Strategies to Fabricate Polypeptide-Based Structures via Ring-Opening Polymerization of N-Carboxyanhydrides


Abstract
:1. Introduction
2. Synthesis of α-Amino Acid N-Carboxyanhydrides
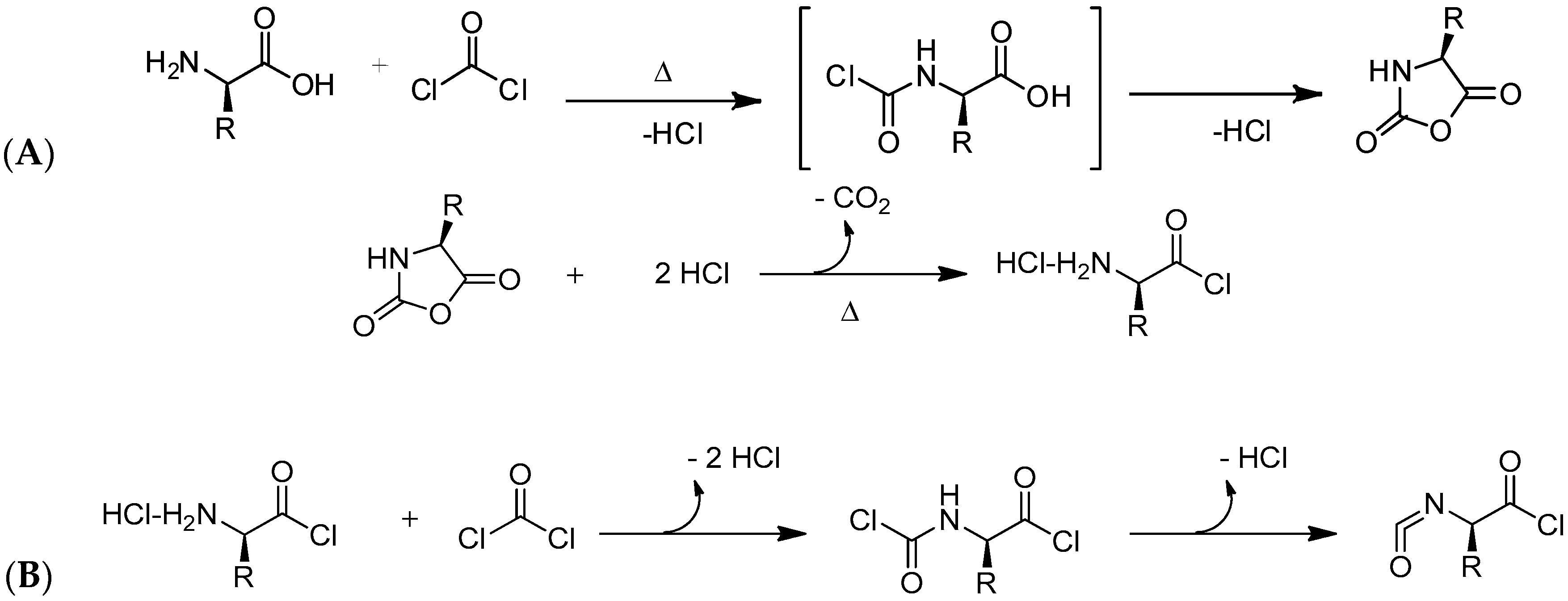
2.1. Pioneer Works about the Cyclization of Aminoacids to Synthesize NCA and Currently Employed Methodologies
2.2. Functional NCA Monomers
2.2.1. Protected Functional Groups Using Natural Occurring Amino Acids
2.2.2. Other Side Chain Functionalized NCAs
3. Polymerization of α-Amino Acid N-Carboxyanhydrides
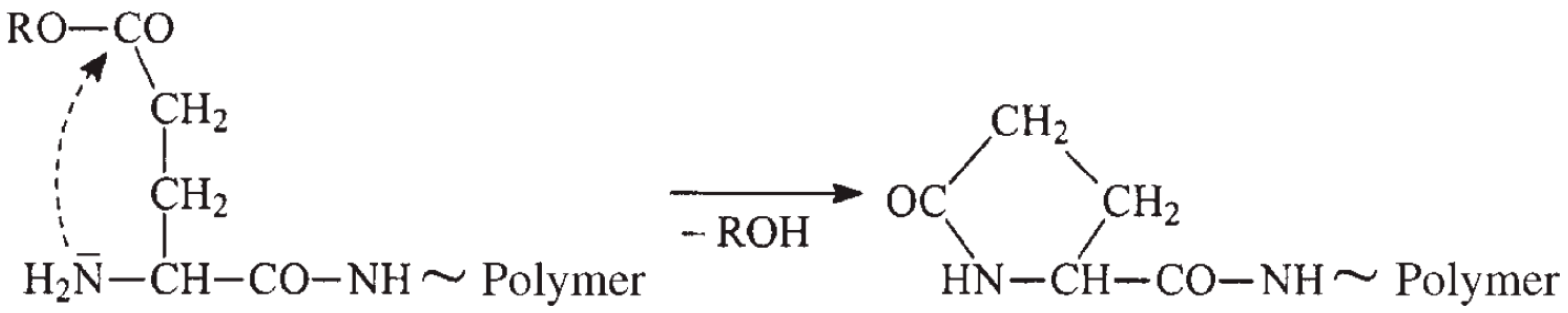
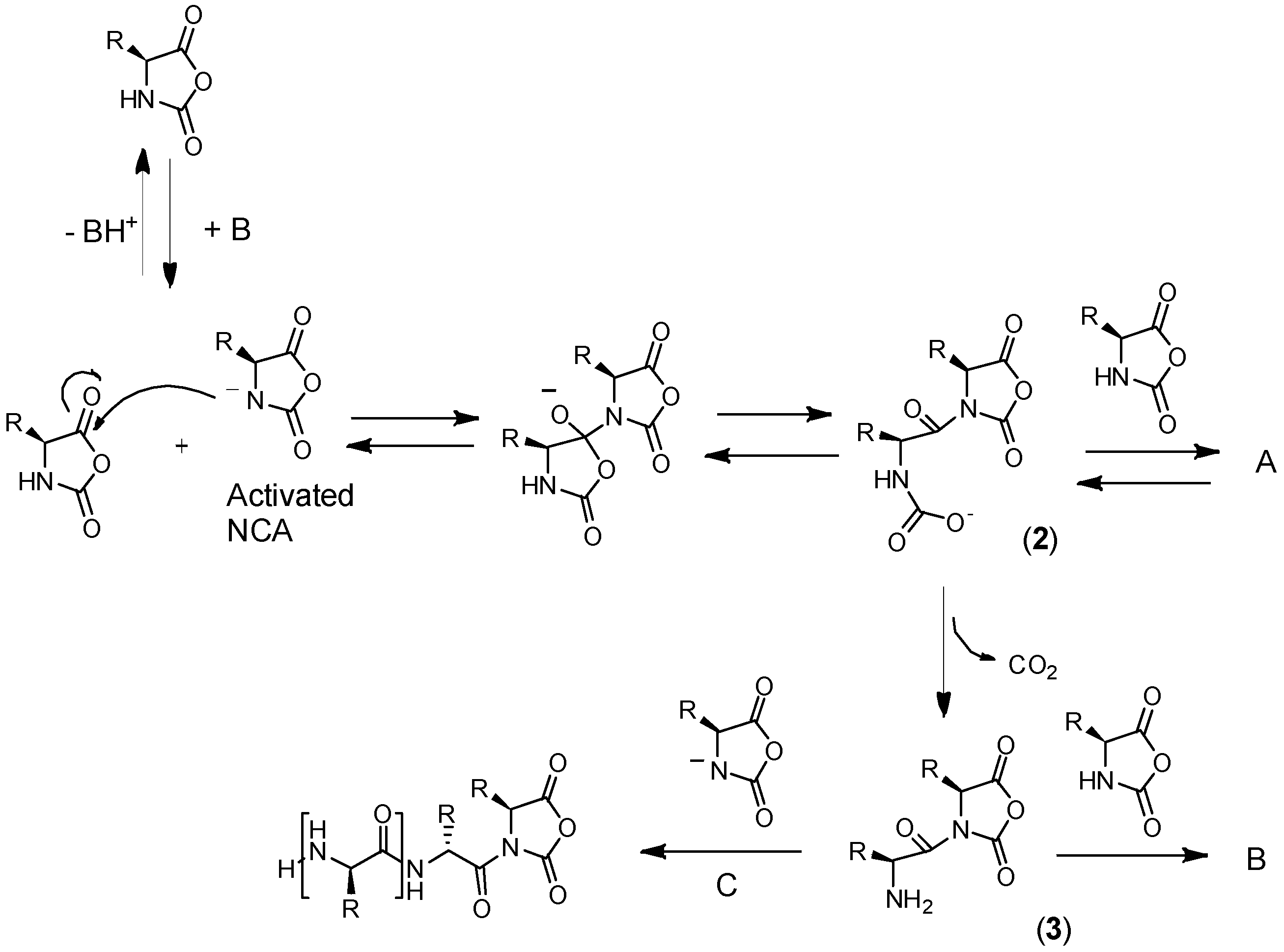
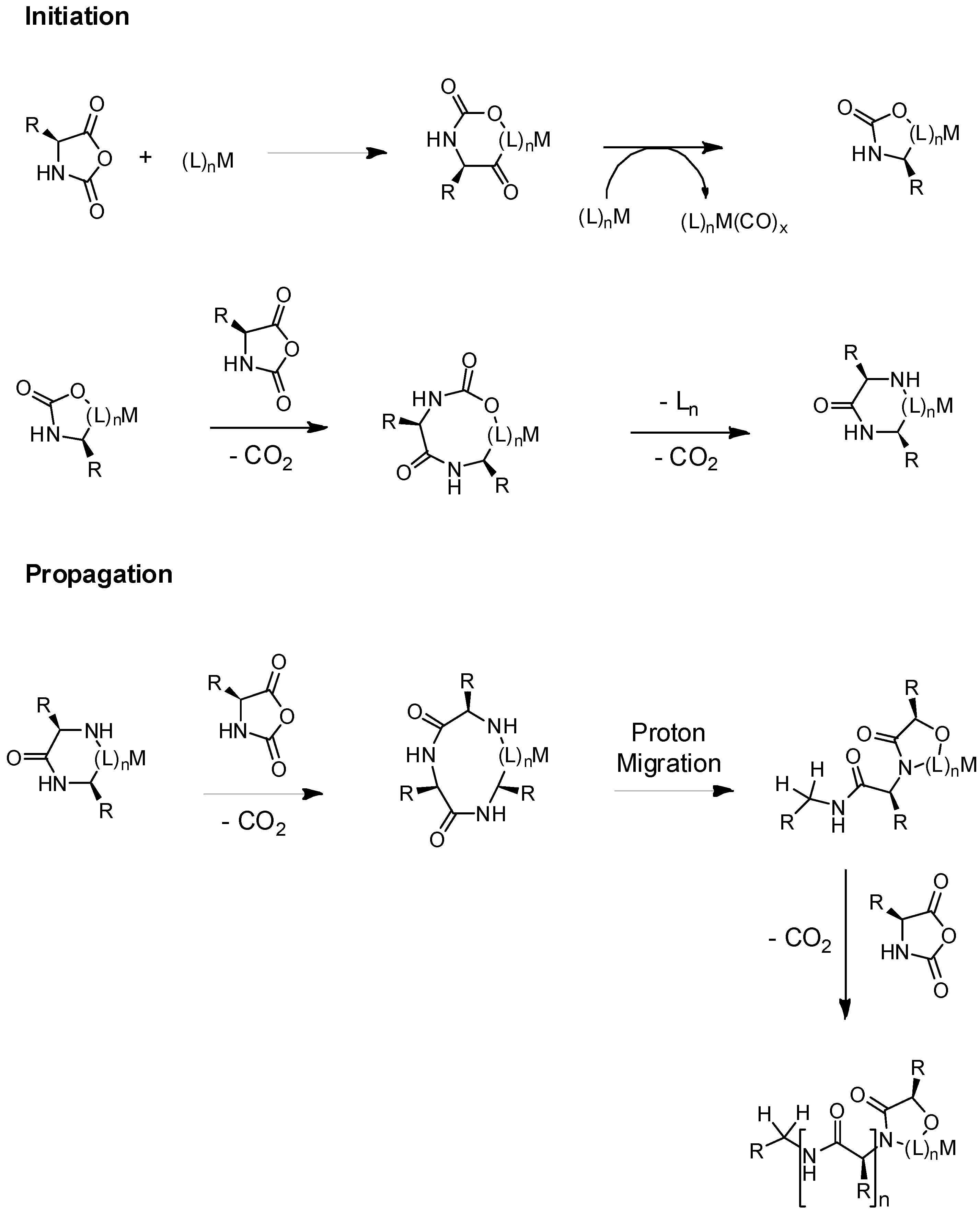
3.1. General Mechanisms and Historical View
3.2. Improvements in the Polymerization of α-Amino Acid N-Carboxyanhydrides
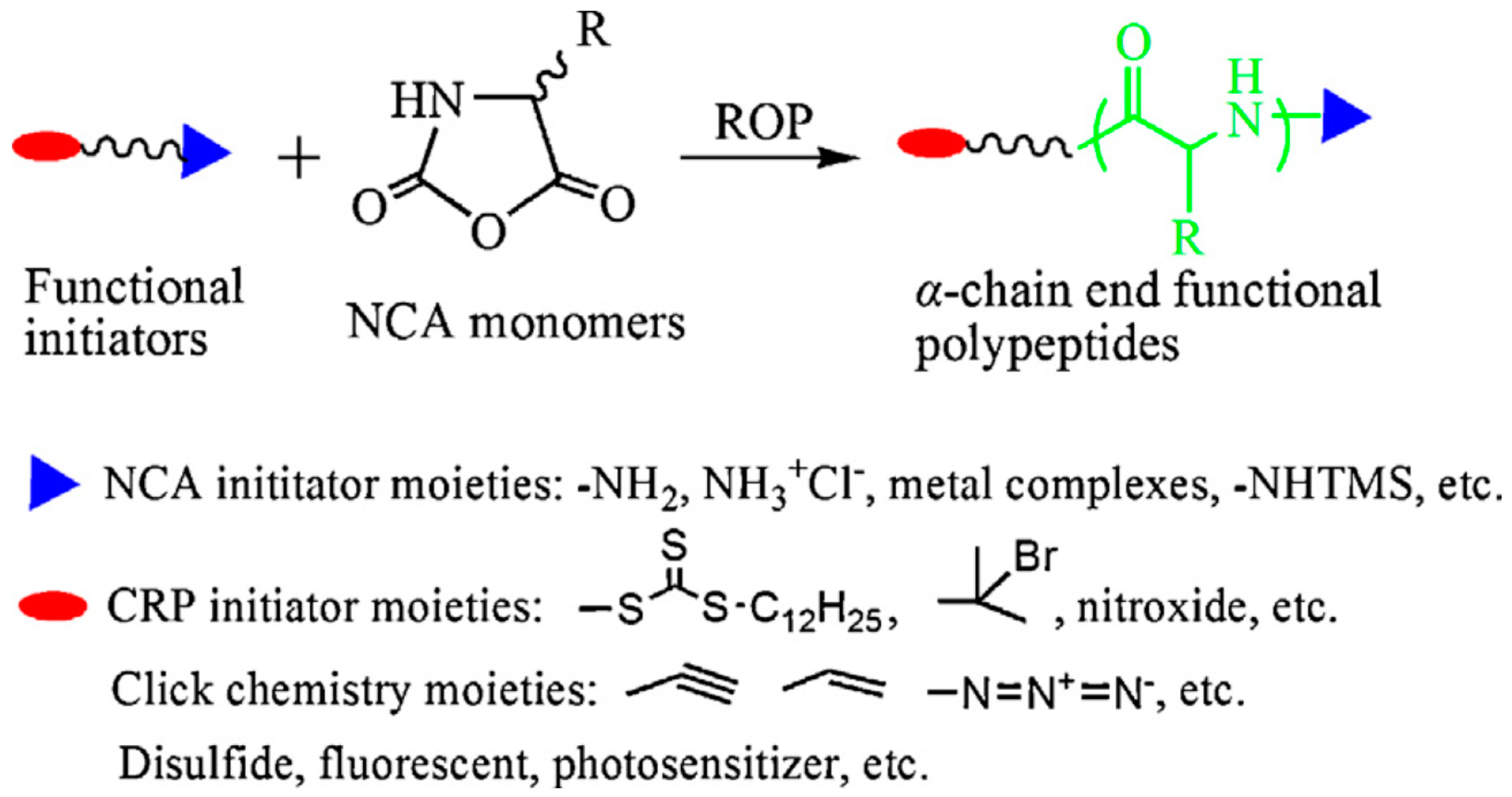
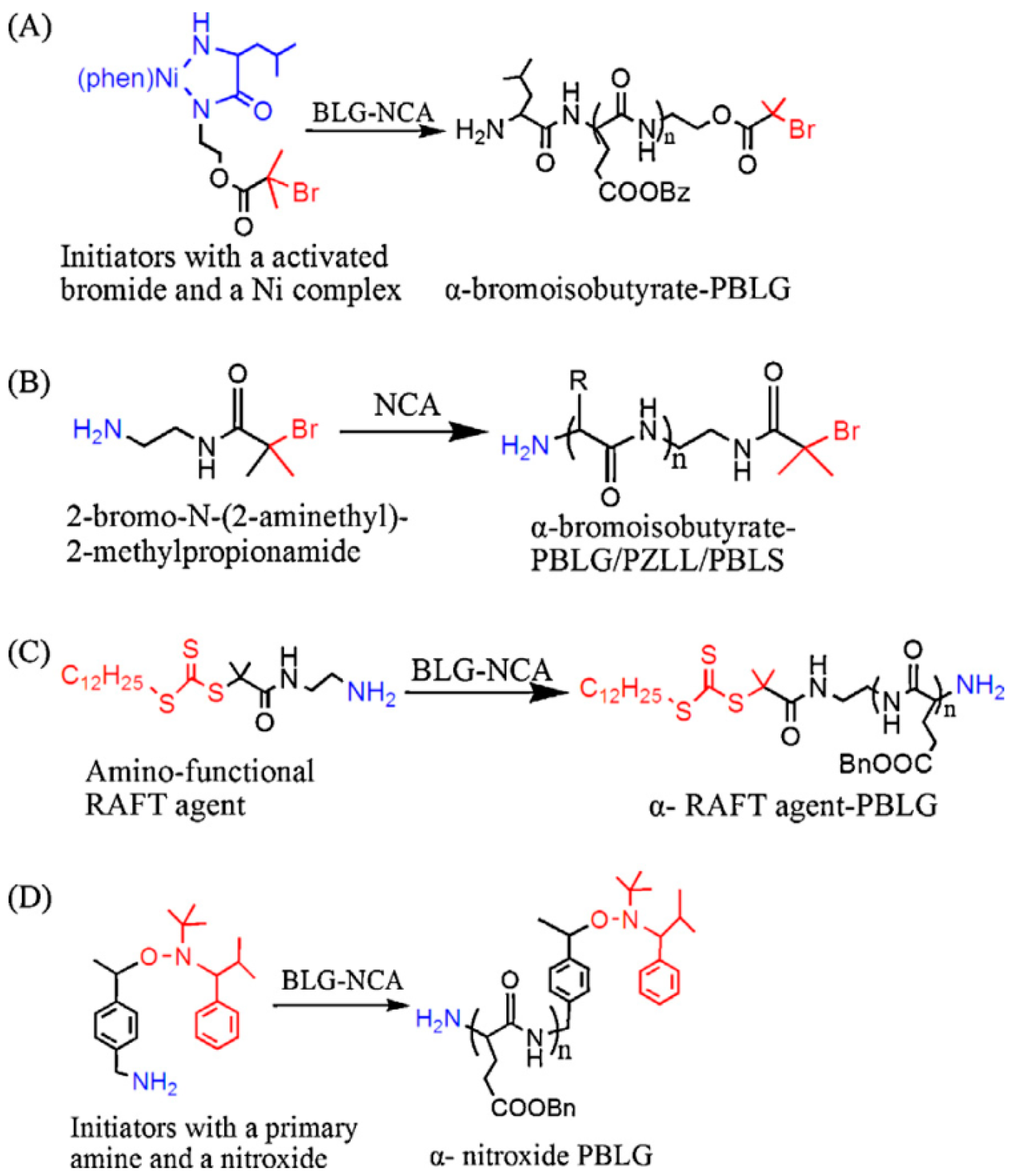
3.2.1. Strategies to Improve Control over Chain Length and Dispersity: Role of the Initiator and Mediation of the Ring-Opening Polymerization
3.2.2. Optimization of the Experimental Conditions for the NCA Polymerization
3.2.3. Alternative Cyclic Monomers to NCAs for the Fabrication of Polypeptides
3.2.4. Improvement of the Polymerization Kinetics While Maintaining the Living Features
4. Linear Side Chain-Functionalized Polypeptides
4.1. Homo and Copolypeptides Bearing Side-Chain Functional Groups
4.1.1. Chemical Modification of Homopolypeptides
4.1.2. Synthesis and Post-Modification of Random Copolypeptides
4.2. Strategies for the Preparation of Block Copolymers Using NCAs
4.2.1. Block Copolymers Comprising Exclusively Polypeptides: Block Copolypeptides
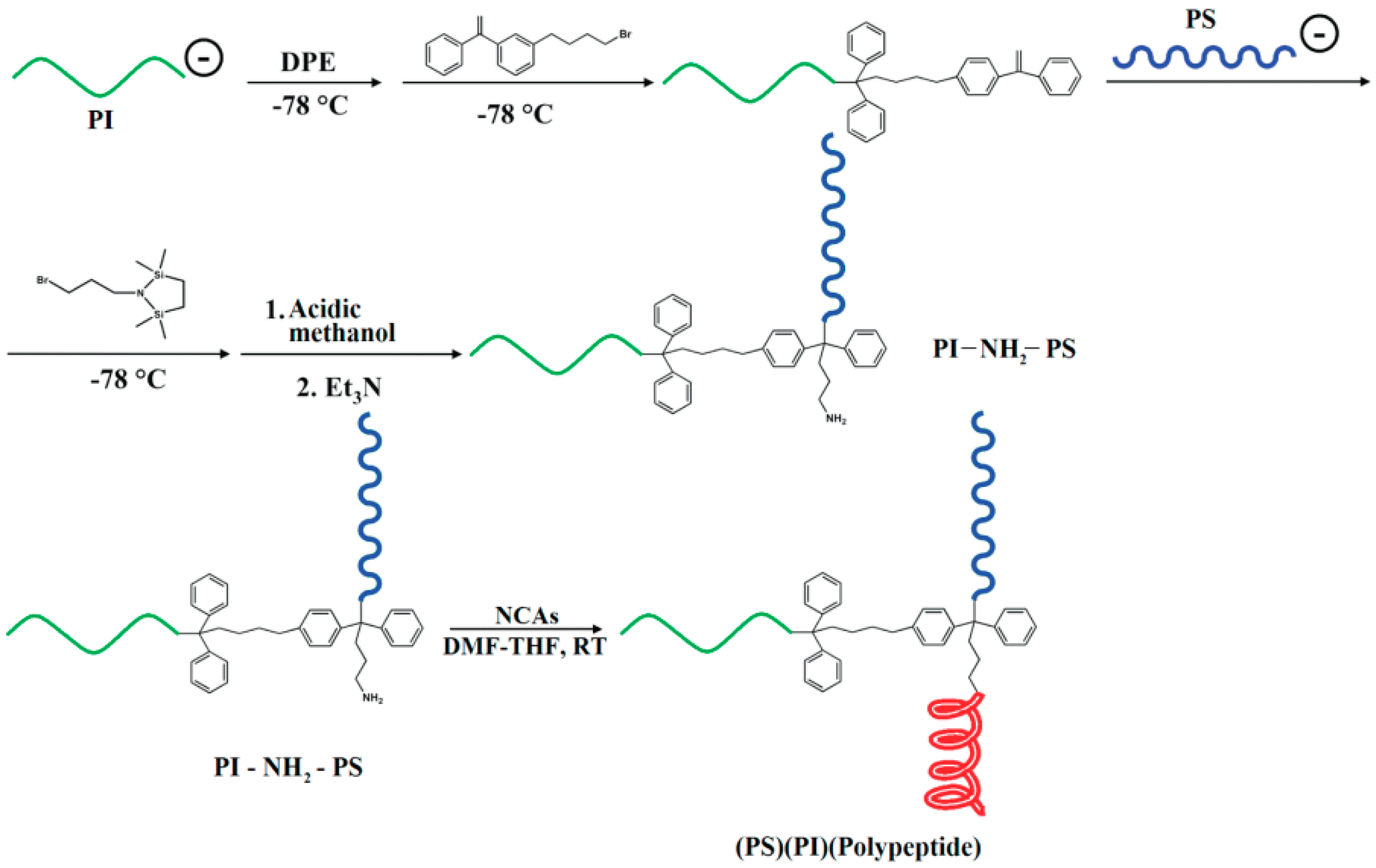
4.2.2. Preparation of Linear Hybrid-Polypeptide Block Copolymers
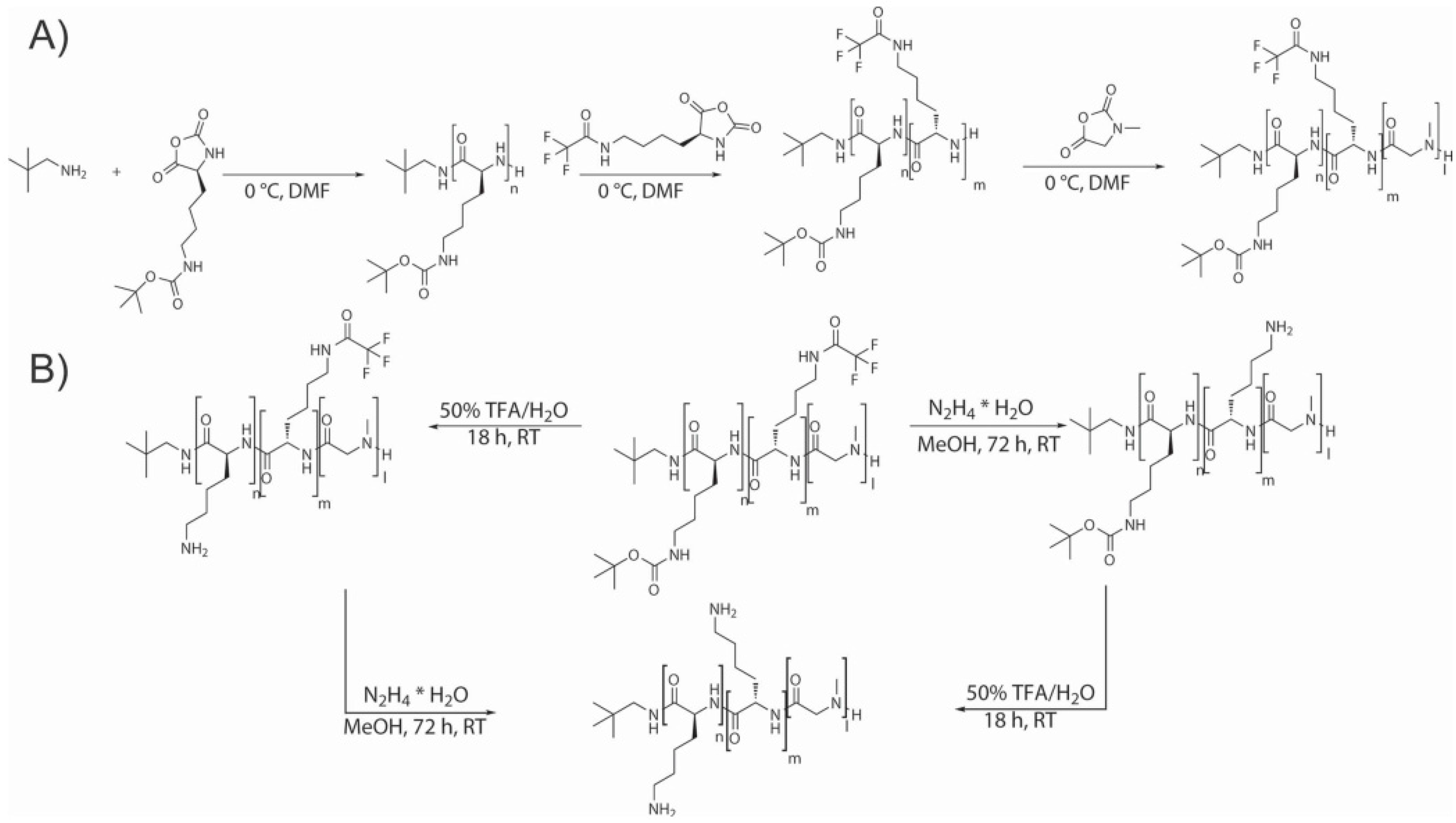
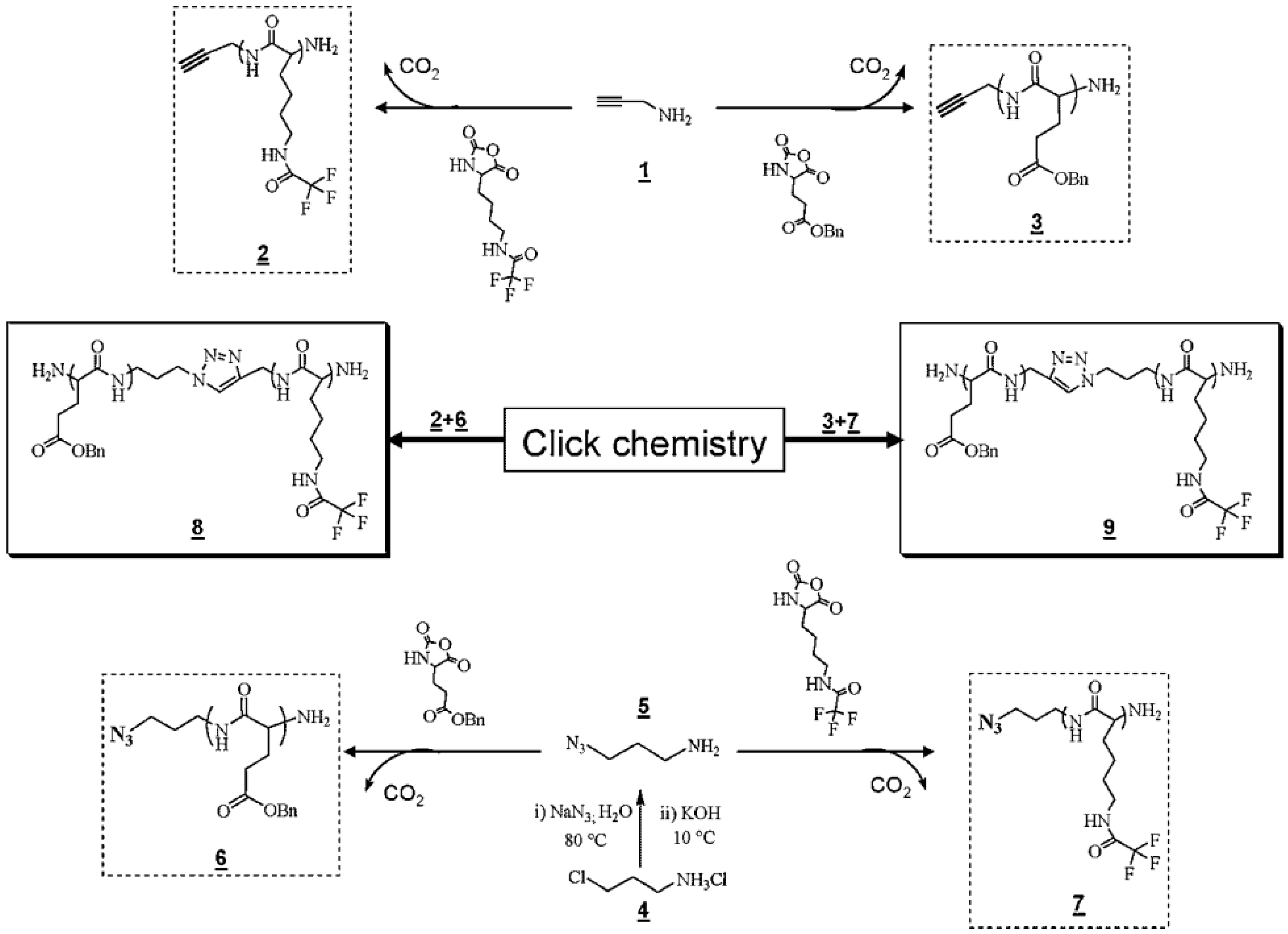
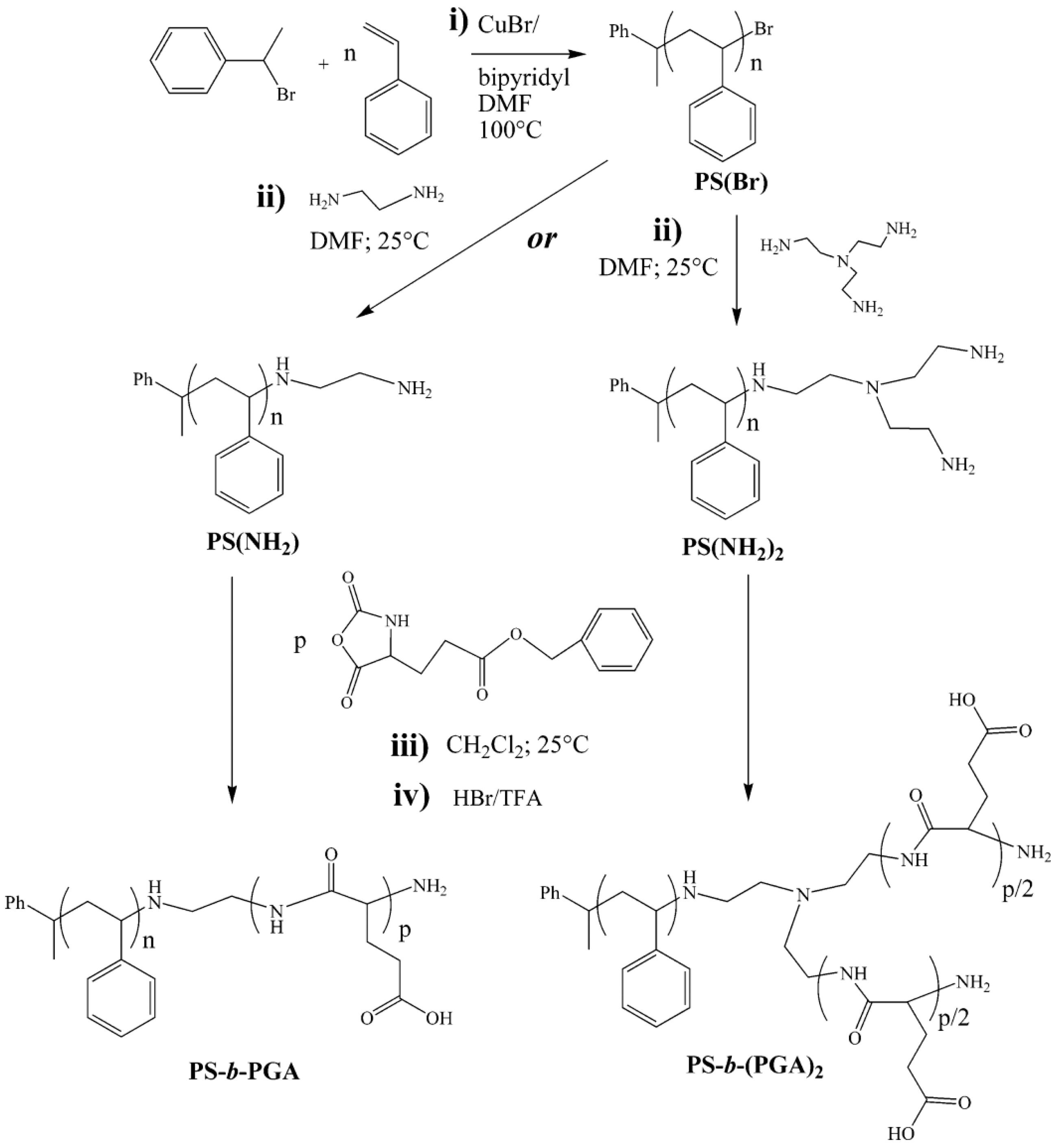
Chain-End Modification of Polypeptides
Fabrication of Hybrid Block Polypeptides
4.2.3. Post-Modification of Block Copolypeptides
5. Strategies to Fabricate Cyclic Polypeptides
6. Non-Linear Polypeptide Architectures: Star, Brush and Highly Branched Polypeptides
6.1. Synthesis of Star-Shaped Polypeptides
6.2. Grafted Polypeptides and Other Highly Branched Structures
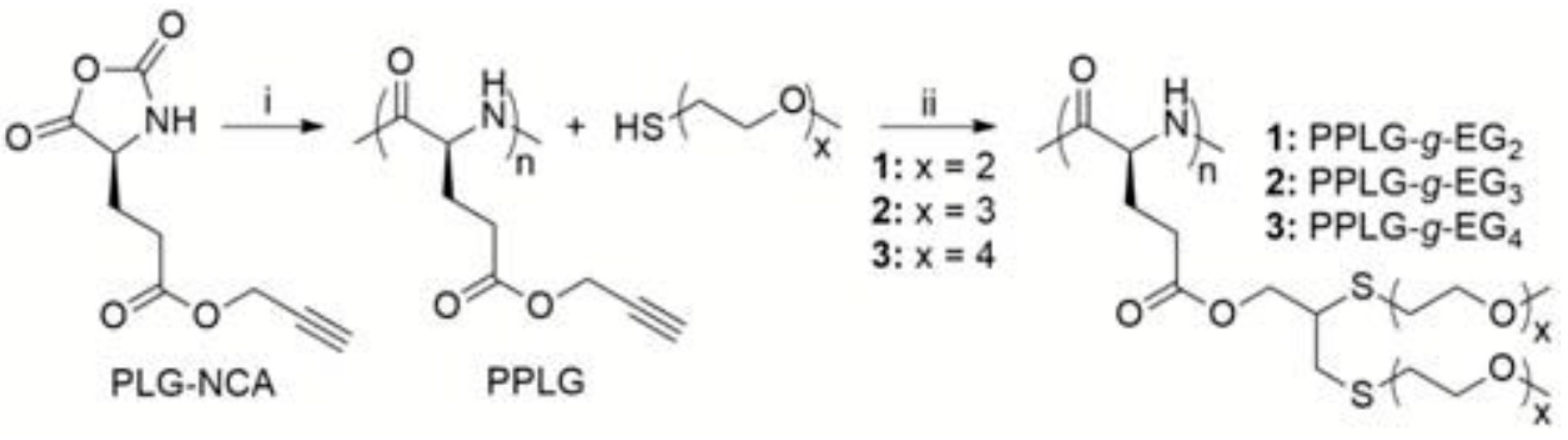
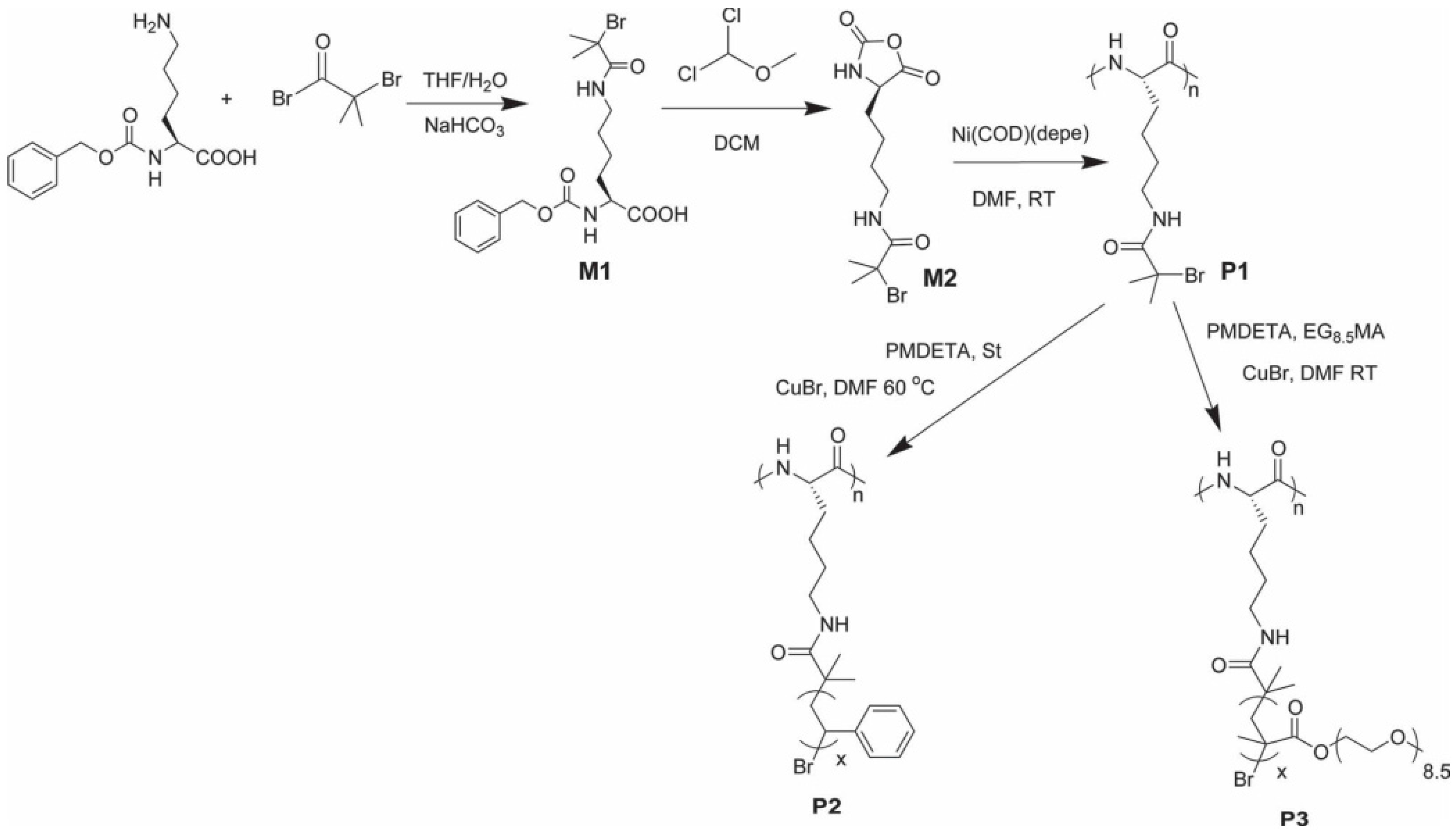
6.2.1. Methods for the Preparation of Polypeptide Brushes
6.2.2. Dendritic Graft, Arborescent and Hyperbranched Polypeptide Architectures
7. Summary and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Leuchs, H.; Manasse, W. Über die Isomerie der Carbäthoxyl—Glycyl glycinester. Eur. J. Inorg. Chem. 1907, 40, 3235–3249. [Google Scholar] [CrossRef]
- Deming, T.J. Methodologies for preparation of synthetic block copolypeptides: Materials with future promise in drug delivery. Adv. Drug Deliv. Rev. 2002, 54, 1145–1155. [Google Scholar] [CrossRef]
- Deming, T.J. Synthetic polypeptides for biomedical applications. Prog. Polym. Sci. 2007, 32, 858–875. [Google Scholar] [CrossRef]
- Hu, H.; Gopinadhan, M.; Osuji, C.O. Directed self-assembly of block copolymers: A tutorial review of strategies for enabling nanotechnology with soft matter. Soft Matter 2014, 10, 3867–3889. [Google Scholar] [CrossRef] [PubMed]
- Matyjaszewski, K. Macromolecular engineering: From rational design through precise macromolecular synthesis and processing to targeted macroscopic material properties. Prog. Polym. Sci. 2005, 30, 858–875. [Google Scholar] [CrossRef]
- Rodriguez, A.R.; Choe, U.-J.; Kamei, D.T.; Deming, T.J. Fine Tuning of Vesicle Assembly and Properties Using Dual Hydrophilic Triblock Copolypeptides. Macromol. Biosci. 2012, 12, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-F.; Sheu, H.-S.; Jeng, U.-S.; Huang, C.-F.; Chang, F.-C. Preparation and Supramolecular Self-Assembly of a Polypeptide-b lock-polypseudorotaxane. Macromolecules 2005, 38, 6551–6558. [Google Scholar] [CrossRef]
- Kopeček, J.; Yang, J. Peptide-directed self-assembly of hydrogels. Acta Biomater. 2009, 5, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Dhaware, V.; Shaikh, A.Y.; Kar, M.; Hotha, S.; Gupta, S.S. Synthesis and self-assembly of amphiphilic homoglycopolypeptide. Langmuir 2013, 29, 5659–5667. [Google Scholar] [CrossRef] [PubMed]
- Caillol, S.; Lecommandoux, S.; Mingotaud, A.-F.; Schappacher, M.; Soum, A.; Bryson, N.; Meyrueix, R. Synthesis and Self-Assembly Properties of Peptide-Polylactide Block Copolymers. Macromolecules 2003, 36, 1118–1124. [Google Scholar] [CrossRef]
- Babin, J.; Taton, D.; Brinkmann, M.; Lecommandoux, S. Synthesis and Self-Assembly in Bulk of Linear and Mikto-Arm Star Block Copolymers Based on Polystyrene and Poly(glutamic acid). Macromolecules 2008, 41, 1384–1392. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Iatrou, H.; Pitsikalis, M.; Sakellariou, G. Synthesis of well-defined polypeptide-based materials via the ring-opening polymerization of α-amino acid N-carboxyanhydrides. Chem. Rev. 2009, 109, 5528–5578. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Wang, J.; Song, Z.; Yin, L.; Zhang, Y.; Tang, H.; Tu, C.; Lin, Y.; Cheng, J. Recent advances in amino acid N-carboxyanhydrides and synthetic polypeptides: Chemistry, self-assembly and biological applications. Chem. Commun. 2014, 50, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Huesmann, D.; Klinker, K.; Barz, M. Orthogonally reactive amino acids and end groups in NCA polymerization. Polym. Chem. 2017, 8, 957–971. [Google Scholar] [CrossRef]
- Byrne, M.; Murphy, R.; Kapetanakis, A.; Ramsey, J.; Cryan, S.A.; Heise, A. Star—Shaped Polypeptides: Synthesis and Opportunities for Delivery of Therapeutics. Macromol. Rapid Commun. 2015, 36, 1862–1876. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, L.; Liu, M.; Le, Y.; Lv, S.; Wang, J.; Chen, J.-F. Dual-responsive star-shaped polypeptides for drug delivery. RSC Adv. 2016, 6, 6368–6377. [Google Scholar] [CrossRef]
- Leuchs, H. Ueber die Glycin-carbonsäure. Berichte der Deutschen Chemischen Gesellschaft 1906, 39, 857–861. [Google Scholar] [CrossRef]
- Kricheldorf, H.R. α-Aminoacid-N-Carboxy-Anhydrides and Related Heterocycles: Syntheses, Properties, Peptide Synthesis, Polymerization; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Leuchs, H.; Geiger, W. Über die Anhydride von α-Amino-N-carbonsäuren und die von α-Aminosäuren. Berichte der Deutschen Chemischen Gesellschaft 1908, 41, 1721–1726. [Google Scholar] [CrossRef]
- Green, M.; Stahmann, M.A. Synthesis and enzymatic hydrolisis of glutamic acid polypeptides. J. Biol. Chem. 1952, 197, 771–782. [Google Scholar] [PubMed]
- Ben-Ishai, D.; Katchalski, E. Synthesis of N-Carboxy-α-amino Acid Anhydrides from N-Carbalkoxy-α-amino Acids by the Use of Phosphorus Tribromide. J. Am. Chem. Soc. 1952, 74, 3688–3689. [Google Scholar] [CrossRef]
- Fuchs, F. On N-carbonic acid-anhydride. Berichte der Deutschen Chemischen Gesellschaft 1922, 55, 2943. [Google Scholar] [CrossRef]
- Brown, C.J.; Coleman, D.; Farthing, A.C. Further studies in synthetic polypeptides. Nature 1949, 163, 834–835. [Google Scholar] [CrossRef] [PubMed]
- Fuller, W.D.; Verlander, M.S.; Goodman, M. Procedure for facile synthesis of amino-acid N-carboxyanhydrides. Biopolymers 1976, 15, 1869–1871. [Google Scholar] [CrossRef] [PubMed]
- Katakai, R.; Iizuka, Y. An improved rapid method for the synthesis of N-carboxy alpha-amino acid anhydrides using trichloromethyl chloroformate. J. Org. Chem. 1985, 50, 715–716. [Google Scholar] [CrossRef]
- Wilder, R.; Mobashery, S. The use of triphosgene in preparation of N-carboxy alpha-amino acid anhydrides. J. Org. Chem. 1992, 57, 2755–2756. [Google Scholar] [CrossRef]
- Penczek, S. Models of Biopolymers by Ring-Opening Polymerization; CRC Press: Boca Raton, FL, USA, 1989. [Google Scholar]
- Poche, D.S.; Moore, M.J.; Bowles, J.L. An unconventional method for purifying the N-carboxyanhydride derivatives of γ-alkyl-l-glutamates. Synth. Commun. 1999, 29, 843–854. [Google Scholar] [CrossRef]
