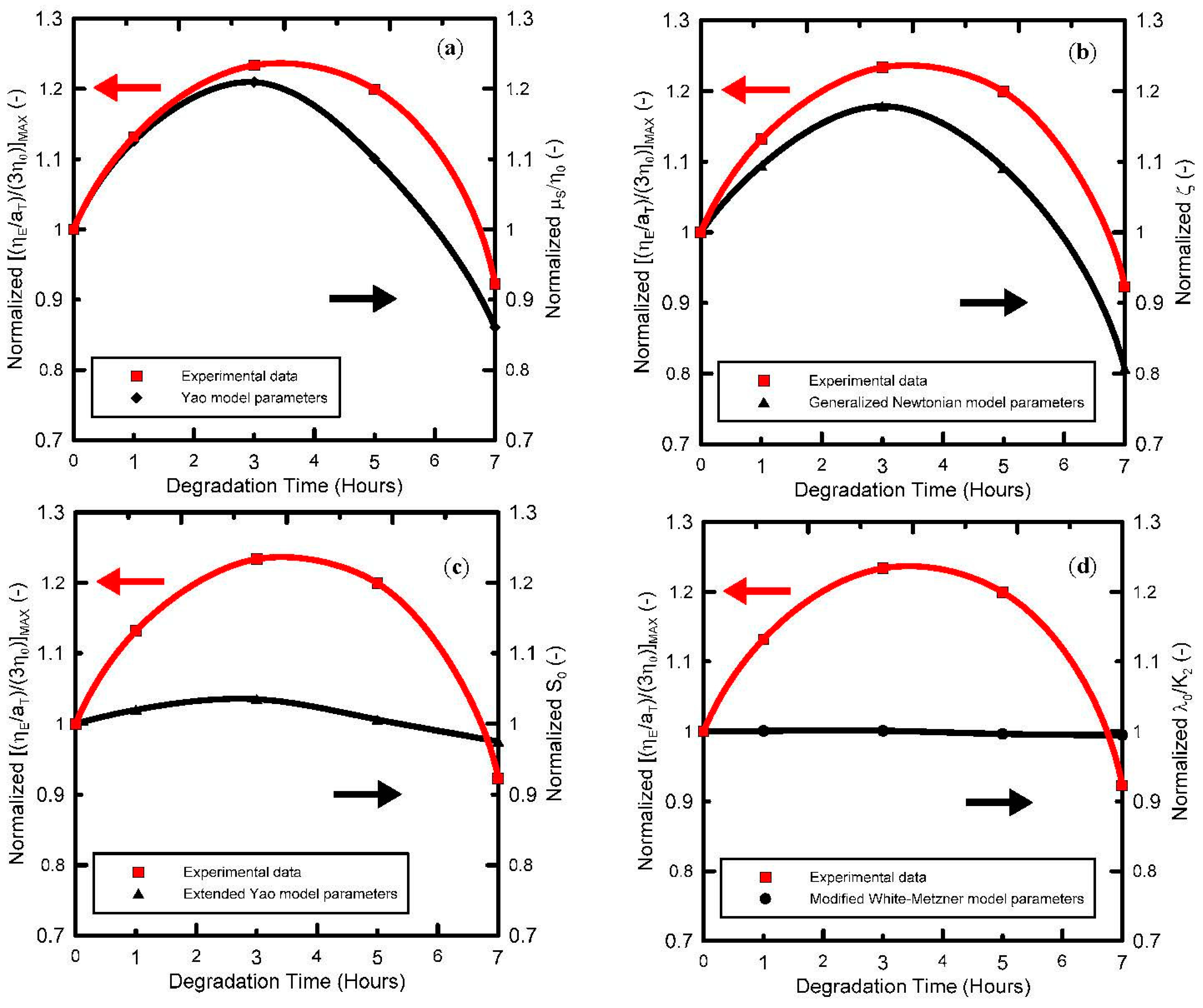
Figure 9. As can be seen, all four models have the capability to describe the measured data very well. Detailed analysis of model parameters has revealed that, firstly, the model macroscopic relaxation time plotted as the function of degradation time (see
Figure 10) follows the same non-monotonic trend with local maximum at 5 hours, which was observed experimentally in basic shear flow characteristics as well as in the
Mz and
Mz+1 molecular weight averages, as discussed above. This suggests that during the thermal degradation of branched PP, the size and weight of the characteristic polymer coil follows the same non-monotonic trend.