- Smeets, N.; van der Weide, P.; Meuldijk, J.; Vekemans, J.; Hulshof, L. A Scalable Synthesis of l-Leucine-N-carboxyanhydride. Org. Process Res. Dev. 2005, 9, 757–763. [Google Scholar] [CrossRef]
- Kramer, J.R.; Deming, T.J. General method for purification of α-amino acid-N-carboxyanhydrides using flash chromatography. Biomacromolecules 2010, 11, 3668–3672. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Wu, J.; Cheng, R.; Meng, F.; Klok, H.-A.; Zhong, Z. Functional polypeptide and hybrid materials: Precision synthesis via α-amino acid N-carboxyanhydride polymerization and emerging biomedical applications. Prog. Polym. Sci. 2014, 39, 330–364. [Google Scholar] [CrossRef]
- Hernández, J.R.; Klok, H.A. Synthesis and ring-opening (co) polymerization of L-lysine N-carboxyanhydrides containing labile side-chain protective groups. J. Polym. Sci. Part A 2003, 41, 1167–1187. [Google Scholar] [CrossRef]
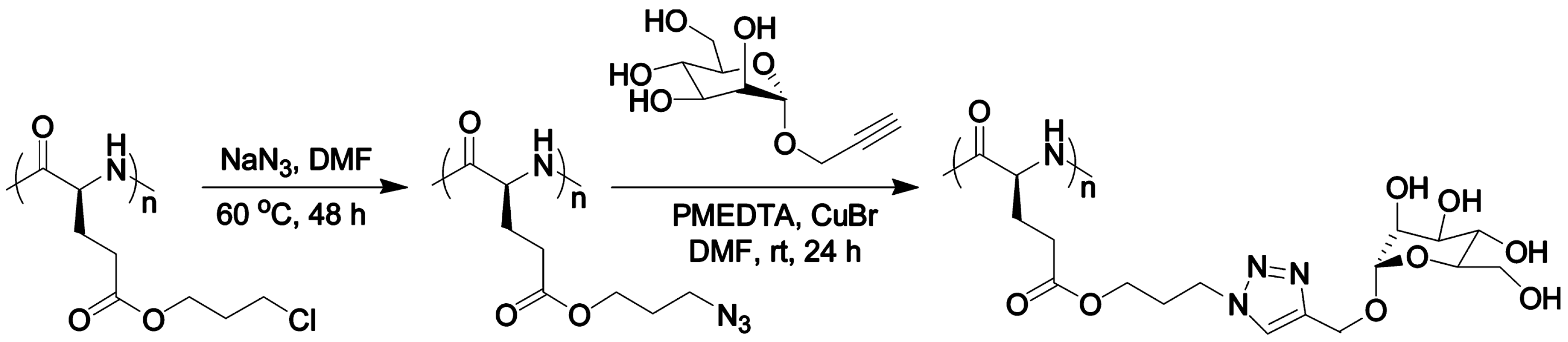
- Engler, A.C.; Lee, H.i.; Hammond, P.T. Highly efficient “grafting onto” a polypeptide backbone using click chemistry. Angew. Chem. Int. Ed. 2009, 48, 9334–9338. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zeng, Y.; Yang, J.; Zeng, Z.; Zhu, F.; Chen, X. Facile functionalization of polypeptides by thiol-yne photochemistry for biomimetic materials synthesis. Chem. Commun. 2011, 47, 7509–7511. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Zhao, C.; He, P.; Tang, Z.; Chen, X.; Jing, X. Facile Synthesis of Glycopolypeptides by Combination of Ring-Opening Polymerization of an Alkyne-Substituted N-carboxyanhydride and Click “GlycosylatioN”. Macromol. Rapid Commun. 2010, 31, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Borase, T.; Ninjbadgar, T.; Kapetanakis, A.; Roche, S.; O’Connor, R.; Kerskens, C.; Heise, A.; Brougham, D.F. Stable Aqueous Dispersions of Glycopeptide-Grafted Selectably Functionalized Magnetic Nanoparticles. Angew. Chem. Int. Ed. 2013, 52, 3164–3167. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Bonduelle, C.; Thévenot, J.; Lecommandoux, S.B.; Heise, A. Biologically active polymersomes from amphiphilic glycopeptides. J. Am. Chem. Soc. 2011, 134, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Habraken, G.; Audouin, F.; Heise, A. Hydrolytically stable bioactive synthetic glycopeptide homo-and copolymers by combination of NCA polymerization and click reaction. Macromolecules 2010, 43, 6050–6057. [Google Scholar] [CrossRef]
- Tang, H.; Zhang, D. General Route toward Side-Chain-Functionalized α-Helical Polypeptides. Biomacromolecules 2010, 11, 1585–1592. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, A.J.; Deming, T.J. Soluble, clickable polypeptides from azide-containing N-carboxyanhydride monomers. ACS Macro Lett. 2013, 2, 351–354. [Google Scholar] [CrossRef]
- Sun, J.; Schlaad, H. Thiol-ene clickable polypeptides. Macromolecules 2010, 43, 4445–4448. [Google Scholar] [CrossRef]
- Krannig, K.-S.; Schlaad, H. pH-responsive bioactive glycopolypeptides with enhanced helicity and solubility in aqueous solution. J. Am. Chem. Soc. 2012, 134, 18542–18545. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Zhang, D. Multi-functionalization of helical block copoly (α-peptide) s by orthogonal chemistry. Polym. Chem. 2011, 2, 1542–1551. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, H.; Lin, Y.; Cheng, J. Water-soluble polypeptides with elongated, charged side chains adopt ultrastable helical conformations. Macromolecules 2011, 44, 6641–6644. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J.R.; Deming, T.J. Glycopolypeptides via living polymerization of glycosylated-l-lysine N-carboxyanhydrides. J. Am. Chem. Soc. 2010, 132, 15068–15071. [Google Scholar] [CrossRef] [PubMed]
- Pati, D.; Shaikh, A.Y.; Das, S.; Nareddy, P.K.; Swamy, M.J.; Hotha, S.; Gupta, S.S. Controlled synthesis of O-glycopolypeptide polymers and their molecular recognition by lectins. Biomacromolecules 2012, 13, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Pati, D.; Shaikh, A.Y.; Hotha, S.; Gupta, S.S. Synthesis of glycopolypeptides by the ring opening polymerization of O-glycosylated-α-amino acid N-carboxyanhydride (NCA). Polym. Chem. 2011, 2, 805–811. [Google Scholar] [CrossRef]
- Chen, C.; Wang, Z.; Li, Z. Thermoresponsive polypeptides from pegylated poly-l-glutamates. Biomacromolecules 2011, 12, 2859–2863. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Shen, Y.; Fu, W.; Li, Z. Thermoresponsive Oligo (ethylene glycol) Functionalized Poly-l-cysteine. Macromolecules 2013, 46, 3753–3760. [Google Scholar] [CrossRef]
- Liu, G.; Dong, C.-M. Photoresponsive poly (S-(o-nitrobenzyl)-l-cysteine)-b-PEO from a l-cysteine N-carboxyanhydride monomer: Synthesis, self-assembly, and phototriggered drug release. Biomacromolecules 2012, 13, 1573–1583. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Tang, H.; Kim, K.H.; Zheng, N.; Song, Z.; Gabrielson, N.P.; Lu, H.; Cheng, J. Light-Responsive Helical Polypeptides Capable of Reducing Toxicity and Unpacking DNA: Toward Nonviral Gene Delivery. Angew. Chem. Int. Ed. 2013, 52, 9182–9186. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, P.; Li, Z. Molecular Bottlebrushes with Polypeptide Backbone Prepared via Ring-Opening Polymerization of NCA and ATRP. Macromol. Rapid Commun. 2012, 33, 287–295. [Google Scholar] [CrossRef] [PubMed]
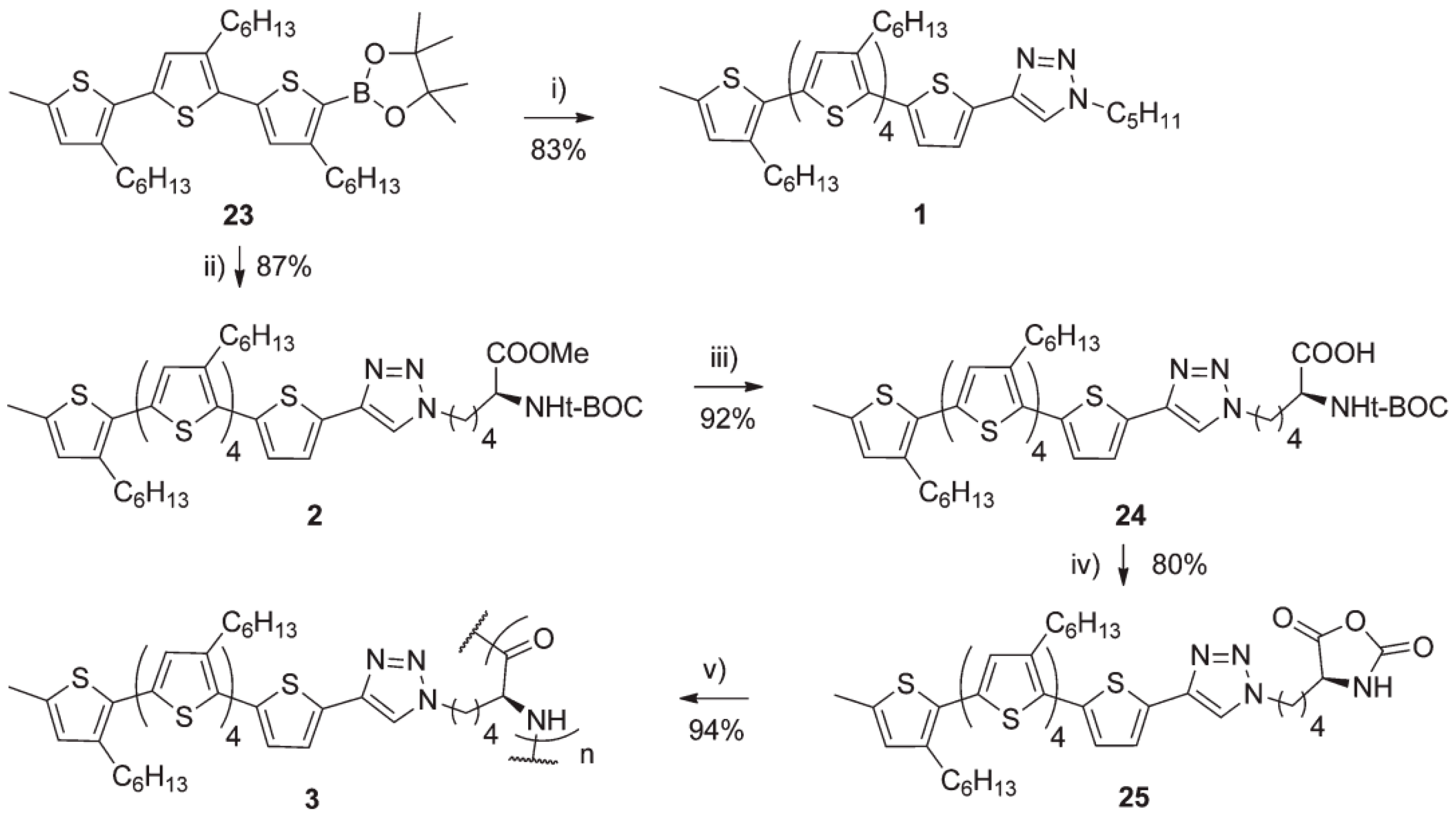
- Kumar, R.J.; MacDonald, J.M.; Singh, T.B.; Waddington, L.J.; Holmes, A.B. Hierarchical self-assembly of semiconductor functionalized peptide α-helices and optoelectronic properties. J. Am. Chem. Soc. 2011, 133, 8564–8573. [Google Scholar] [CrossRef] [PubMed]
- Daly, W.H.; Poche, D. The preparation of N-carboxyanhydrides of alpha-amino-acids using bis(trichloromethyl)carbonate. Tetrahedron Lett. 1988, 29, 5859–5862. [Google Scholar] [CrossRef]
- Kricheldorf, H.R. Models of Biopolymers by Ring-Opening Polymerization; CRC Press Inc.: Boca Raton, FL, USA, 1990. [Google Scholar]
- Stahmann, M.A. Polyamino Acids, Polypeptides, and Proteins; University of Wisconsin Press: Madison, WI, USA, 1962. [Google Scholar]
- Kricheldorf, H.R. Oligomerization and Polymerization of NCAs: Chemical Aspects. In α-Aminoacid-N-Carboxy-Anhydrides and Related Heterocycles: Syntheses, Properties, Peptide Synthesis, Polymerization; Kricheldorf, H.R., Ed.; Springer: Berlin/Heidelberg, Germany, 1987; pp. 59–157. [Google Scholar]
- Kricheldorf, H.R. Polypeptides and 100 Years of Chemistry of α-Amino Acid N-Carboxyanhydrides. Angew. Chem. Int. Ed. 2006, 45, 5752–5784. [Google Scholar] [CrossRef] [PubMed]
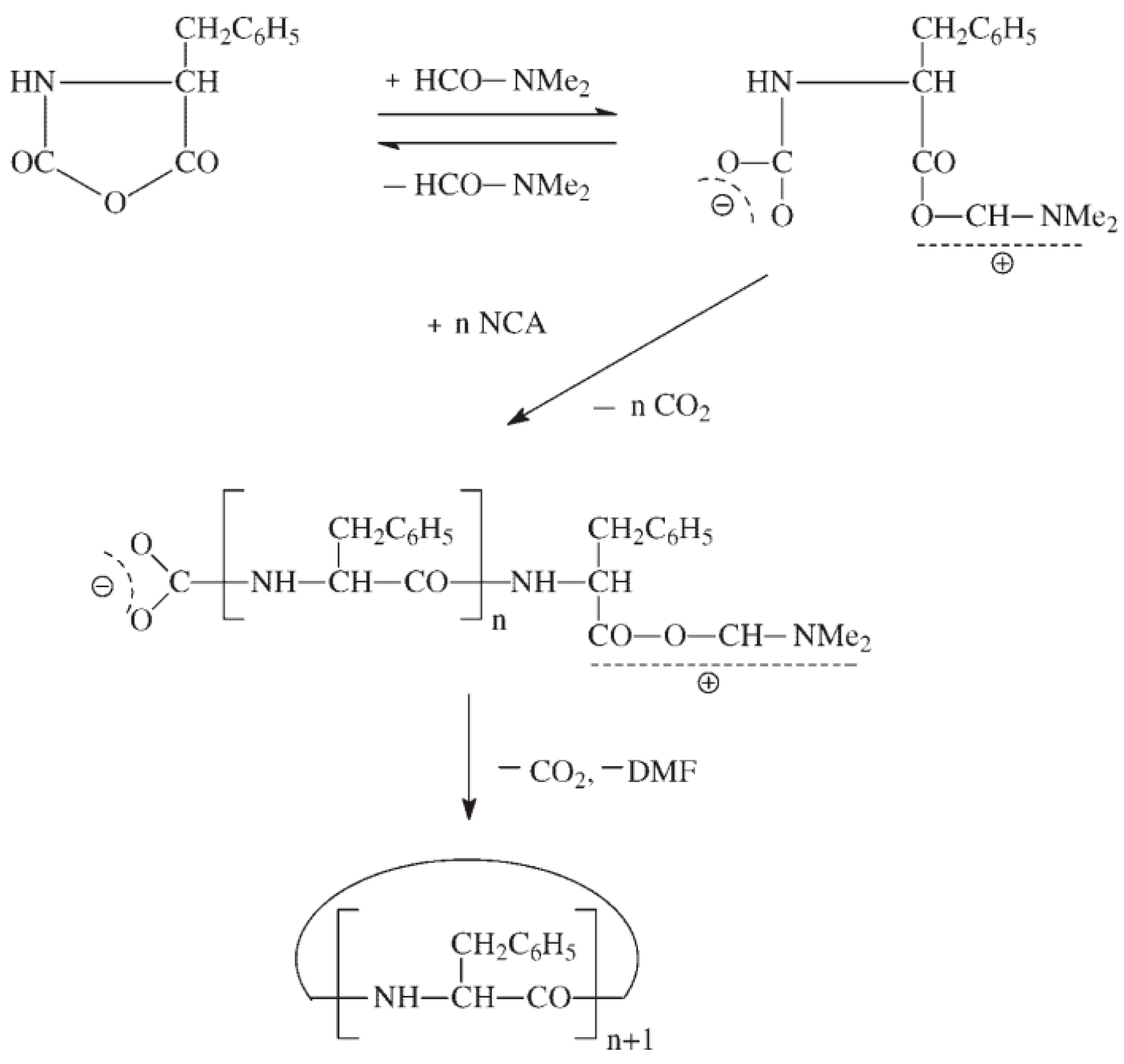
- Bamford, C.H.; Block, H. The initiation step in the polymerization of N-carboxy-[small alpha]-amino-acid anhydrides. Part I. Catalysis by tertiary bases. J. Chem. Soc. (Resumed) 1961, 981, 4989–4991. [Google Scholar] [CrossRef]
- Bamford, C.H.; Block, H. The initiation step in the polymerization of N-carboxy-[small alpha]-amino-acid anhydrides. Part II. Effects related to the structure of amine initiators. J. Chem. Soc. (Resumed) 1961, 982, 4992–4995. [Google Scholar] [CrossRef]
- Blout, E.R.; Karlson, R.H. Polypeptides. III. The Synthesis of High Molecular Weight Poly-γ-benzyl-l-glutamates1. J. Am. Chem. Soc. 1956, 78, 941–946. [Google Scholar] [CrossRef]
- Katchalski, E.; Sela, M. Synthesis and chemical properties of poly-α-amino acids. Adv. Protein Chem. 1958, 13, 243–492. [Google Scholar] [PubMed]
- Sela, M.; Katchalski, E. Biological properties of poly-α-amino acids. Adv. Protein Chem. 1959, 14, 391–478. [Google Scholar] [PubMed]
- Deming, T.J. Cobalt and Iron Initiators for the Controlled Polymerization of α-Amino Acid-N-Carboxyanhydrides. Macromolecules 1999, 32, 4500–4502. [Google Scholar] [CrossRef]
- Goodwin, A.A.; Bu, X.; Deming, T.J. Reactions of α-amino acid-N-carboxyanhydrides (NCAs) with organometallic palladium (0) and platinum (0) compounds: Structure of a metallated NCA product and its role in polypeptide synthesis. J. Organomet. Chem. 1999, 589, 111–114. [Google Scholar] [CrossRef]
- Deming, T.J. Living polymerization of α-amino acid-N-carboxyanhydrides. J. Polym. Sci. Part A 2000, 38, 3011–3018. [Google Scholar] [CrossRef]
- Deming, T.J.; Curtin, S.A. Chain Initiation Efficiency in Cobalt- and Nickel-Mediated Polypeptide Synthesis. J. Am. Chem. Soc. 2000, 122, 5710–5717. [Google Scholar] [CrossRef]
- Dimitrov, I.; Schlaad, H. Synthesis of nearly monodisperse polystyrene-polypeptide block copolymers via polymerisation of N-carboxyanhydrides. Chem. Commun. 2003, 23, 2944–2945. [Google Scholar] [CrossRef]
- Knobler, Y.; Bittner, S.; Frankel, M. Reaction of N-carboxy-[small alpha]-amino-acid anhydrides with hydrochlorides of hydroxylamine, O-alkylhydroxylamines, and amines; syntheses of amino-hydroxamic acids, amido-oxy-peptides, and [small alpha]-amino-acid amides. J. Chem. Soc. (Resumed) 1964, 749, 3941–3951. [Google Scholar] [CrossRef]
- Knobler, Y.; Bittner, S.; Virov, D.; Frankel, M. Alpha-Aminoacyl Derivatives of Aminobenzoic Acid and of Amino-Oxy-Acids by Reaction of Their Hydrochlorides with Amino-Acid N-Carboxyanhydrides. J. Chem. Soc. C Org. 1969, 14, 1821–1824. [Google Scholar] [CrossRef]
- Conejos-Sánchez, I.; Duro-Castano, A.; Birke, A.; Barz, M.; Vicent, M.J. A controlled and versatile NCA polymerization method for the synthesis of polypeptides. Polym. Chem. 2013, 4, 3182–3186. [Google Scholar] [CrossRef]
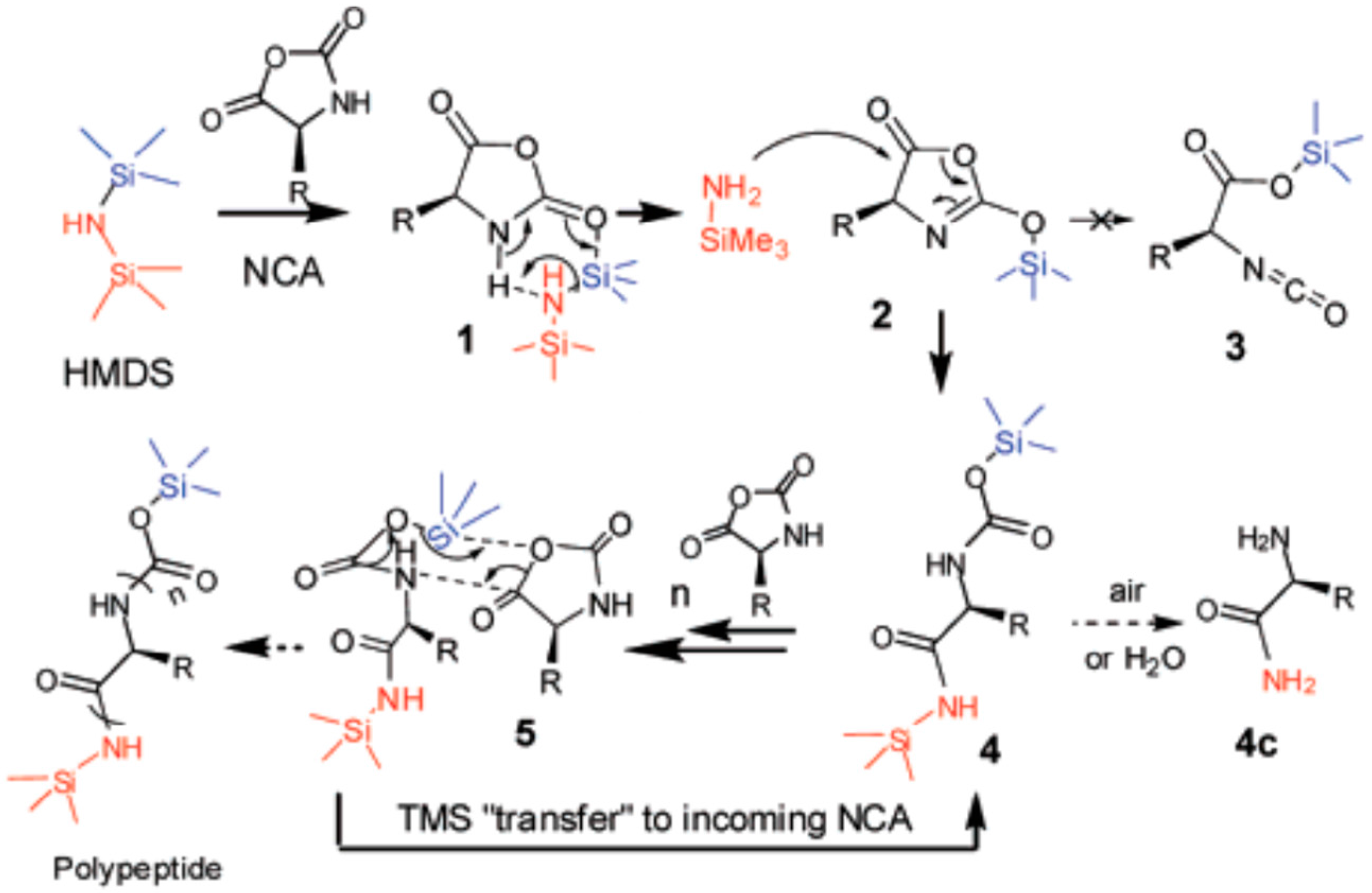
- Lu, H.; Cheng, J. Hexamethyldisilazane-Mediated Controlled Polymerization of α-Amino Acid N-Carboxyanhydrides. J. Am. Chem. Soc. 2007, 129, 14114–14115. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Cheng, J. N-trimethylsilyl amines for controlled ring-opening polymerization of amino acid N-carboxyanhydrides and facile end group functionalization of polypeptides. J. Am. Chem. Soc. 2008, 130, 12562–12563. [Google Scholar] [CrossRef] [PubMed]
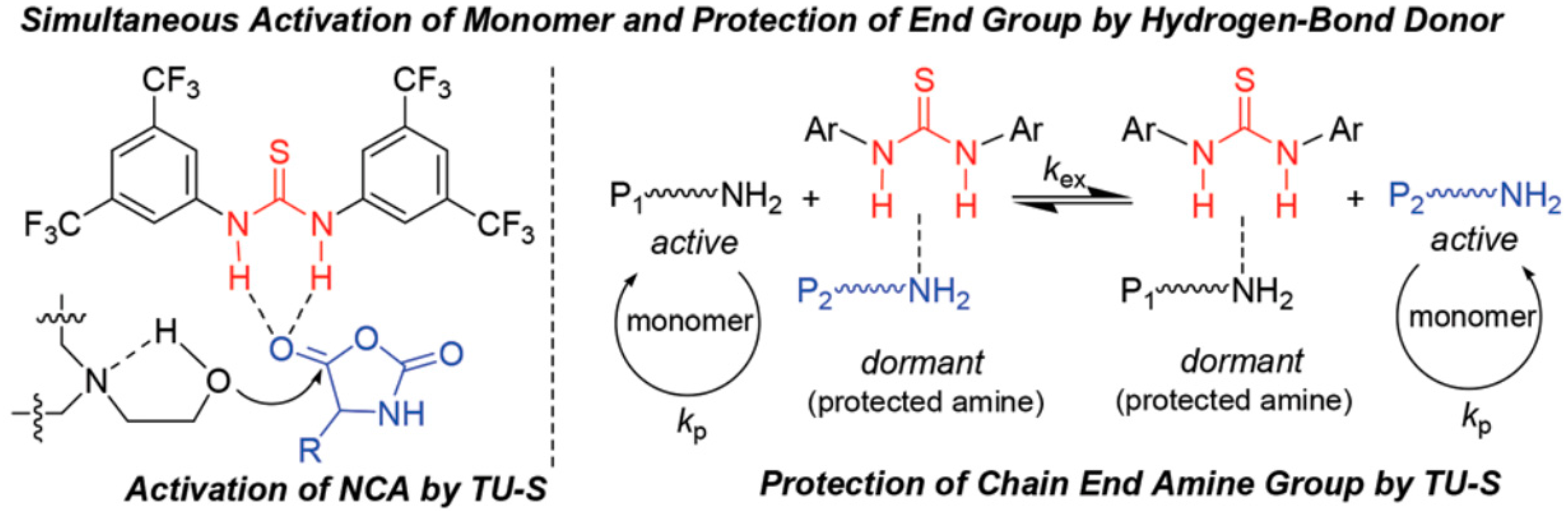
- Zhao, W.; Gnanou, Y.; Hadjichristidis, N. Organocatalysis by hydrogen-bonding: A new approach to controlled/living polymerization of α-amino acid N-carboxyanhydrides. Polym. Chem. 2015, 6, 6193–6201. [Google Scholar] [CrossRef]
- Yuan, J.; Sun, Y.; Wang, J.; Lu, H. Phenyl Trimethylsilyl Sulfide-Mediated Controlled Ring-Opening Polymerization of α-Amino Acid N-Carboxyanhydrides. Biomacromolecules 2016, 17, 891–896. [Google Scholar] [CrossRef] [PubMed]
- Aliferis, T.; Iatrou, H.; Hadjichristidis, N. Living Polypeptides. Biomacromolecules 2004, 5, 1653–1656. [Google Scholar] [CrossRef] [PubMed]
- Pickel, D.L.; Politakos, N.; Avgeropoulos, A.; Messman, J.M. A Mechanistic Study of α-(Amino acid)-N-carboxyanhydride Polymerization: Comparing Initiation and Termination Events in High-Vacuum and Traditional Polymerization Techniques. Macromolecules 2009, 42, 7781–7788. [Google Scholar] [CrossRef]
- Vayaboury, W.; Giani, O.; Cottet, H.; Deratani, A.; Schué, F. Living Polymerization of α-Amino Acid N-Carboxyanhydrides (NCA) upon Decreasing the Reaction Temperature. Macromol. Rapid Commun. 2004, 25, 1221–1224. [Google Scholar] [CrossRef]
- Habraken, G.J.; Wilsens, K.H.; Koning, C.E.; Heise, A. Optimization of N-carboxyanhydride (NCA) polymerization by variation of reaction temperature and pressure. Polym. Chem. 2011, 2, 1322–1330. [Google Scholar] [CrossRef]
- Habraken, G.J.; Peeters, M.; Dietz, C.H.; Koning, C.E.; Heise, A. How controlled and versatile is N-carboxy anhydride (NCA) polymerization at 0 C? Effect of temperature on homo-, block-and graft (co) polymerization. Polym. Chem. 2010, 1, 514–524. [Google Scholar] [CrossRef]
- Stukenkemper, T.; Jansen, J.F.G.A.; Lavilla, C.; Dias, A.A.; Brougham, D.F.; Heise, A. Polypeptides by light: Photo-polymerization of N-carboxyanhydrides (NCA). Polym. Chem. 2017, 8, 828–832. [Google Scholar] [CrossRef]
- Cao, J.; Siefker, D.; Chan, B.A.; Yu, T.; Lu, L.; Saputra, M.A.; Fronczek, F.R.; Xie, W.; Zhang, D. Interfacial Ring-Opening Polymerization of Amino-Acid-Derived N-Thiocarboxyanhydrides Toward Well-Defined Polypeptides. ACS Macro Lett. 2017, 6, 836–840. [Google Scholar] [CrossRef]
- Zou, J.; Fan, J.; He, X.; Zhang, S.; Wang, H.; Wooley, K.L. A Facile Glovebox-Free Strategy To Significantly Accelerate the Syntheses of Well-Defined Polypeptides by N-Carboxyanhydride (NCA) Ring-Opening Polymerizations. Macromolecules 2013, 46, 4223–4226. [Google Scholar] [CrossRef] [PubMed]
- Thunig, D.; Semen, J.; Elias, H.G. Carbon dioxide influence on NCA polymerizations. Macromol. Chem. Phys. 1977, 178, 603–607. [Google Scholar] [CrossRef]
- Zhao, W.; Gnanou, Y.; Hadjichristidis, N. From competition to cooperation: A highly efficient strategy towards well-defined (co) polypeptides. Chem. Commun. 2015, 51, 3663–3666. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Gnanou, Y.; Hadjichristidis, N. Fast and Living Ring-Opening Polymerization of α-Amino Acid N-Carboxyanhydrides Triggered by an “Alliance” of Primary and Secondary Amines at Room Temperature. Biomacromolecules 2015, 16, 1352–1357. [Google Scholar] [CrossRef] [PubMed]
- Engler, A.C.; Bonner, D.K.; Buss, H.G.; Cheung, E.Y.; Hammond, P.T. The synthetic tuning of clickable pH responsive cationic polypeptides and block copolypeptides. Soft Matter 2011, 7, 5627–5637. [Google Scholar] [CrossRef]
- Zhao, X.; Poon, Z.; Engler, A.C.; Bonner, D.K.; Hammond, P.T. Enhanced stability of polymeric micelles based on postfunctionalized poly (ethylene glycol)-b-poly (γ-propargyl l-glutamate): The substituent effect. Biomacromolecules 2012, 13, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zheng, N.; Song, Z.; Yin, L.; Cheng, J. The effect of side-chain functionality and hydrophobicity on the gene delivery capabilities of cationic helical polypeptides. Biomaterials 2014, 35, 3443–3454. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J.R.; Deming, T.J. Preparation of Multifunctional and Multireactive Polypeptides via Methionine Alkylation. Biomacromolecules 2012, 13, 1719–1723. [Google Scholar] [CrossRef] [PubMed]
- Gharakhanian, G.E.; Deming, T.J. Versatile Synthesis of Stable, Functional Polypeptides via Reaction with Epoxides. Biomacromolecules 2015, 16, 1802–1806. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chen, P.; Deng, C.; Meng, F.; Cheng, R.; Zhong, Z. A simple and versatile synthetic strategy to functional polypeptides via vinyl sulfone-substituted l-cysteine N-carboxyanhydride. Macromolecules 2013, 46, 6723–6730. [Google Scholar] [CrossRef]
- Deng, Y.; Xu, Y.; Wang, X.; Yuan, Q.; Ling, Y.; Tang, H. Water-Soluble Thermoresponsive α-Helical Polypeptide with an Upper Critical Solution Temperature: Synthesis, Characterization, and Thermoresponsive Phase Transition Behaviors. Macromol. Rapid Commun. 2015, 36, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Yakovlev, I.; Deming, T.J. Controlled synthesis of phosphorylcholine derivatives of poly (serine) and poly (homoserine). J. Am. Chem. Soc. 2015, 137, 4078–4081. [Google Scholar] [CrossRef] [PubMed]
- Wamsley, A.; Jasti, B.; Phiasivongsa, P.; Li, X. Synthesis of random terpolymers and determination of reactivity ratios of N-carboxyanhydrides of leucine, β-benzyl aspartate, and valine. J. Polym. Sci. Part A 2004, 42, 317–325. [Google Scholar] [CrossRef]
- Hill, D.J.T.; Cardinaux, F.; Scheraga, H.A. On the amino-acid-sequence distribution in benzylglutamate-methionine copolymers prepared from N-carboxyanhydrides. Biopolymers 1977, 16, 2469–2480. [Google Scholar] [CrossRef] [PubMed]
- Sederel, W.; Deshmane, S.; Hayashi, T.; Anderson, J. The random copolymerization of γ-benzyl-l-glutamate and l-valine N-carboxyanhydrides: Reactivity ratio and heteogeneity studies. Biopolymers 1978, 17, 2835–2849. [Google Scholar] [CrossRef]