Secondly, it has been found that the strain-hardening parameter ζ in the Generalized Newtonian model, strain-hardening
λ0/
K2 parameter in the modified White Metzner model as well as μ
S/η
0 parameter in the Yao model and the ceiling stretch for disentanglement
S0 in the extended Yao model firstly increases with the degradation time, reaching the local maximum at 3 h of thermal degradation, and then decreases, i.e., all the parameters follow a non-monotonic trend in the extensional strain-hardening level with respect to degradation time as observed experimentally. This suggests that all four models can be used to quantify the level of the branching for polypropylenes via their parameters (ζ,
λ0/
K2, μ
S/η
0 and
S0) in a very simple way due to availability of analytical solutions for extensional viscosities and analytical (or in the case of the Extended Yao model, nearly analytical) solutions for steady-state shear viscosity, which makes the experimental data a fitting procedure, both simple and straightforward. It is interesting to note that Yao and Generalized Newtonian models are able to quantify level of extensional strain hardening (i.e., the maximum steady-state uniaxial extensional viscosity divided by three-fold Newtonian viscosity) as a function of degradation time via their parameters, not only quantitatively but also qualitatively as shown in
Figure 11. It is important to mention that Yao model parameter μ
S/η
0 represents the relative resistance against the slip of polymer coils, which depends on their overlap as well as on their capability to create entanglements during the flow. As it has already been shown, the size and the weight of polymer coils during thermal degradation decrease during the first 3 h. Thus, the observed increase in the Yao parameter μ
S/η
0 during the first 3 h of thermal degradation can be explained via enhanced capability to create entanglements between the coils due to increased number of branches but having reduced molecular weight in comparison with the virgin polymer. On the other hand, decrease in μ
S/η
0 for degradation lasting more than 3 h can be interpreted as a decrease in number of branches.
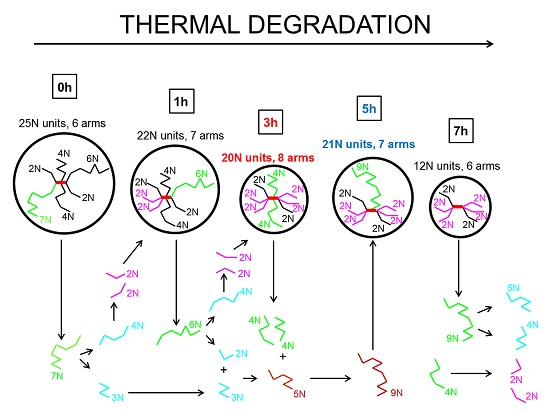
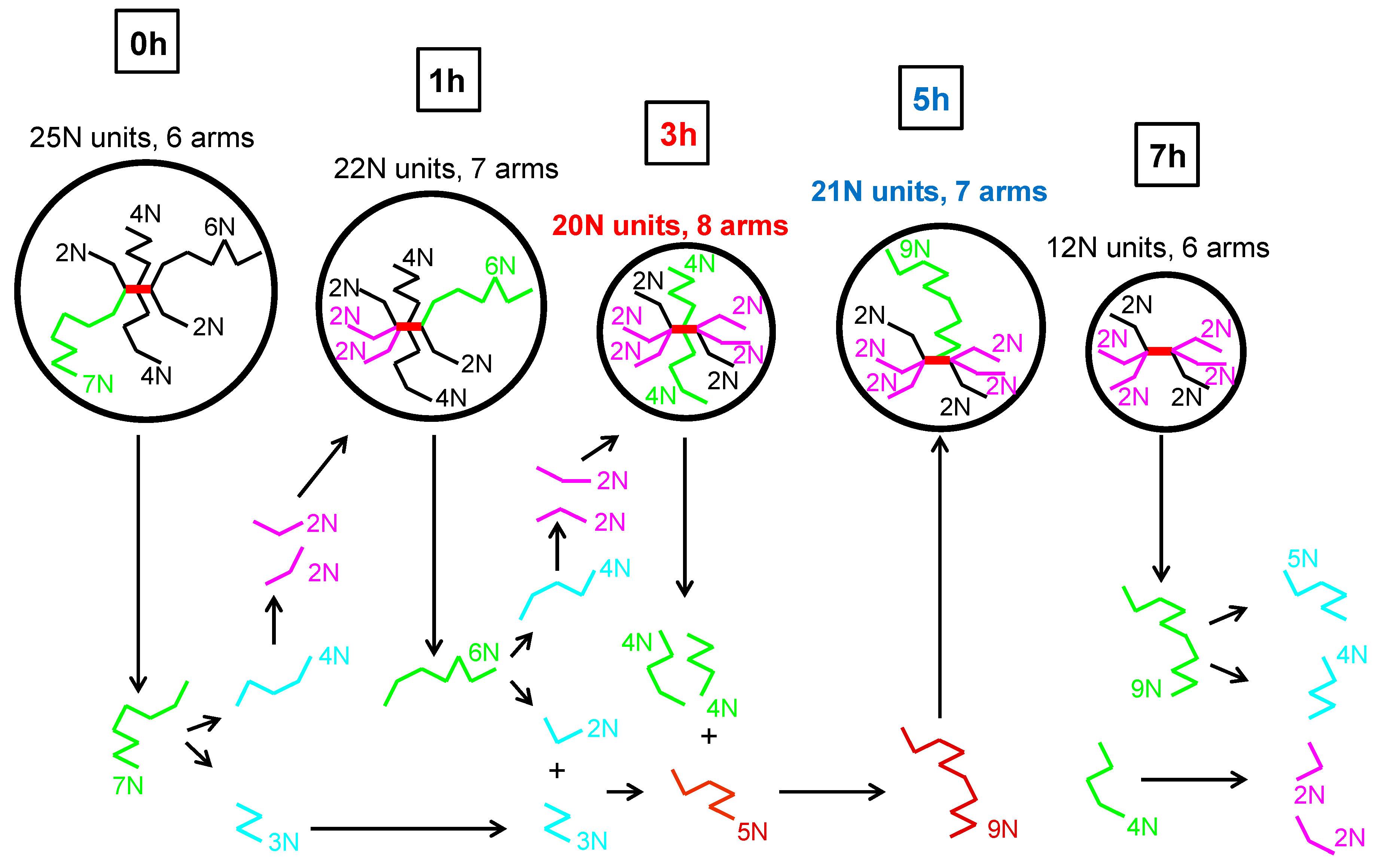
Therefore, local increase in shear viscosity, elasticity, relaxation time and molecular weight of the coil at 5 h of thermal degradation could be explained by the presence of a lower number of branches where some of them have higher molecular weight in comparison with the 3 h degraded sample. Simplified visualization of possible changes in “characteristic” star polymer coil during thermal degradation according to collected experimental data and the above-described hypothesis is provided in
Figure 12. Here, the coil size is proportional to the molecular weight, the longest branches are represented by the green colour, and branches formed via branch scission and recombination reactions are represented here by blue/purple and brown colours, respectively. It should also be mentioned that the presented image of molecular changes during thermal degradation of branched polypropylene is consistent with the Bueche degradation theory and conclusions from [
56], i.e., that the longest chains break first.
5. Conclusions