- Saudek, V.; Stejskal, J.; Schmidt, P.; Zimmermann, K.; Škarda, V.; Kratochvil, P.; Drobnik, J. Correlation between the sequence-length distribution and structure of statistical copolymers of l-valine and γ-benzyl-l-glutamate. Biopolymers 1987, 26, 705–725. [Google Scholar] [CrossRef]
- Kricheldorf, H.R.; Hull, W.E. 15N NMR spectroscopy 14. Neighboring residue effects in glycine-containing polypeptides. Die Makromol. Chem. 1979, 180, 161–174. [Google Scholar] [CrossRef]
- Kricheldorf, H.R.; Müller, D.; Hull, W.E. Secondary structure of peptides: 19. Primary/secondary structure relationships of leucine/valine copolypeptides prepared from α-amino acid N-carboxyanhydrides. Int. J. Biol. Macromolecules 1986, 8, 20–26. [Google Scholar] [CrossRef]
- Kricheldorf, H.R.; Mang, T. 13C NMR sequence analysis, 20. Stereospecificity of the polymerization of d,l-Leu-NCA and d,l-Val-NCA. Die Makromol. Chem. 1981, 182, 3077–3098. [Google Scholar] [CrossRef]
- Ōya, M.; Takahashi, T. Copolymerization of N-carboxy Nϵ-carbobenzoxy l-lysine anhydride with N-carboxy β-benzyl l-aspartate anhydride in acetonitrile. J. Polym. Sci. Polym. Chem. Ed. 1982, 20, 529–539. [Google Scholar] [CrossRef]
- Atreyi, M.; Rao, M.; Kumar, S. Copolymerization kinetics of N-carboxyanhydrides of some α-amino acids: Influence of nature of amino acids. Biopolymers 1983, 22, 747–753. [Google Scholar] [CrossRef]
- Kricheldorf, H.R.; Müller, D.; Hull, W.E. Secondary structure of peptides. 18. 15N-nmr and 13C-nmr CP/MAS investigation of the primary and secondary structure of (Ala/Val) copolypeptides. Biopolymers 1985, 24, 2113–2129. [Google Scholar] [CrossRef]
- Kumar, A. Copolymerization Kinetics of N-Carboxyanhydrides of ϵ-Benzyloxycarbonyl-l-Lysine and l-Valine. J. Macromol. Sci. Chem. 1987, 24, 707–710. [Google Scholar] [CrossRef]
- Volpe, R.; Frisch, H. Template effect on the copolymerization of l-alanine NCA and sarcosine NCA. Macromolecules 1987, 20, 1747–1752. [Google Scholar] [CrossRef]
- Goury, V.; Jhurry, D.; Bhaw-Luximon, A.; Novak, B.M.; Belleney, J. Synthesis and Characterization of Random and Block Copolypeptides Derived from γ-Methylglutamate and Leucine N-Carboxyanhydrides. Biomacromolecules 2005, 6, 1987–1991. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Deming, T.J. Synthetic polypeptide mimics of marine adhesives. Macromolecules 1998, 31, 4739–4745. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Hwang, J.; Deming, T.J. Role of l-3,4-dihydroxyphenylalanine in mussel adhesive proteins. J. Am. Chem. Soc. 1999, 121, 5825–5826. [Google Scholar] [CrossRef]
- Hayashi, S.; Ohkawa, K.; Yamamoto, H. Random and Sequential Copolypeptides Containing O-Phospho-l-Threonine and l-Aspartic Acid; Roles in CaCO3 Biomineralization. Macromol. Biosci. 2006, 6, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Wyrsta, M.D.; Cogen, A.L.; Deming, T.J. A parallel synthetic approach for the analysis of membrane interactive copolypeptides. J. Am. Chem. Soc. 2001, 123, 12919–12920. [Google Scholar] [CrossRef] [PubMed]
- Kakinoki, S.; Kitamura, M.; Yuge, M.; Furuta, M.; Oka, M.; Hirano, Y.; Kono, K.; Kaetsu, I. Solution property and irradiation effect of random copolypeptides composed of Ala and Pro residues. Polym. Bull. 2007, 58, 393–400. [Google Scholar] [CrossRef]
- Higuchi, M.; Takizawa, A.; Kinoshita, T.; Tsujita, Y.; Okochi, K. Simple method for the preparation of an amphiphilic sequential polypeptide. Macromolecules 1990, 23, 361–365. [Google Scholar] [CrossRef]
- Minoura, N.; Higuchi, M.; Kinoshita, T. Stimuli-responsive formation of helical polypeptide rod assemblies. Mater. Sci. Eng. C 1997, 4, 249–254. [Google Scholar] [CrossRef]
- Hull, W.E.; Kricheldorf, H.R. 15N NMR spectroscopy, 25. Terpolymerization of glycine-NCA, leucine-NCA, and valine-NCA. Die Makromol. Chem. 1980, 181, 1949–1966. [Google Scholar] [CrossRef]
- Zelzer, M.; Heise, A. Terpolymerization kinetics of amino acid N-carboxy anhydrides. J. Polym. Sci. Part A 2014, 52, 1228–1236. [Google Scholar] [CrossRef]
- Guo, J.; Huang, Y.; Jing, X.; Chen, X. Synthesis and characterization of functional poly (γ-benzyl-l-glutamate) (PBLG) as a hydrophobic precursor. Polymer 2009, 50, 2847–2855. [Google Scholar] [CrossRef]
- Zhu, M.; Wu, Y.; Ge, C.; Ling, Y.; Tang, H. SO2-Induced Solution Phase Transition of Water-Soluble and α-Helical Polypeptides. Macromolecules 2016, 49, 3542–3549. [Google Scholar] [CrossRef]
- Aoi, K.; Tsutsumiuchi, K.; Okada, M. Glycopeptide Synthesis by an.alpha.-Amino Acid N-Carboxyanhydride (NCA) Method: Ring-Opening Polymerization of a Sugar-Substituted NCA. Macromolecules 1994, 27, 875–877. [Google Scholar] [CrossRef]
- Guillermain, C.; Gallot, B. Homopolymers and copolymers of Nε-4-phenylbenzamido-L-lysine and Nε-trifluoroacetyl-L-lysine: Synthesis and liquid-crystalline properties. Macromol. Chem. Phys. 2002, 203, 1346–1356. [Google Scholar] [CrossRef]
- Higashi, N.; Koga, T.; Niwa, M. Helical Superstructures from a Poly(γ-benzyl-l-glutamate)−Poly(l-glutamic acid) Amphiphilic Diblock Copolymer: Monolayer Formation on Water and Its Specific Binding of Amino Acids. Langmuir 2000, 16, 3482–3486. [Google Scholar] [CrossRef]
- Papadopoulos, P.; Floudas, G.; Schnell, I.; Aliferis, T.; Iatrou, H.; Hadjichristidis, N. Nanodomain-Induced Chain Folding in Poly(γ-benzyl-l-glutamate)-b-polyglycine Diblock Copolymers. Biomacromolecules 2005, 6, 2352–2361. [Google Scholar] [CrossRef] [PubMed]
- Hanski, S.; Houbenov, N.; Ruokolainen, J.; Chondronicola, D.; Iatrou, H.; Hadjichristidis, N.; Ikkala, O. Hierarchical Ionic Self-Assembly of Rod-Comb Block Copolypeptide−Surfactant Complexes. Biomacromolecules 2006, 7, 3379–3384. [Google Scholar] [CrossRef] [PubMed]
- Heller, P.; Weber, B.; Birke, A.; Barz, M. Synthesis and Sequential Deprotection of Triblock Copolypept(o)ides Using Orthogonal Protective Group Chemistry. Macromol. Rapid Commun. 2015, 36, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Agut, W.; Agnaou, R.; Lecommandoux, S.; Taton, D. Synthesis of Block Copolypeptides by Click Chemistry. Macromol. Rapid Commun. 2008, 29, 1147–1155. [Google Scholar] [CrossRef]
- Minoura, N.; Aiba, S.; Fujiwara, Y. Spontaneous oscillation of the electrical membrane potential in tri-block copolypeptide membranes composed of l-glutamic acid and l-leucine. J. Am. Chem. Soc. 1993, 115, 5902–5906. [Google Scholar] [CrossRef]
- Minoura, N.; Higuchi, M. Microphase-separated structure in triblock copolypeptide membranes composed of l-glutamic acid and l-leucine. Macromolecules 1997, 30, 1023–1027. [Google Scholar] [CrossRef]
- Breedveld, V.; Nowak, A.P.; Sato, J.; Deming, T.J.; Pine, D.J. Rheology of Block Copolypeptide Solutions: Hydrogels with Tunable Properties. Macromolecules 2004, 37, 3943–3953. [Google Scholar] [CrossRef]
- Nowak, A.P.; Sato, J.; Breedveld, V.; Deming, T.J. Hydrogel Formation in Amphiphilic Triblock Copolypeptides. Supramol. Chem. 2006, 18, 423–427. [Google Scholar] [CrossRef]
- Deming, T.J. Facile synthesis of block copolypeptides of defined architecture. Nature 1997, 390, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Euliss, L.E.; Grancharov, S.G.; O’Brien, S.; Deming, T.J.; Stucky, G.D.; Murray, C.B.; Held, G.A. Cooperative Assembly of Magnetic Nanoparticles and Block Copolypeptides in Aqueous Media. Nano Lett. 2003, 3, 1489–1493. [Google Scholar] [CrossRef]
- Nowak, A.P.; Breedveld, V.; Pine, D.J.; Deming, T.J. Unusual salt stability in highly charged diblock co-polypeptide hydrogels. J. Am. Chem. Soc. 2003, 125, 15666–15670. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.-Y.; Yusufoglu, Y.; Kanapathipillai, M.; Yang, C.-Y.; Wu, Y.; Thiyagarajan, P.; Deming, T.; Akinc, M.; Schmidt-Rohr, K.; Mallapragada, S. Self-assembled calcium phosphate nanocomposites using block copolypeptide templates. Soft Matter 2009, 5, 4311–4320. [Google Scholar] [CrossRef]
- Pakstis, L.M.; Ozbas, B.; Hales, K.D.; Nowak, A.P.; Deming, T.J.; Pochan, D. Effect of Chemistry and Morphology on the Biofunctionality of Self-Assembling Diblock Copolypeptide Hydrogels. Biomacromolecules 2004, 5, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Jan, J.S.; Shantz, D.F. Biomimetic Silica Formation: Effect of Block Copolypeptide Chemistry and Solution Conditions on Silica Nanostructure. Adv. Mater. 2007, 19, 2951–2956. [Google Scholar] [CrossRef]
- Jain, R.; Forciniti, D. Adsorption of Diblock Polypeptides on Polystyrene Latex. Langmuir 2012, 28, 15323–15335. [Google Scholar] [CrossRef] [PubMed]
- Gaspard, J.; Silas, J.A.; Shantz, D.F.; Jan, J.-S. Supramolecular assembly of lysine-b-glycine block copolypeptides at different solution conditions. Supramol. Chem. 2010, 22, 178–185. [Google Scholar] [CrossRef]
- Lai, J.-K.; Chuang, T.-H.; Jan, J.-S.; Wang, S.S.-S. Efficient and stable enzyme immobilization in a block copolypeptide vesicle-templated biomimetic silica support. Colloids Surfaces B Biointerfaces 2010, 80, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Huang, Y.; Shi, Q.; Chen, X.; Jing, X. Oxygen carrier based on hemoglobin/poly (l-lysine)-block-poly (l-phenylalanine) vesicles. Langmuir 2009, 25, 13726–13729. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.R.; Kramer, J.R.; Deming, T.J. Enzyme-Triggered Cargo Release from Methionine Sulfoxide Containing Copolypeptide Vesicles. Biomacromolecules 2013, 14, 3610–3614. [Google Scholar] [CrossRef] [PubMed]
- Iatrou, H.; Frielinghaus, H.; Hanski, S.; Ferderigos, N.; Ruokolainen, J.; Ikkala, O.; Richter, D.; Mays, J.; Hadjichristidis, N. Architecturally induced multiresponsive vesicles from well-defined polypeptides. Formation of gene vehicles. Biomacromolecules 2007, 8, 2173–2181. [Google Scholar] [CrossRef] [PubMed]
- Brzezinska, K.R.; Curtin, S.A.; Deming, T.J. Polypeptide End-Capping Using Functionalized Isocyanates: Preparation of Pentablock Copolymers. Macromolecules 2002, 35, 2970–2976. [Google Scholar] [CrossRef]
- Kros, A.; Jesse, W.; Metselaar, G.A.; Cornelissen, J.J.L.M. Synthesis and Self-Assembly of Rod–Rod Hybrid Poly(γ-benzyl l-glutamate)-block-Polyisocyanide Copolymers. Angew. Chem. Int. Ed. 2005, 44, 4349–4352. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.-B.; Xia, W.-J.; Dong, H.-Q.; Li, Y.-Y. Sheddable micelles based on disulfide-linked hybrid PEG-polypeptide copolymer for intracellular drug delivery. Polymer 2011, 52, 3580–3586. [Google Scholar] [CrossRef]
- Cheon, J.-B.; Jeong, Y.-I.; Cho, C.-S. Effects of temperature on diblock copolymer micelle composed of poly (γ-benzyl l-glutamate) and poly (N-isopropylacrylamide). Polymer 1999, 40, 2041–2050. [Google Scholar] [CrossRef]
- Brzezinska, K.R.; Deming, T.J. Synthesis of AB Diblock Copolymers by Atom-Transfer Radical Polymerization (ATRP) and Living Polymerization of α-Amino Acid-N-Carboxyanhydrides. Macromol. Biosci. 2004, 4, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Steig, S.; Cornelius, F.; Witte, P.; Staal, B.B.; Koning, C.E.; Heise, A.; Menzel, H. Synthesis of polypeptide based rod–coil block copolymers. Chem. Commun. 2005, 43, 5420–5422. [Google Scholar] [CrossRef] [PubMed]
- Habraken, G.J.; Koning, C.E.; Heise, A. Peptide block copolymers by N-carboxyanhydride ring-opening polymerization and atom transfer radical polymerization: The effect of amide macroinitiators. J. Polym. Sci. Part A 2009, 47, 6883–6893. [Google Scholar] [CrossRef]
- Knoop, R.J.; Habraken, G.J.; Gogibus, N.; Steig, S.; Menzel, H.; Koning, C.E.; Heise, A. Synthesis of poly (benzyl glutamate-b-styrene) rod-coil block copolymers by dual initiation in one pot. J. Polym. Sci. Part A 2008, 46, 3068–3077. [Google Scholar] [CrossRef]
- Habraken, G.J.; Peeters, M.; Thornton, P.D.; Koning, C.E.; Heise, A. Selective enzymatic degradation of self-assembled particles from amphiphilic block copolymers obtained by the combination of N-carboxyanhydride and nitroxide-mediated polymerization. Biomacromolecules 2011, 12, 3761–3769. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, J.; Li, W.; Zhang, A. Synthesis and characterization of thermo-and pH-responsive double-hydrophilic diblock copolypeptides. Biomacromolecules 2007, 8, 3557–3567. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.; Gathergood, N.; Heise, A. Synthesis of Polypeptide Block Copolymer Hybrids by the Combination of N-Carboxyanhydride Polymerization and RAFT. Macromol. Rapid Commun. 2013, 34, 1325–1329. [Google Scholar] [CrossRef] [PubMed]
- Karatzas, A.; Bilalis, P.; Iatrou, H.; Pitsikalis, M.; Hadjichristidis, N. Synthesis of well-defined functional macromolecular chimeras based on poly (ethylene oxide) or poly (N-vinyl pyrrolidone). React. Funct. Polym. 2009, 69, 435–440. [Google Scholar] [CrossRef]
- Langer, R. Biomaterials in drug delivery and tissue engineering: One laboratory’s experience. Acc. Chem. Res. 2000, 33, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Gombotz, W.R.; Pettit, D.K. Biodegradable polymers for protein and peptide drug delivery. Bioconjugate Chem. 1995, 6, 332–351. [Google Scholar] [CrossRef]
- Rong, G.; Deng, M.; Deng, C.; Tang, Z.; Piao, L.; Chen, X.; Jing, X. Synthesis of poly (ε-caprolactone)-b-poly (γ-benzyl-l-glutamic acid) block copolymer using amino organic calcium catalyst. Biomacromolecules 2003, 4, 1800–1804. [Google Scholar] [CrossRef] [PubMed]
- Gotsche, M.; Keul, H.; Höcker, H. Amino-termined poly (l-lactide) s as initiators for the polymerization of N-carboxyanhydrides: Synthesis of poly (l-lactide)-block-poly (α-amino acid) s. Macromol. Chem. Phys. 1995, 196, 3891–3903. [Google Scholar] [CrossRef]
- Arimura, H.; Ohya, Y.; Ouchi, T. The formation of biodegradable polymeric micelles from newly synthesized poly (aspartic acid)-block-polylactide AB-type diblock copolymers. Macromol. Rapid Commun. 2004, 25, 743–747. [Google Scholar] [CrossRef]
- Klok, H.-A.; Langenwalter, J.F.; Lecommandoux, S. Self-assembly of peptide-based diblock oligomers. Macromolecules 2000, 33, 7819–7826. [Google Scholar] [CrossRef]
- Schlaad, H.; Smarsly, B.; Losik, M. The role of chain-length distribution in the formation of solid-state structures of polypeptide-based rod-coil block copolymers. Macromolecules 2004, 37, 2210–2214. [Google Scholar] [CrossRef]
- Kong, X.; Jenekhe, S.A. Block copolymers containing conjugated polymer and polypeptide sequences: Synthesis and self-assembly of electroactive and photoactive nanostructures. Macromolecules 2004, 37, 8180–8183. [Google Scholar] [CrossRef]
- Schatz, C.; Louguet, S.; le Meins, J.F.; Lecommandoux, S. Polysaccharide-block-polypeptide Copolymer Vesicles: Towards Synthetic Viral Capsids. Angew. Chem. Int. Ed. 2009, 48, 2572–2575. [Google Scholar] [CrossRef] [PubMed]
- Agut, W.; Brûlet, A.; Schatz, C.; Taton, D.; Lecommandoux, S. pH and temperature responsive polymeric micelles and polymersomes by self-assembly of poly [2-(dimethylamino) ethyl methacrylate]-b-poly (glutamic acid) double hydrophilic block copolymers. Langmuir 2010, 26, 10546–10554. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.-S.; Nah, J.-W.; Jeong, Y.-I.; Cheon, J.-B.; Asayama, S.; Ise, H.; Akaike, T. Conformational transition of nanoparticles composed of poly (γ-benzyl l-glutamate) as the core and poly (ethylene oxide) as the shell. Polymer 1999, 40, 6769–6775. [Google Scholar] [CrossRef]
- Lutz, J.F.; Schütt, D.; Kubowicz, S. Preparation of Well-Defined Diblock Copolymers with Short Polypeptide Segments by Polymerization of N-Carboxy Anhydrides. Macromol. Rapid Commun. 2005, 26, 23–28. [Google Scholar] [CrossRef]
- Cho, I.; Kim, J.-B.; Jung, H.-J. Synthesis and characterization of di-and triblock copolymers of poly (ethylene oxide) and poly (dl-valine-co-dl-leucine). Polymer 2003, 44, 5497–5500. [Google Scholar] [CrossRef]
- Degêe, P.; Dubois, P.; Jérôme, R.; Teyssié, P. Synthesis and characterization of biocompatible and biodegradable poly (ϵ-caprolactone-b-γ-benzylglutamate) diblock copolymers. J. Polym. Sci. Part A 1993, 31, 275–278. [Google Scholar] [CrossRef]