In this work, thermal stability of the branched polypropylene was investigated via shear and extensional rheology, Gel Permeation Chromatography (GPC) measurements and constitutive equations. It was shown that the studied polypropylene is thermorheologically simple showing simultaneous occurrence of chain scission and branching during thermal degradation. In more detail, complex viscosity, shear elasticity, Newtonian viscosity and average relaxation time was found to decrease with the degradation time within first 3 h, then increase up to the local maximum at five hours and finally decrease again indicating simultaneous occurrence of the chain scission and recombination reactions. This rheological observation was found to be in good correspondence with the changes in polydispersity index Mw/Mn, Mz and Mz+1 molecular weight averages. Consequent analysis of the virgin and thermally degraded polymer melts in the uniaxial extensional flows has revealed that the level of extensional strain hardening (i.e., the level of chain branching) increases within the first three hours of thermal degradation and then decreases. This indicated dominance of recombination and scission reactions on the chain branches before and after 3 h of thermal degradation, respectively.
It has been found that the utilized Generalized Newtonian law, modified White Metzner model, Yao model and Extended Yao model have the capability to describe the measured steady state shear and extensional rheology of the virgin as well as degraded branched polypropylenes. Analysis of the rheological data via the utilized model parameters has revealed that, firstly, the model macroscopic relaxation time plotted as the function of degradation time has been found to follow the same non-monotonic trend with local maximum at 5 h, which was observed experimentally in basic shear flow and molecular weight characteristics (Mw/Mn, Mz, Mz+1); Secondly, it has been found that the strain hardening parameter ζ in the Generalized Newtonian model, strain hardening λ0/K2 parameter in the modified White Metzner model as well as μS/η0 parameter in the Yao model and the ceiling stretch for disentanglement S0 in the extended Yao model follows the non-monotonic trend in the extensional strain hardening level with respect to degradation time as observed experimentally, i.e., all four models were found to have the capability to quantify the level of the branching for virgin as well as degraded polypropylenes. Interestingly, Yao and Generalized Newtonian models showed the capability to quantify level of extensional strain hardening (i.e., the maximum steady-state uniaxial extensional viscosity divided by three-fold Newtonian viscosity) as a function of degradation time not only quantitatively but also qualitatively. Finally, based on the Yao’s μS/η0 parameter (characterizing the resistance against the slip of the coil), it was suggested that the local maximum at 5 h in shear rheological characteristics and molecular weight could be explained by the presence of high molecular weight branches. On the other hand, the local minimum in shear rheology characteristics and molecular weights with simultaneous occurrence of maximum in extensional strain hardening observed at 3 h of thermal degradation was suggested to be due to the presence of a high number of short branches.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}