- Kricheldorf, H.R.; Hauser, K. Polylactones. 55. A-b-A Triblock Copolymers of Various Polypeptides. Syntheses Involving 4-Aminobenzoyl-Terminated Poly (ε-caprolactone) as B Block. Biomacromolecules 2001, 2, 1110–1115. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.T.; Vandermeulen, G.W.; Winnik, M.A.; Manners, I. Organometallic-Polypeptide Block Copolymers: Synthesis and Properties of Poly (ferrocenyldimethylsilane)-b-poly-(γ-benzyl-l-glutamate). Macromolecules 2005, 38, 4958–4961. [Google Scholar] [CrossRef]
- Tsutsumiuchi, K.; Aoi, K.; Okada, M. Synthesis of Polyoxazoline-(Glyco) peptide Block Copolymers by Ring-Opening Polymerization of (Sugar-Substituted) α-Amino Acid N-Carboxyanhydrides with Polyoxazoline Macroinitiators. Macromolecules 1997, 30, 4013–4017. [Google Scholar] [CrossRef]
- Naka, K.; Yamashita, R.; Nakamura, T.; Ohki, A.; Maeda, S.; Aoi, K.; Tsutsumiuchi, K.; Okada, M. Aggregates of peptide-containing block copolymers and their interactions with a lipase in aqueous solution. Macromol. Chem. Phys. 1997, 198, 89–100. [Google Scholar] [CrossRef]
- Matsubara, T.; Shinohara, H.; Sisido, M. Synthesis and conformation of poly (L-2-anthraquinonylalanine). Macromolecules 1997, 30, 2651–2656. [Google Scholar] [CrossRef]
- Cho, C.S.; Kim, S.W.; Komoto, T. Synthesis and structural study of an ABA block copolymer consisting of poly (γ-benzyl l-glutamate) as the a block and poly (ethylene oxide) as the B block. Macromol. Chem. Phys. 1990, 191, 981–991. [Google Scholar] [CrossRef]
- Izunobi, J.U.; Higginbotham, C.L. Microstructure characterization and thermal analysis of hybrid block copolymer alpha-methoxy-poly(ethylene glycol)-block-poly epsilon-(benzyloxycarbonyl)-l-lysine for biomedical applications. J. Mol. Struct. 2010, 977, 153–164. [Google Scholar] [CrossRef]
- Yang, Z.; Yuan, J.; Cheng, S. Self-assembling of biocompatible BAB amphiphilic triblock copolymers PLL (Z)–PEG–PLL (Z) in aqueous medium. Eur. Polym. J. 2005, 41, 267–274. [Google Scholar] [CrossRef]
- Deng, C.; Rong, G.; Tian, H.; Tang, Z.; Chen, X.; Jing, X. Synthesis and characterization of poly (ethylene glycol)-b-poly (L-lactide)-b-poly (L-glutamic acid) triblock copolymer. Polymer 2005, 46, 653–659. [Google Scholar] [CrossRef]
- Karatzas, A.; Iatrou, H.; Hadjichristidis, N.; Inoue, K.; Sugiyama, K.; Hirao, A. Complex macromolecular chimeras. Biomacromolecules 2008, 9, 2072–2080. [Google Scholar] [CrossRef] [PubMed]
- Kricheldorf, H.R.; von Lossow, C.; Schwarz, G. Tertiary amine catalyzed polymerizations of α-amino acid N-carboxyanhydrides: The role of cyclization. J. Polym. Sci. Part A 2006, 44, 4680–4695. [Google Scholar] [CrossRef]
- Kricheldorf, H.R.; von Lossow, C.; Schwarz, G. Imidazole-initiated polymerizations of α-amino acid N-carboxyanhydrides-simultaneous chain-growth and step-growth polymerization. J. Polym. Sci. Part A 2005, 43, 5690–5698. [Google Scholar] [CrossRef]
- Kricheldorf, H.R.; Lossow, C.V.; Lomadze, N.; Schwarz, G. Cyclic polypeptides by thermal polymerization of α-amino acid N-carboxyanhydrides. J. Polym. Sci. Part A 2008, 46, 4012–4020. [Google Scholar] [CrossRef]
- Kricheldorf, H.R.; Schwarz, G. Primary Amine and Solvent-Induced Polymerizations of L-or d,l-Phenylalanine N-Carboxyanhydride. Macromol. Chem. Phys. 2005, 206, 282–290. [Google Scholar] [CrossRef]
- Klok, H.A.; Hernández, J.R.; Becker, S.; Müllen, K. Star-shaped fluorescent polypeptides. J. Polym. Sci. Part A 2001, 39, 1572–1583. [Google Scholar] [CrossRef]
- Inoue, K.; Sakai, H.; Ochi, S.; Itaya, T.; Tanigaki, T. Preparation and Conformation of Hexaarmed Star Poly (beta.-benzyl-l-aspartates) Synthesized Utilizing Hexakis (4-aminophenoxy) cyclotriphosphazene. J. Am. Chem. Soc. 1994, 116, 10783–10784. [Google Scholar] [CrossRef]
- Inoue, K.; Horibe, S.; Fukae, M.; Muraki, T.; Ihara, E.; Kayama, H. Synthesis and Conformation of Star-Shaped Poly (γ-benzyl-l-glutamate) s on a Cyclotriphosphazene Core. Macromol. Biosci. 2003, 3, 26–33. [Google Scholar] [CrossRef]
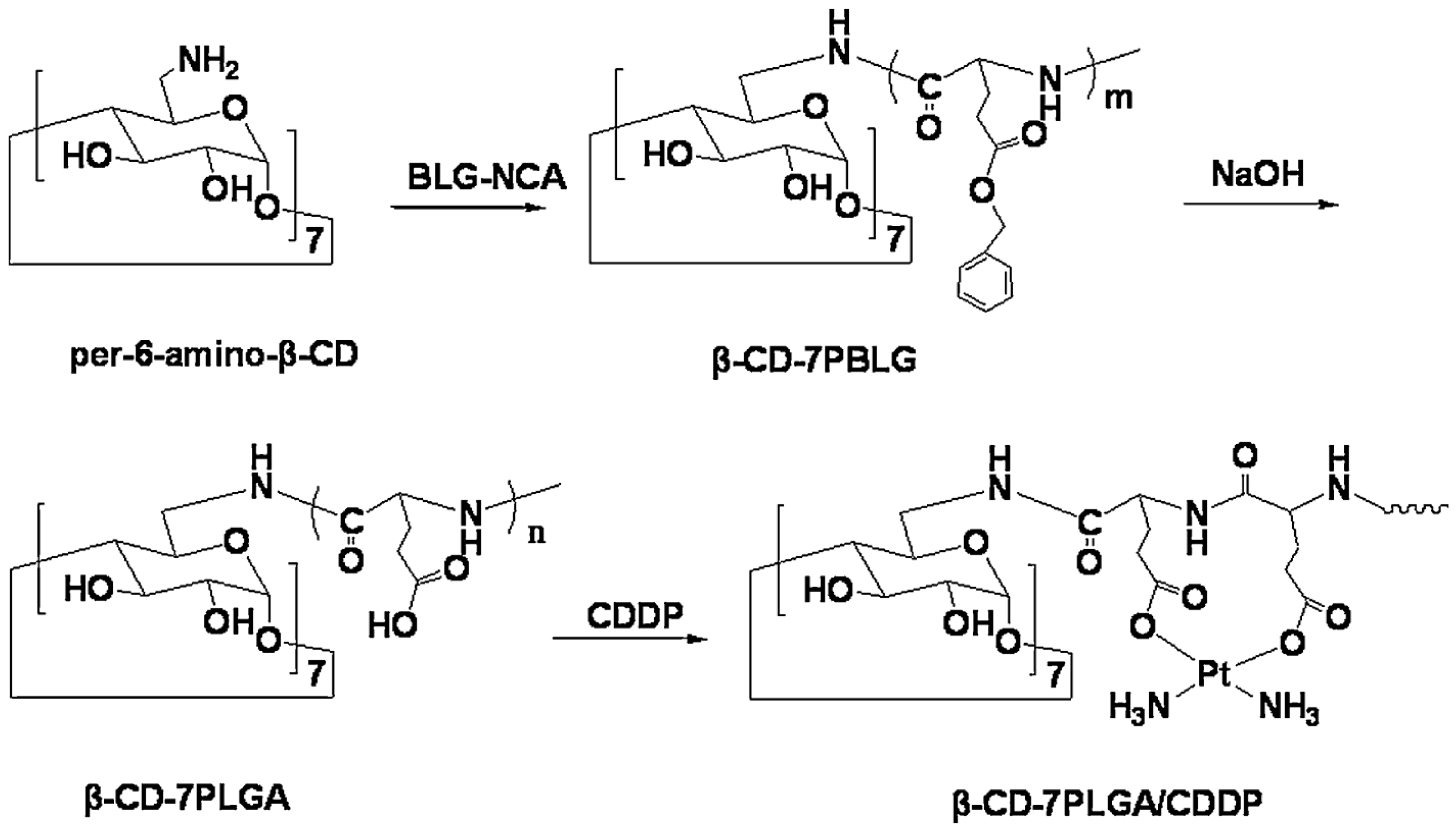
- Yong, D.; Luo, Y.; Du, F.; Huang, J.; Lu, W.; Dai, Z.; Yu, J.; Liu, S. CDDP supramolecular micelles fabricated from adamantine terminated mPEG and β-cyclodextrin based seven-armed poly (l-glutamic acid)/CDDP complexes. Colloids Surfaces B Biointerfaces 2013, 105 (Suppl. C), 31–36. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Ren, T.; Wang, A.; Yuan, W.; Ren, J. Amphiphilic star-shaped poly (ε-caprolactone)-block-poly (l-lysine) copolymers with porphyrin core: Synthesis, self-assembly, and cell viability assay. J. Appl. Polym. Sci. 2014, 131, 40097. [Google Scholar] [CrossRef]
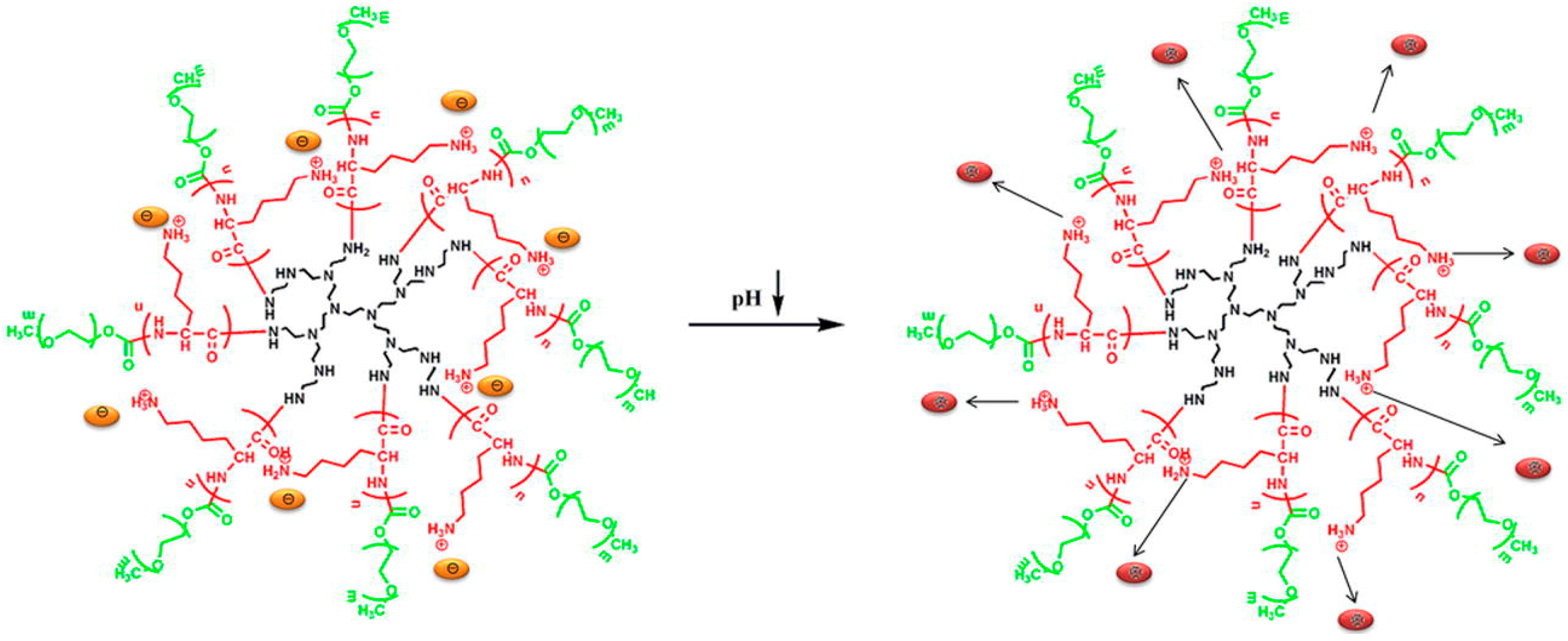
- Tian, H.; Deng, C.; Lin, H.; Sun, J.; Deng, M.; Chen, X.; Jing, X. Biodegradable cationic PEG–PEI–PBLG hyperbranched block copolymer: Synthesis and micelle characterization. Biomaterials 2005, 26, 4209–4217. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Chen, X.; Lin, H.; Deng, C.; Zhang, P.; Wei, Y.; Jing, X. Micellization and Reversible Ph-Sensitive Phase Transfer of the Hyperbranched Multiarm PEI–PBLG Copolymer. Chem. A Eur. J. 2006, 12, 4305–4312. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Xiong, W.; Wei, J.; Wang, Y.; Chen, X.; Jing, X.; Zhu, Q. Gene transfection of hyperbranched PEI grafted by hydrophobic amino acid segment PBLG. Biomaterials 2007, 28, 2899–2907. [Google Scholar] [CrossRef] [PubMed]
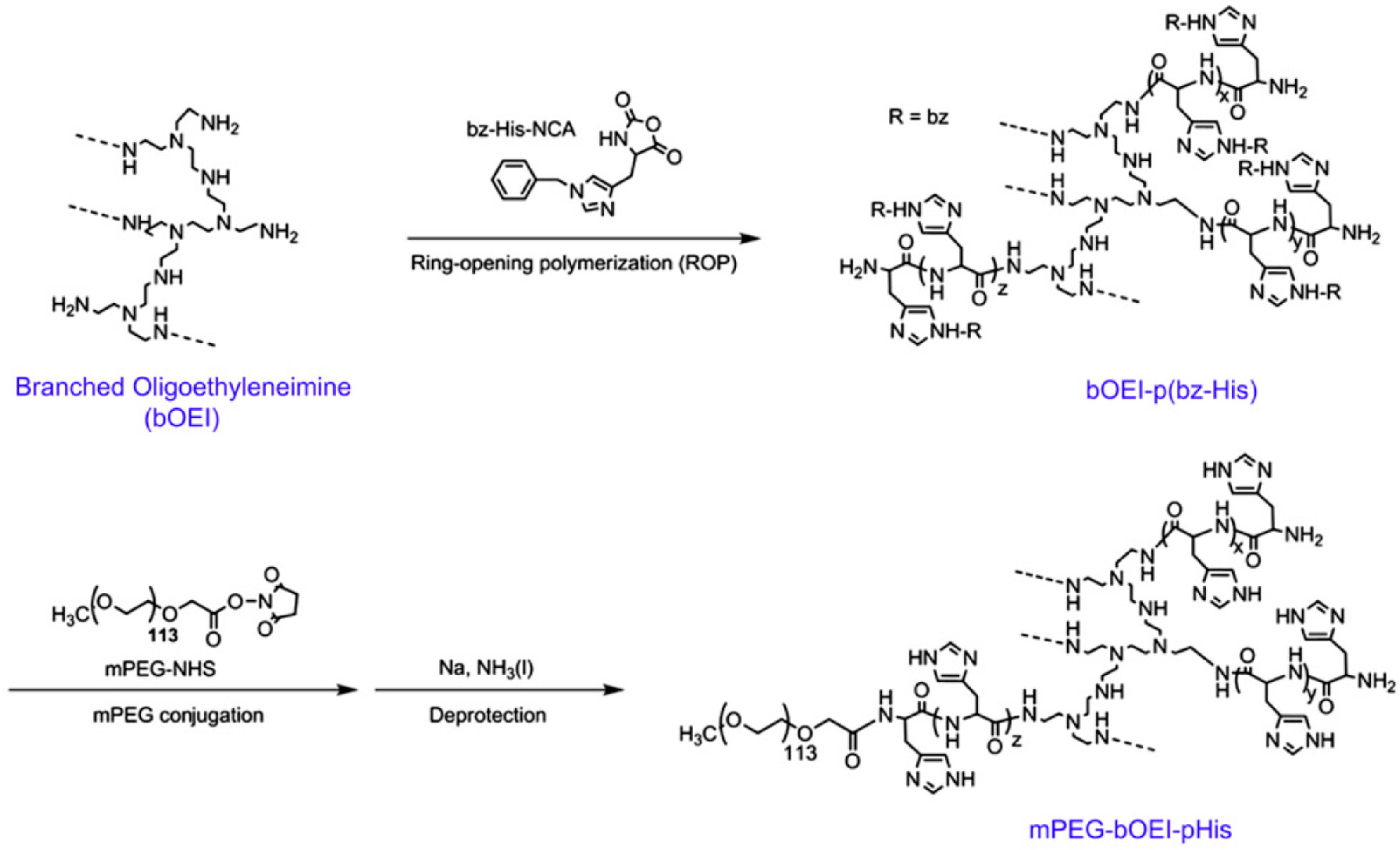
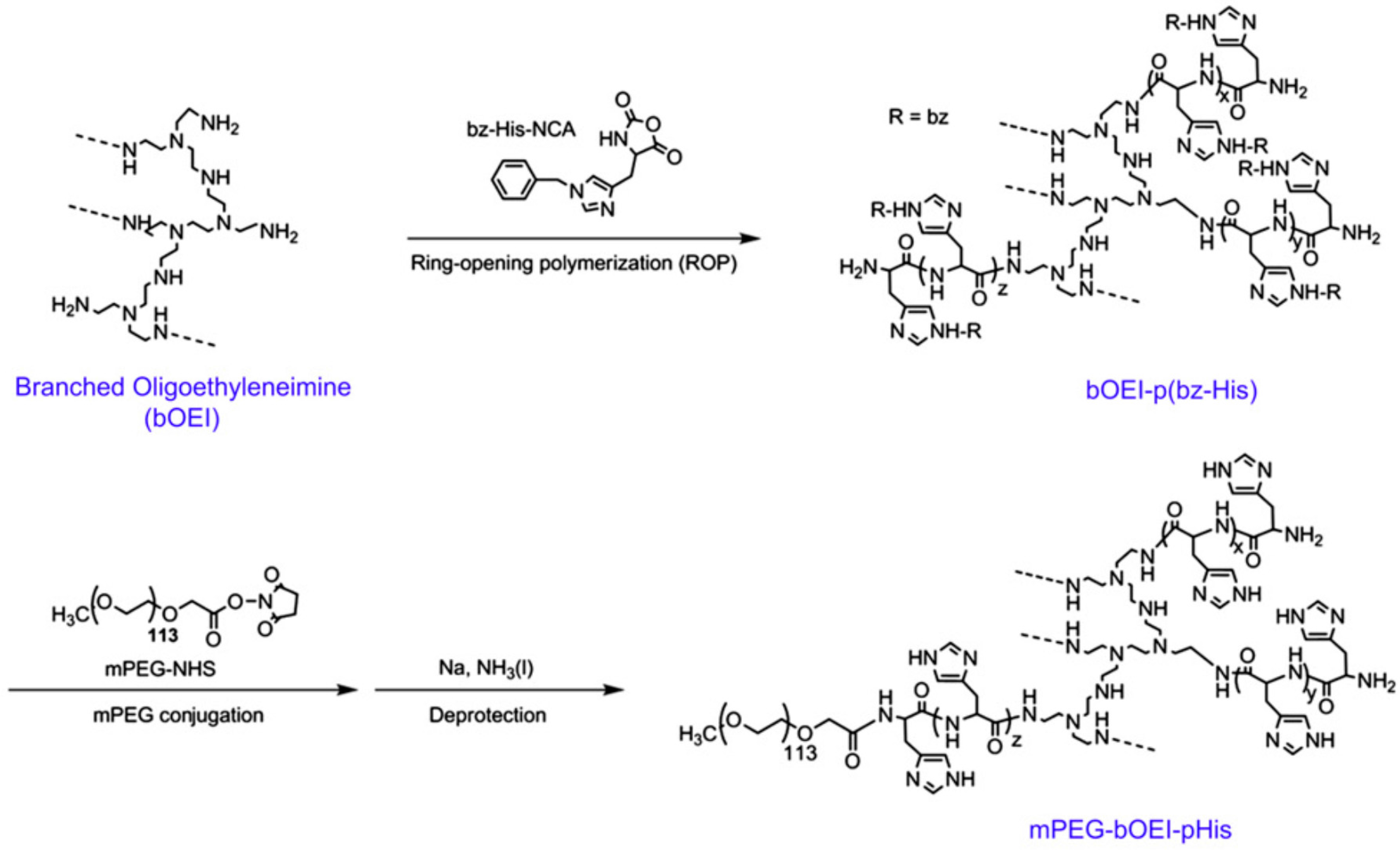
- Park, W.; Kim, D.; Kang, H.C.; Bae, Y.H.; Na, K. Multi-arm histidine copolymer for controlled release of insulin from poly (lactide-co-glycolide) microsphere. Biomaterials 2012, 33, 8848–8857. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yu, J.; Yan, Y.; Xu, S.; Wang, F.; Li, Q.; Wang, J.; Zhang, X.; Liu, D. Controlled biomimetic silica formation using star-shaped poly (l-lysine). Polym. Chem. 2012, 3, 1284–1290. [Google Scholar] [CrossRef]
- Yan, Y.; Wei, D.; Li, J.; Zheng, J.; Shi, G.; Luo, W.; Pan, Y.; Wang, J.; Zhang, L.; He, X. A poly (l-lysine)-based hydrophilic star block co-polymer as a protein nanocarrier with facile encapsulation and pH-responsive release. Acta Biomater. 2012, 8, 2113–2120. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Li, J.; Liao, L.; Li, J.; Wu, L.; Dong, C.; Lai, P.; Liu, D. Poly (l-glutamic acid)-based star-block copolymers as pH-responsive nanocarriers for cationic drugs. Eur. Polym. J. 2012, 48, 696–704. [Google Scholar] [CrossRef]
- Li, J.; Xu, S.; Zheng, J.; Pan, Y.; Wang, J.; Zhang, L.; He, X.; Liu, D. Polypeptide-based star-block quadripolymers as unimolecular nanocarriers for the simultaneous encapsulation of hydrophobic and hydrophilic guests. Eur. Polym. J. 2012, 48, 1696–1708. [Google Scholar] [CrossRef]
- Aoi, K.; Tsutsumiuchi, K.; Yamamoto, A.; Okada, M. Globular carbohydrate macromolecule “sugar balls” 3.“Radial-growth polymerization” of sugar-substituted α-amino acid N-carboxyanhydrides (glycoNCAs) with a dendritic initiator. Tetrahedron 1997, 53, 15415–15427. [Google Scholar] [CrossRef]
- Harada, A.; Kawamura, M.; Matsuo, T.; Takahashi, T.; Kono, K. Synthesis and Characterization of a Head−Tail Type Polycation Block Copolymer as a Nonviral Gene Vector. Bioconjug. Chem. 2006, 17, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Nakanishi, K.; Ichimura, S.; Kojima, C.; Kono, K. Spontaneous formation of narrowly-distributed self-assembly from polyamidoamine dendron-poly (L-lysine) block copolymers through helix-coil transition of poly (l-lysine) block. J. Polym. Sci. Part A 2009, 47, 1217–1223. [Google Scholar] [CrossRef]
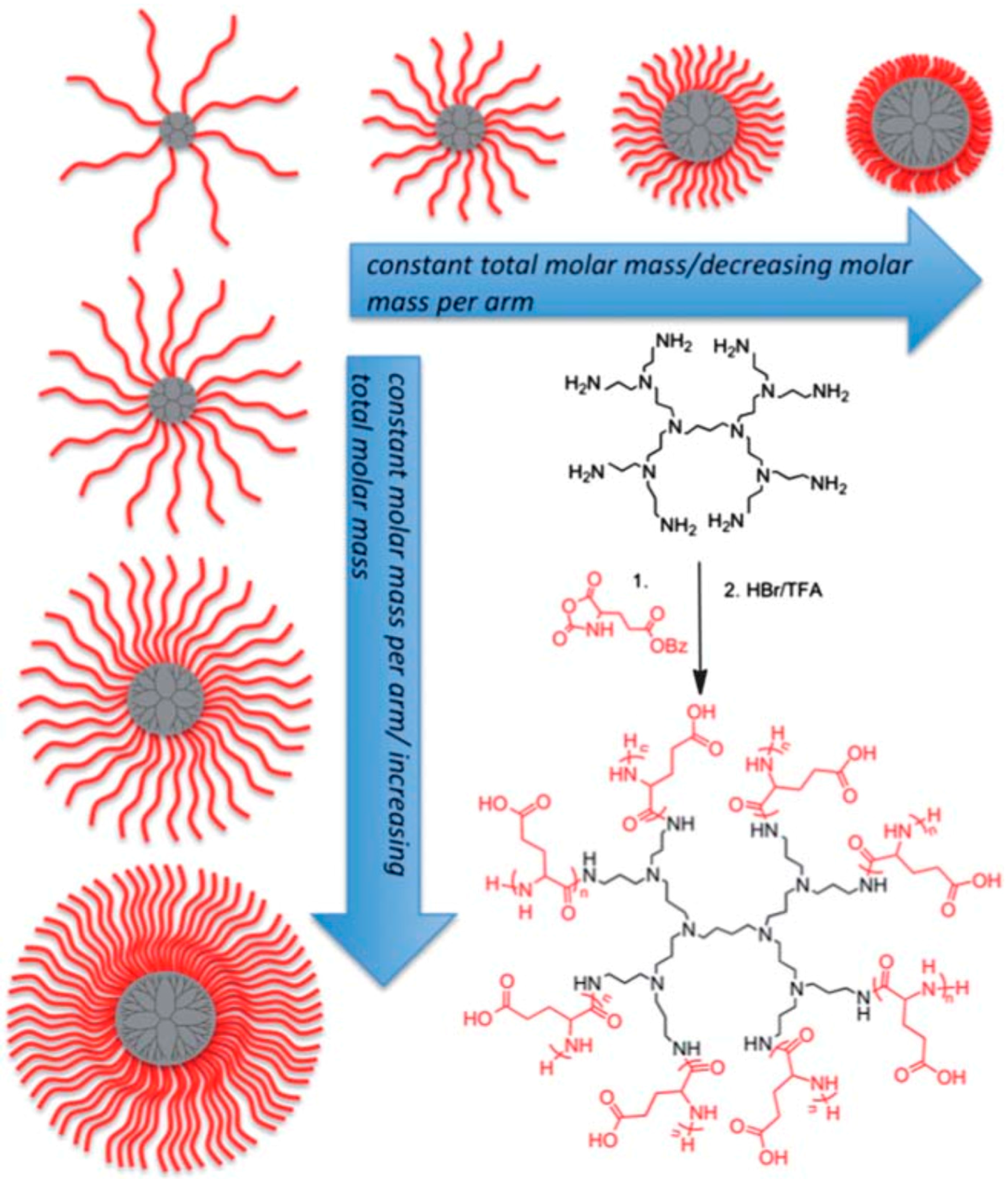
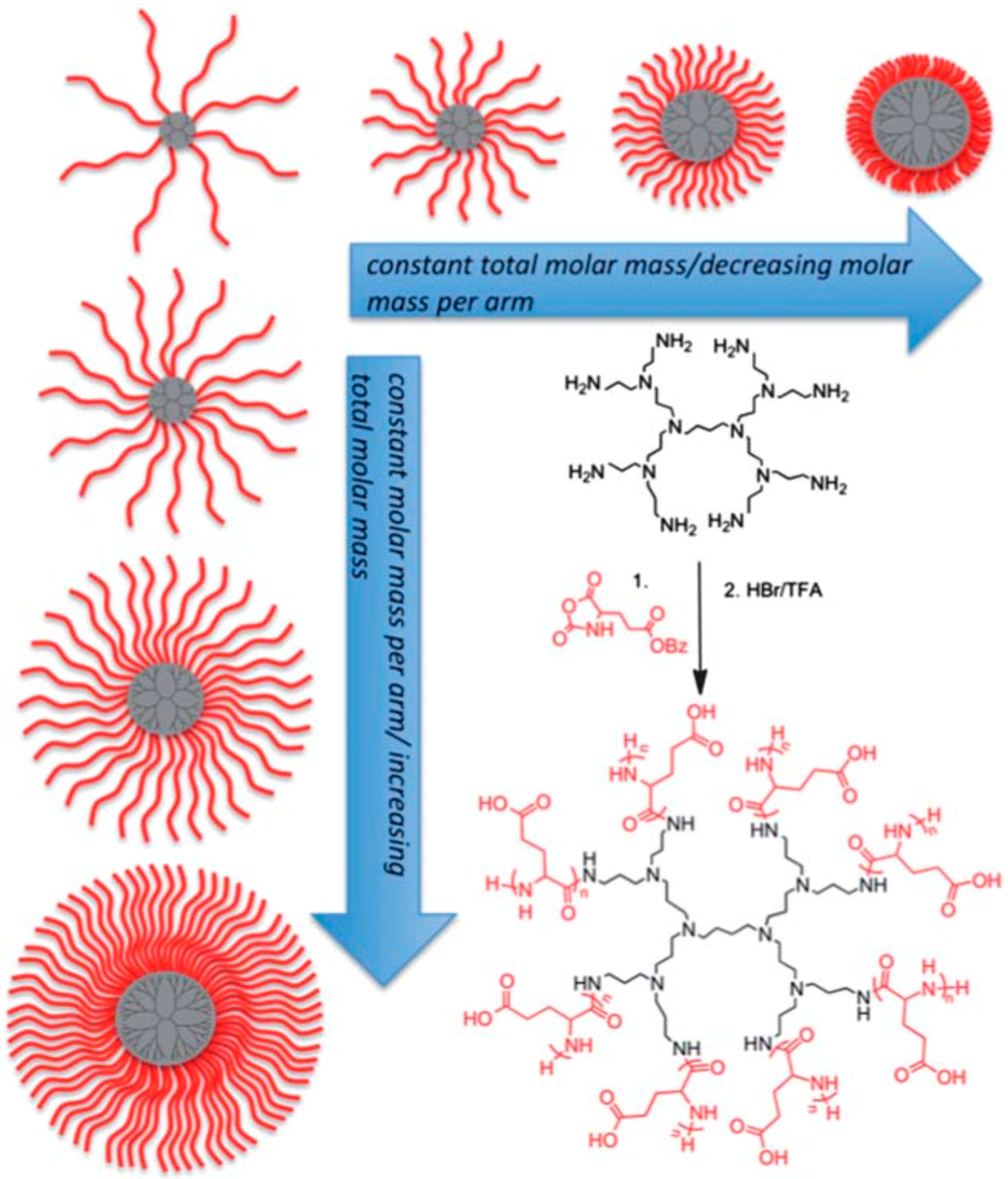
- Byrne, M.; Victory, D.; Hibbitts, A.; Lanigan, M.; Heise, A.; Cryan, S.-A. Molecular weight and architectural dependence of well-defined star-shaped poly (lysine) as a gene delivery vector. Biomater. Sci. 2013, 1, 1223–1234. [Google Scholar] [CrossRef]
- Byrne, M.; Thornton, P.D.; Cryan, S.-A.; Heise, A. Star polypeptides by NCA polymerisation from dendritic initiators: Synthesis and enzyme controlled payload release. Polym. Chem. 2012, 3, 2825–2831. [Google Scholar] [CrossRef]
- Byrne, M.; Mildner, R.; Menzel, H.; Heise, A. Glycosylated star polypeptides from NCA polymerization: Selective binding as a function of degree of branching and glycosylation. Macromol. Biosci. 2015, 15, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Aliferis, T.; Iatrou, H.; Hadjichristidis, N. Well-defined linear multiblock and branched polypeptides by linking chemistry. J. Polym. Sci. Part A 2005, 43, 4670–4673. [Google Scholar] [CrossRef]
- Sulistio, A.; Widjaya, A.; Blencowe, A.; Zhang, X.; Qiao, G. Star polymers composed entirely of amino acid building blocks: A route towards stereospecific, biodegradable and hierarchically functionalized stars. Chem. Commun. 2011, 47, 1151–1153. [Google Scholar] [CrossRef] [PubMed]
- Sulistio, A.; Blencowe, A.; Widjaya, A.; Zhang, X.; Qiao, G. Development of functional amino acid-based star polymers. Polym. Chem. 2012, 3, 224–234. [Google Scholar] [CrossRef]
- Sulistio, A.; Lowenthal, J.; Blencowe, A.; Bongiovanni, M.N.; Ong, L.; Gras, S.L.; Zhang, X.; Qiao, G.G. Folic acid conjugated amino acid-based star polymers for active targeting of cancer cells. Biomacromolecules 2011, 12, 3469–3477. [Google Scholar] [CrossRef] [PubMed]
- Knoop, R.J.; de Geus, M.; Habraken, G.J.; Koning, C.E.; Menzel, H.; Heise, A. Stimuli responsive peptide conjugated polymer nanoparticles. Macromolecules 2010, 43, 4126–4132. [Google Scholar] [CrossRef]
- Audouin, F.; Knoop, R.J.; Huang, J.; Heise, A. Star polymers by cross-linking of linear poly (benzyl-l-glutamate) macromonomers via free-radical and RAFT polymerization. A simple route toward peptide-stabilized nanoparticles. J. Polym. Sci. Part A 2010, 48, 4602–4610. [Google Scholar] [CrossRef]
- Kuo, S.-W.; Tsai, H.-T. Control of peptide secondary structure on star shape polypeptides tethered to polyhedral oligomeric silsesquioxane nanoparticle through click chemistry. Polymer 2010, 51, 5695–5704. [Google Scholar] [CrossRef]
- Babin, J.; Leroy, C.; Lecommandoux, S.; Borsali, R.; Gnanou, Y.; Taton, D. Towards an easy access to amphiphilic rod-coil miktoarm star copolymers. Chem. Commun. 2005, 15, 1993–1995. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.-S.; Jeong, Y.-I.; Kim, S.-H.; Nah, J.-W.; Kubota, M.; Komoto, T. Thermoplastic hydrogel based on hexablock copolymer composed of poly (γ-benzyl l-glutamate) and poly (ethylene oxide). Polymer 2000, 41, 5185–5193. [Google Scholar] [CrossRef]
- Sun, J.; Chen, X.; Guo, J.; Shi, Q.; Xie, Z.; Jing, X. Synthesis and self-assembly of a novel Y-shaped copolymer with a helical polypeptide arm. Polymer 2009, 50, 455–461. [Google Scholar] [CrossRef]
- Sugimoto, H.; Nakanishi, E.; Hanai, T.; Yasumura, T.; Inomata, K. Aggregate formation and release behaviour of hydrophobic drugs with graft copolypeptide-containing tryptophan. Polym. Int. 2004, 53, 972–983. [Google Scholar] [CrossRef]
- Boulahia, J.; Carrière, F.; Sekiguchi, H. Synthesis and characterization of copolymers from acrylamide and methacrylamide carrying peptide side chains. Macromol. Chem. Phys. 1989, 190, 1975–1986. [Google Scholar] [CrossRef]
- Aoyama, M.; Youda, A.; Watanabe, J.; Inoue, S. Synthesis and circular dichroic and photoresponsive properties of a graft copolymer containing an azoaromatic polypeptide branch and its membrane. Macromolecules 1990, 23, 1458–1463. [Google Scholar] [CrossRef]
- Aoyama, M.; Watanabe, J.; Inoue, S. Photoregulation of permeability across a membrane from a graft copolymer containing a photoresponsive polypeptide branch. J. Am. Chem. Soc. 1990, 112, 5542–5545. [Google Scholar] [CrossRef]
- Fu, X.; Ma, Y.; Sun, J.; Li, Z. Biodegradable thermal-and redox-responsive poly (l-glutamate) with Y-shaped oligo (ethylene glycol) side-chain and tunable phase transition temperature. RSC Adv. 2016, 6, 70243–70250. [Google Scholar] [CrossRef]
- Zhang, B.; Fischer, K.; Schmidt, M. Cylindrical polypeptide brushes. Macromol. Chem. Phys. 2005, 206, 157–162. [Google Scholar] [CrossRef]
- Xiang, Y.; Si, J.; Zhang, Q.; Liu, Y.; Guo, H. Homogeneous graft copolymerization and characterization of novel artificial glycoprotein: Chitosan-poly(l-tryptophan) copolymers with secondary structural side chains. J. Polym. Sci. Part A 2009, 47, 925–934. [Google Scholar] [CrossRef]
- Rhodes, A.J.; Deming, T.J. Tandem Catalysis for the Preparation of Cylindrical Polypeptide Brushes. J. Am. Chem. Soc. 2012, 134, 19463–19467. [Google Scholar] [CrossRef] [PubMed]
- Takaki, M.; Asami, R.; Hanada, Y.; Ochiai, N. Preparation of (vinylbenzylimino) oligo (dl-phenylalanine NCA) macromer with narrow molecular weight distribution. Polym. Bull. 1987, 18, 105–110. [Google Scholar] [CrossRef]
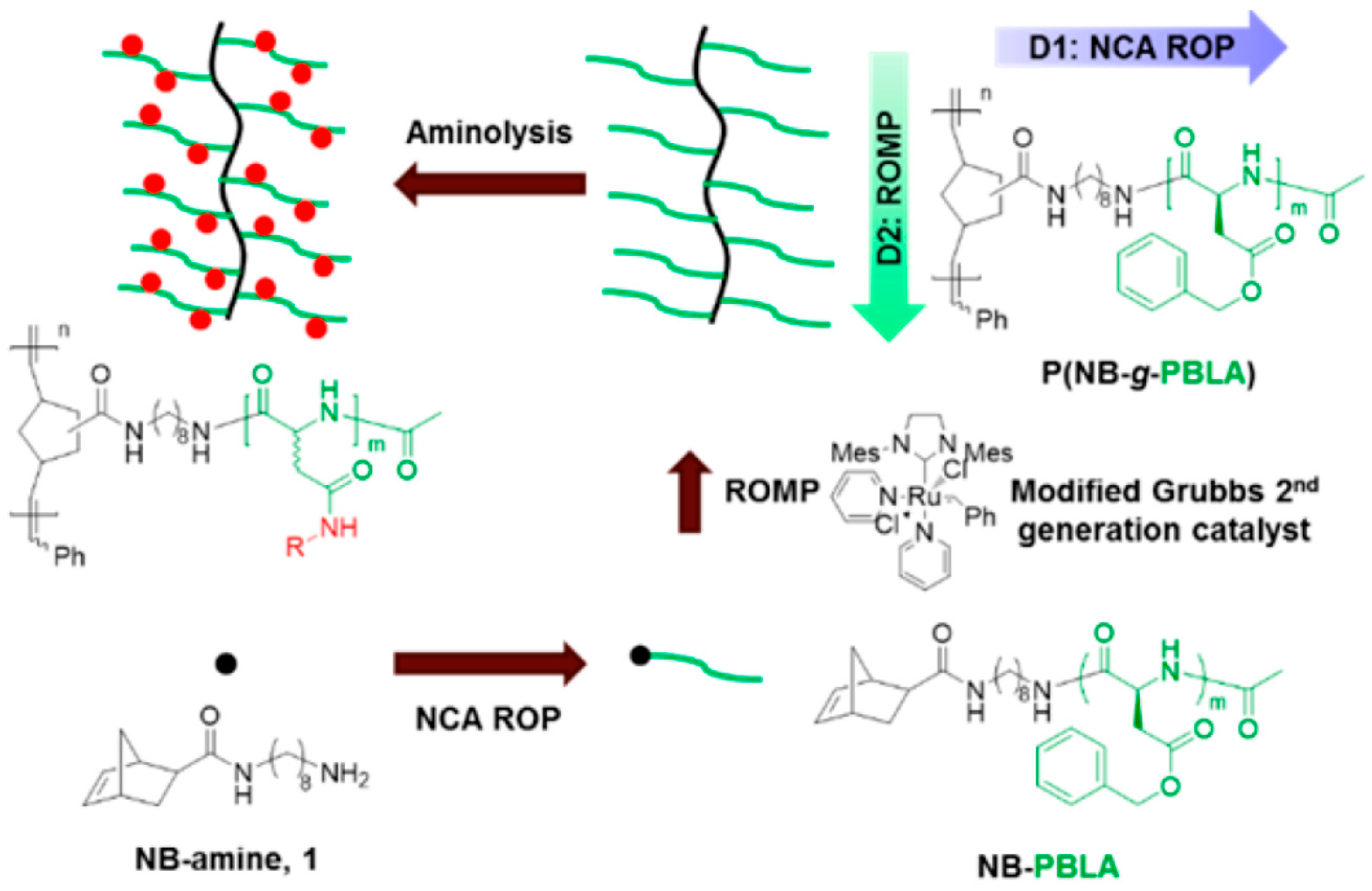
- Fan, J.; Borguet, Y.P.; Su, L.; Nguyen, T.P.; Wang, H.; He, X.; Zou, J.; Wooley, K.L. Two-Dimensional Controlled Syntheses of Polypeptide Molecular Brushes via N-Carboxyanhydride Ring-Opening Polymerization and Ring-Opening Metathesis Polymerization. ACS Macro Lett. 2017, 6, 1031–1035. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Xiao, C.; Tang, Z.; Zhuang, X.; Chen, X. Highly Efficient “Grafting From” an α-Helical Polypeptide Backbone by Atom Transfer Radical Polymerization. Macromol. Biosci. 2011, 11, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Xiao, C.; Zhao, L.; Cheng, Y.; Ma, L.; Tang, Z.; Zhuang, X.; Chen, X. Poly (l-glutamic acid) grafted with oligo (2-(2-(2-methoxyethoxy) ethoxy) ethyl methacrylate): Thermal phase transition, secondary structure, and self-assembly. J. Polym. Sci. Part A 2011, 49, 2665–2676. [Google Scholar] [CrossRef]
- Klok, H.-A.; Rodríguez-Hernández, J. Dendritic−graft polypeptides. Macromolecules 2002, 35, 8718–8723. [Google Scholar] [CrossRef]
- Rodríguez-Hernández, J.; Gatti, M.; Klok, H.-A. Highly branched poly (l-lysine). Biomacromolecules 2003, 4, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Dong, C.-M. Phototriggered Ring-Opening Polymerization of a Photocaged l-Lysine N-Carboxyanhydride to Synthesize Hyperbranched and Linear Polypeptides. ACS Macro Lett. 2017, 6, 292–297. [Google Scholar] [CrossRef]

















































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
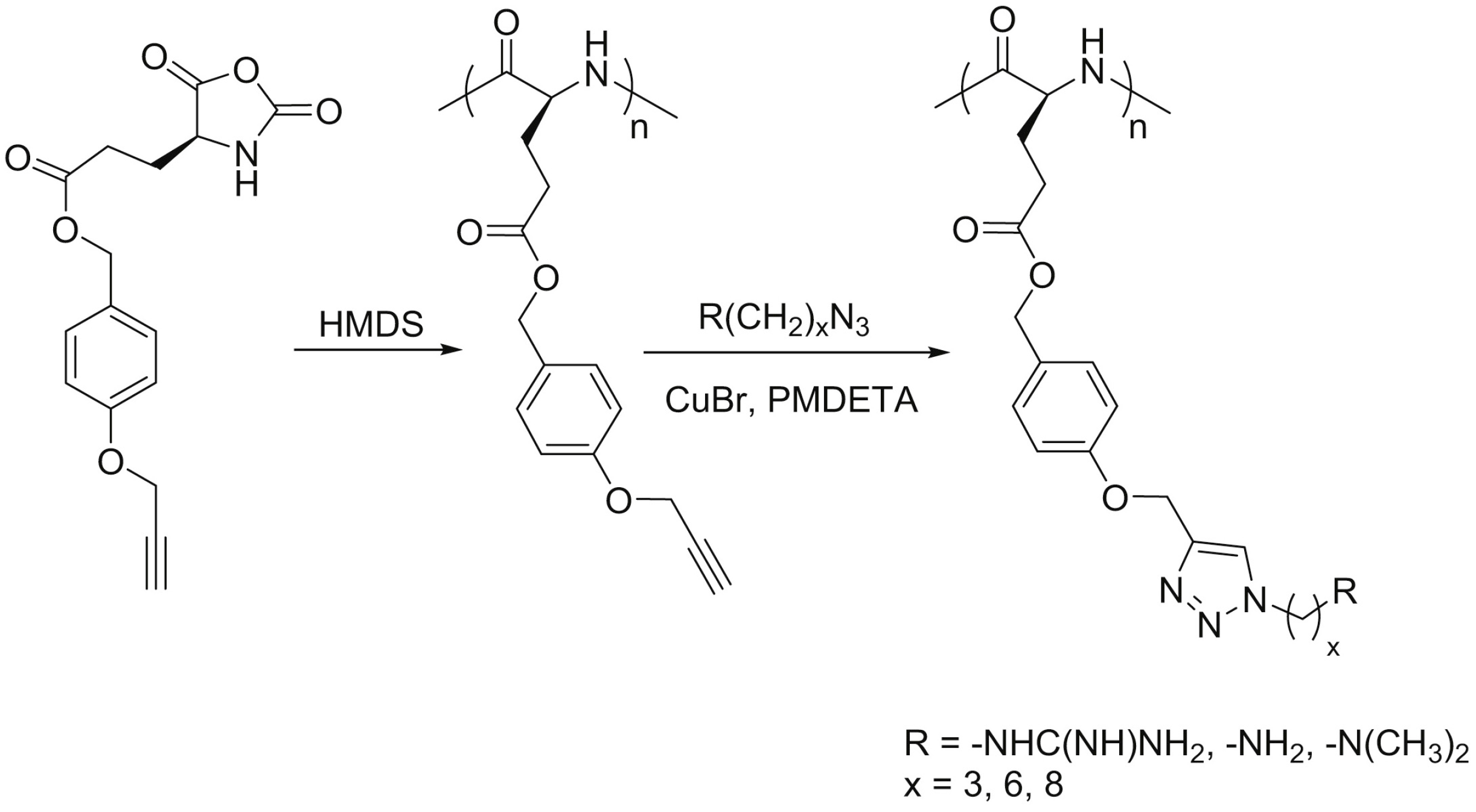{kind=link}
{kind=link}
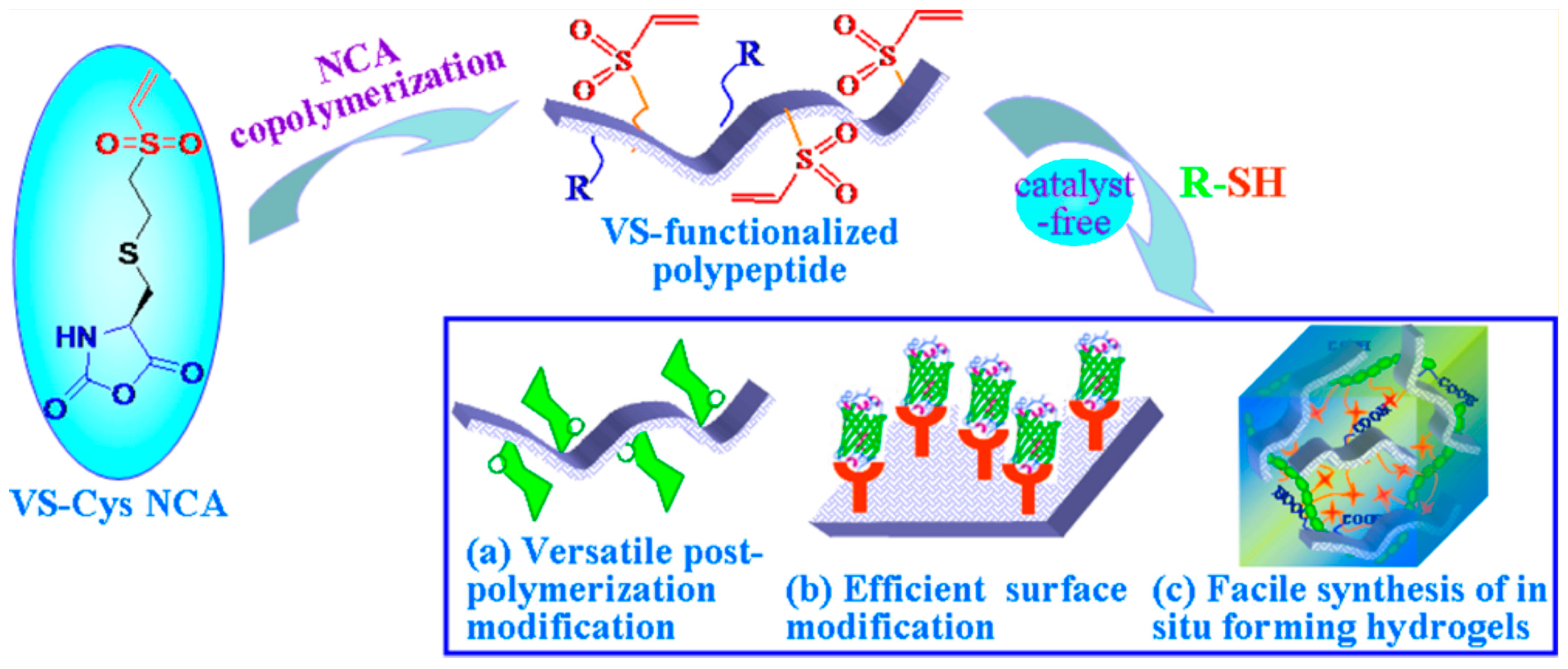{kind=link}
{kind=link}
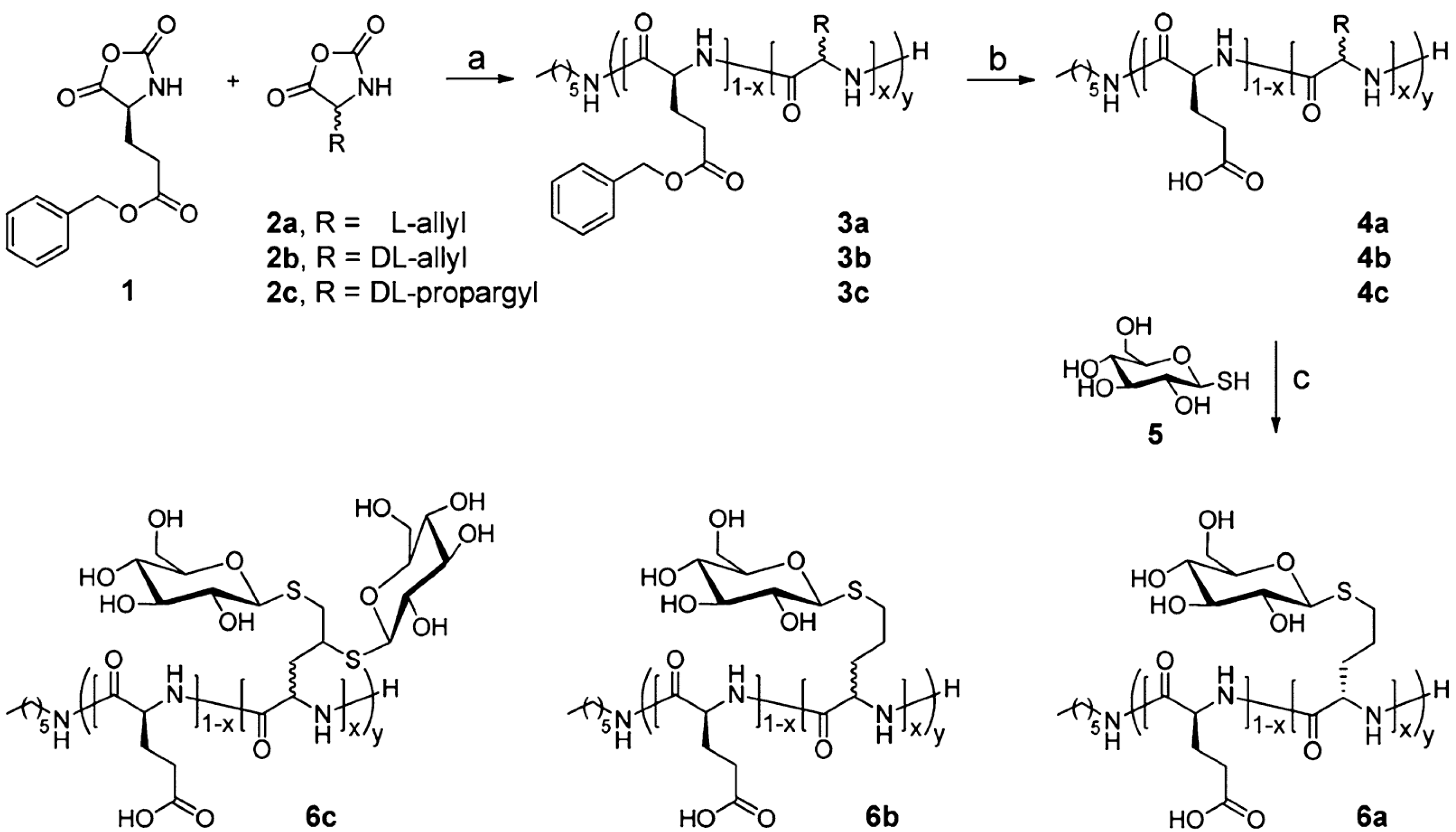{kind=link}
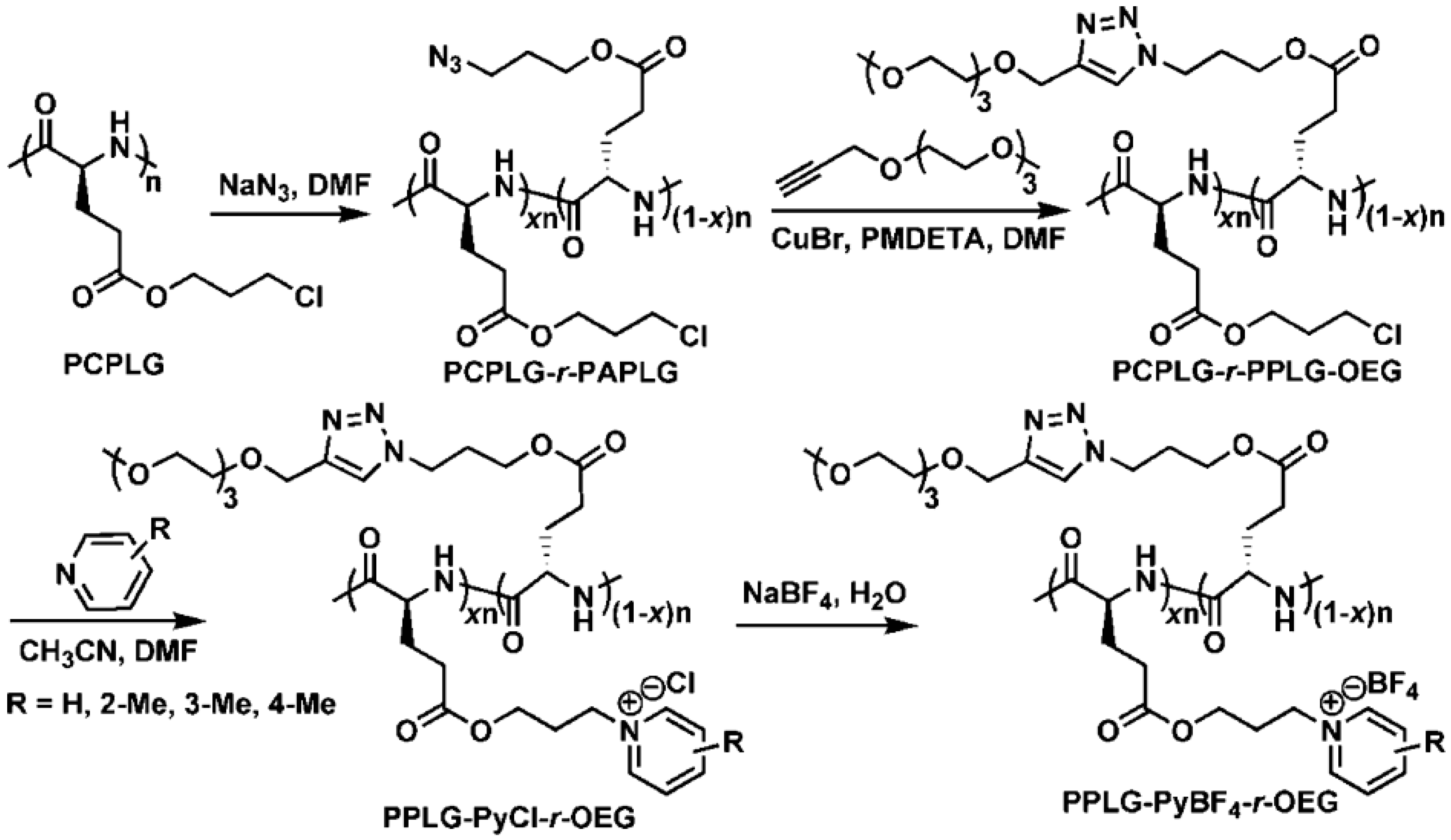{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alkyne-Azide [2 + 3] Huisgen Cycloaddition | |||
PG-NCA [33,34,35,36]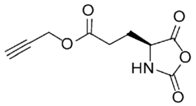 | PG-Gly-NCA [37,38]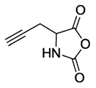 | ||
CP-NCA [39] 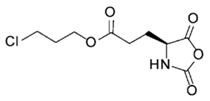 | Anl-NCA and Anv-NCA [40]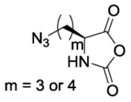 | ||
| Thiol-Ene Reactions | |||
DL-Allylglycine [41,42] | AL-NCA [43] | AOB-Glu-NCA [44]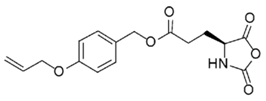 | |
| Mimics of Glycosylated Peptides and Proteins | |||
α-Glyco-Lys-NCAs [45,46,47] | Glyco NCAs (R = Ac4Gal, Ac4Glu or Ac7Lac; X = O or S; R0 = Me or H)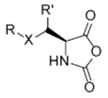 | ||
| Thermo-Responsive Polypeptides | |||
L-EGxGlu-NCA [48] | EGxMA-Cys-NCA (R = CH3) and EGxA-Cys-NCA (R = H) [49]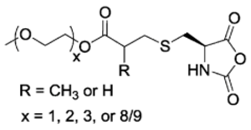 | ||
| Photo-Responsive | |||
NBC-NCA [50]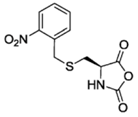 | DMNB-Glu-NCA (R = OMe) [51]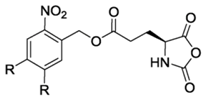 | ||
| ATRP to Prepare Molecular Bottlebrush | Organic Semiconductor Unit; Organic Photovoltaic and Organic Field Effect Transistor | ||
Br-Lys-NCA [52] | Hexithiophene-LysNCA [53] | ||
| M/I | Temperature (°C) | Living Polymers (°C) a | Dead Polymers (Peaks B and C) (%) a |
|---|---|---|---|
| 50 | 50 | 20 | 80 |
| 50 | Room | 22 | 78 |
| 50 | 0 | 99 | 1 |
| Type of Block Copolymer | Block Copolypeptide | Reference |
|---|---|---|
| Diblock | poly[γ-benzyl-l-glutamate-b-(l-glutamic Acid)] | [121] |
| Diblock | Nε-4-phenylbenzamido-l-lysine-b-Nε-trifluoroacetyl-l-lysine | [120] |
| Diblock | o-(tetra-o-acetyl-d-glucopyranosyl)-l-serine-NCA-b-alanine NCA | [119] |
| Diblock | poly(γ-benzyl-l-glutamate)-b-polyglycine | [122] |
| Diblock | poly[γ-benzyl-l-glutamate-b-(poly(Nε-carbobenzoxy-l-lysine)], poly[γ-benzyl-l-glutamate-b-polyglycine], poly[γ-benzyl-l-glutamate-b-poly(l-tyrosine)], poly[γ-benzyl-l-glutamate-b-poly(poly(l-leucine)]. | [76] |
| Diblock | poly(γ-benzyl-l-glutamate)-b-poly(l-lysine) | [123] |
| Diblock | poly[γ-benzyl-l-glutamate-b-(poly(Nε-carbobenzoxy-l-lysine)], and block copolypeptides containing l-leucine and l-proline) | [130] |
| Diblock | poly(Nε-2[2-(2-methoxyethoxy)ethoxy]acetyl-l-lysine)-b-poly(l-aspartic acid, sodium salt) | [131] |
| Diblock | poly(l-lysine Hbr) or poly(l-glutamic acid sodium salt), and helical, hydrophobic segments of poly(l-leucine) | [132,133] |
| Diblock | diblock copolypeptides of hydrophilic l-lysine or l-glutamic acid and hydrophobic leucine or valine | [134] |
| Diblock | poly(l-lysine)-b-poly(l-glycine), poly(l-lysine)-b-poly(l-alanine), poly(l-lysine)-b-poly(l-phenylalanine) | [135] |
| Diblock | poly(S-(o-nitrobenzyl)-l-cysteine)-b-poly(ethylene glycol) (PNBC-b-PEO) block copolymers | [50] |
| Diblock | Hydrophilic (glutamic acid or lysine) and one nonpolar block (alanine) or with both hydrophilic blocks with opposite charges (glutamic acid and lysine) | [136] |
| Diblock | Poly(l-lysine)-b-poly(l-glycine) | [137,138] |
| Diblock | poly(l-lysine)-b-poly(l-phenylalanine) | [139] |
| Diblock | poly(l-methionine)65-b-poly(l-leucine0.5-stat-l-phenylalanine0.5)20 | [140] |
| Diblock/Triblock | poly-l-lysine-b-poly-l-leucine and poly-l-lysine-b-poly-l-leucine-b-poly-l-lysine | [128] |
| Triblock | poly[(γ-benzyl-l-glutamate)-b-(l-leucine)-b-(γ-benzyl-l-glutamate)] and the corresponding poly[(l-Glutamic Acid)-b-(l-leucine)-b-(l-Glutamic Acid)], poly(Nε-carbobenzoxy-l-lysine)-b-(γ-benzyl-l-glutamate)-b-(poly-Nε-carbobenzoxy-l-lysine) | [76,126,127,141] |
| Triblock | poly(l-homoarginine HCl)m-block-poly(Lglutamate Na)n-block-poly(l-leucine)20 | [6] |
| Triblock | poly[(Nε-carbobenzoxy-l-lysine)-b-poly(l-leucine)-b-poly(Nε-carbobenzoxy l-lysine)] | [128,129] |
| Tetrablock | Poly(γ-benzyl-l-glutamate)-b-poly(l-alanine)-b-poly(Nε-benzyloxycarbonyl-l-lysine)-b-poly(β-benzyl-l-aspartate) | [79] |
| Type of Block Copolymer (Diblock, Triblock, Pentablock…) | Non-Polypeptidic Block | Hybrid Copolymer Prepared | References |
|---|---|---|---|
| Polyethylene oxide | polyethyelene oxide-b-poly(γ-benzyl-l-glutamate) | [164,165] | |
| Polyethylene oxide | polyethyelene oxide-b-poly(-benzyl-l-aspartate) | [165] | |
| Polyethylene oxide | polyethyelene oxide-b--b-poly(l-valine/l-leucine) | [166] | |
| Polypseudorotaxane | poly(γ-benzyl-l-glutamate)-b--b-polypseudorotaxane | [7] | |
| Diblock | poly(ε-caprolactone) | poly(ε-caprolactone)-b-poly(γ-benzyl-l-glutamate) | [167] |
| Diblock | Polylactic acid | polylactic acid-b-poly(l-Alanine), polylactic acid-b-poly(l-phenylalanine), polylactic acid-b-poly(l-leucine), polylactic acid-b-poly(γ-benzyl-l-glutamate) and polylactic acid-b-Poly(benzyl-l-aspartate) | [10,157,158] |
| Diblock | Poly(ε-caprolactone) | poly(ε-caprolactone)-b-poly(γ-benzyl-l-glutamate) | [156] |
| Diblock | Poly(ε-caprolactone) | poly(ε-caprolactone)-b-poly(l-Glycine), poly(ε-caprolactone)-b-poly(l-alanine), poly(ε-caprolactone)-b-poly(l-Phenylalanine), poly(ε-caprolactone)-b-poly(γ-benzyl-l-glutamate) | [168] |
| Diblock | Poly(N-isopropylacrylamide)- | poly(N-isopropylacrylamide)-b-poly(γ-benzyl-l-glutamate) | [145,151] |
| Diblock | Poly(2,7-dibromo-9,9-dihexylfluorene) | 2,7-dibromo-9,9-dihexylfluorene-b-poly(γ-benzyl-l-glutamate) | [161] |
| Diblock | Poly(N-Vinylpirrolidone) | poly(N-Vinylpirrolidone)-b-poly(γ-benzyl-l-glutamate), poly(N-Vinylpirrolidone)-b--b-poly(Z-l-Lysine) and poly(N-Vinylpirrolidone)-b-poly(γ-benzyl-l-glutamate)-b-poly(Z-l-Lysine) | [153] |
| Diblock | Polysaccharide | polysaccharide-b-poly(γ-benzyl-l-glutamate) | [162] |
| Diblock | Poly(ferrocenyldimethylsilane) | poly(ferrocenyldimethylsilane)-b-poly(γ-benzyl-l-glutamate) | [169] |
| Diblock | Poly(1-(3-chloropropyl)-2,2,5,5-tetramethyl-1-aza-2,5-disilacyclopentane) | 1-(3-chloropropyl)-2,2,5,5-tetramethyl-1-aza-2,5-disilacyclopentane-b-poly(γ-benzyl-l-glutamate) | [159] |
| Diblock | Poly(2-methyl-2-oxazoline)- | poly(2-methyl-2-oxazoline)-b-poly(γ-benzyl-l-glutamate), poly(2-methyl-2-oxazoline)-b-poly(phenylalanine), poly(2-methyl- 2-oxazoline)-b-poly[O-(tetra-O-acetyl--d-glucopyranosyl)-l-serine], and poly(2-phenyl-2-oxazoline)-b-poly[O-(tetraO-acetyl--d-glucopyranosyl)-l-serine] | [170,171] |
| Diblock | Poly(methyl acrylate) | poly(methyl acrylate)-b-poly(γ-benzyl-l-glutamate) | [146] |
| Diblock | Polystyrene | polystyrene-b-Poly(Z-l-Lysine) | [68,160] |
| Triblock | Poly(l-2-anthraquinonylalanine) | poly(γ-benzyl-l-glutamate)-b-poly(l-2-anthraquinonylalanine)-b-poly(γ-benzyl-l-glutamate) | [172] |
| Triblock copolymer | Polyethylene oxide | poly(γ-benzyl-l-glutamate)-b-polyethyelene oxide-b-poly(γ-benzyl-l-glutamate | [173] |
| Triblock copolymer | Polyethylene oxide | poly(Z-l-Lysine)-b-polyethyelene oxide-b-Poly(Z-l-Lysine) | [174,175] |
| Triblock copolymer | Polyethylene oxide | poly[(l-valine)-co-(l-leucine)]-b-polyethyelene oxide-b-poly[(l-valine)-co-(l-leucine)] | [166] |
| Triblock copolymer | Polyethylene oxide/Polylactic acid | polyethylene oxide-b-Polylactic acid-b-poly(γ-benzyl-l-glutamate) | [176] |
| Triblock terpolymers | Polyethylene oxide | polyethyelene oxide-b-Poly(Z-l-Lysine)-b-poly(γ-benzyl-l-glutamate) | [153] |
| Pentablock | Polystyrene | poly(ε-tert-butyloxycarbonyl-l-lysine)]-b-poly(γ-benzyl-l-glutamate)-b-Polystyrene-b poly(γ-benzyl-l-glutamate)-b-poly(ε-tert-butyloxycarbonyl-l-lysine)] | [177] |
| Amino Acid N-Carboxyanhydride(s) Employed | Conditions | References |
|---|---|---|
| d,l-Leucine-NCA, and d,l-Phenylalanine-NCA | Dioxane at 60 °C/Initiator: imidazole | [179] |
| d,l-alanine N-carboxyanhydride, d,l-phenylalanine N-carboxyanhydride, and d,l-leucine N-carboxyanhydride | Using pyridine as initiator. | [178] |
| Sarcosine-NCA at 120 °C | Thermal polymerization | [180] |
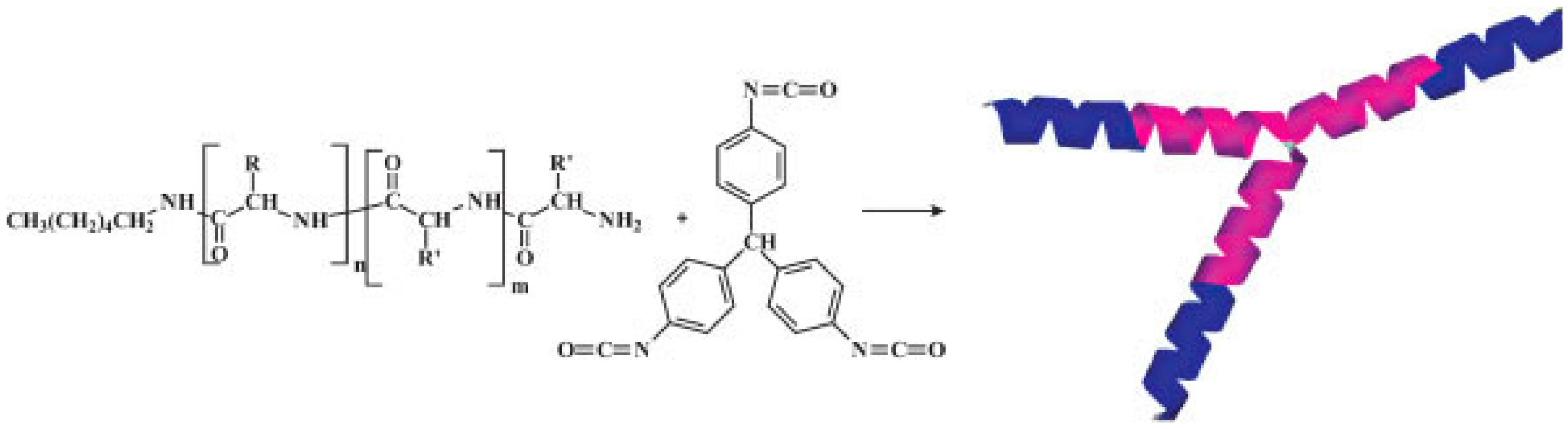
| d,l-Phenylalanine-NCA | Difunctional primary amines (1,12-diaminododecane, DAD, or 1,13-diamino-4,7,10-trioxatridecane, DATT) for the synthesis of linear well-defined telechelic polymers bearing amino end groups. Subsequent reaction of the telechelic polymers with 4,4′-Diisocyanatodiphenylmethane lead to cyclic polypeptides | [181] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Henríquez, C.M.; Sarabia-Vallejos, M.A.; Rodríguez-Hernández, J. Strategies to Fabricate Polypeptide-Based Structures via Ring-Opening Polymerization of N-Carboxyanhydrides. Polymers 2017, 9, 551. https://doi.org/10.3390/polym9110551
González-Henríquez CM, Sarabia-Vallejos MA, Rodríguez-Hernández J. Strategies to Fabricate Polypeptide-Based Structures via Ring-Opening Polymerization of N-Carboxyanhydrides. Polymers. 2017; 9(11):551. https://doi.org/10.3390/polym9110551
Chicago/Turabian StyleGonzález-Henríquez, Carmen M., Mauricio A. Sarabia-Vallejos, and Juan Rodríguez-Hernández. 2017. "Strategies to Fabricate Polypeptide-Based Structures via Ring-Opening Polymerization of N-Carboxyanhydrides" Polymers 9, no. 11: 551. https://doi.org/10.3390/polym9110551
APA StyleGonzález-Henríquez, C. M., Sarabia-Vallejos, M. A., & Rodríguez-Hernández, J. (2017). Strategies to Fabricate Polypeptide-Based Structures via Ring-Opening Polymerization of N-Carboxyanhydrides. Polymers, 9(11), 551. https://doi.org/10.3390/polym9110551