
New Mechanism Proposed for the Base-Catalyzed Urea–Formaldehyde Condensation Reactions: A Theoretical Study


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Quantum Chemistry Calculations
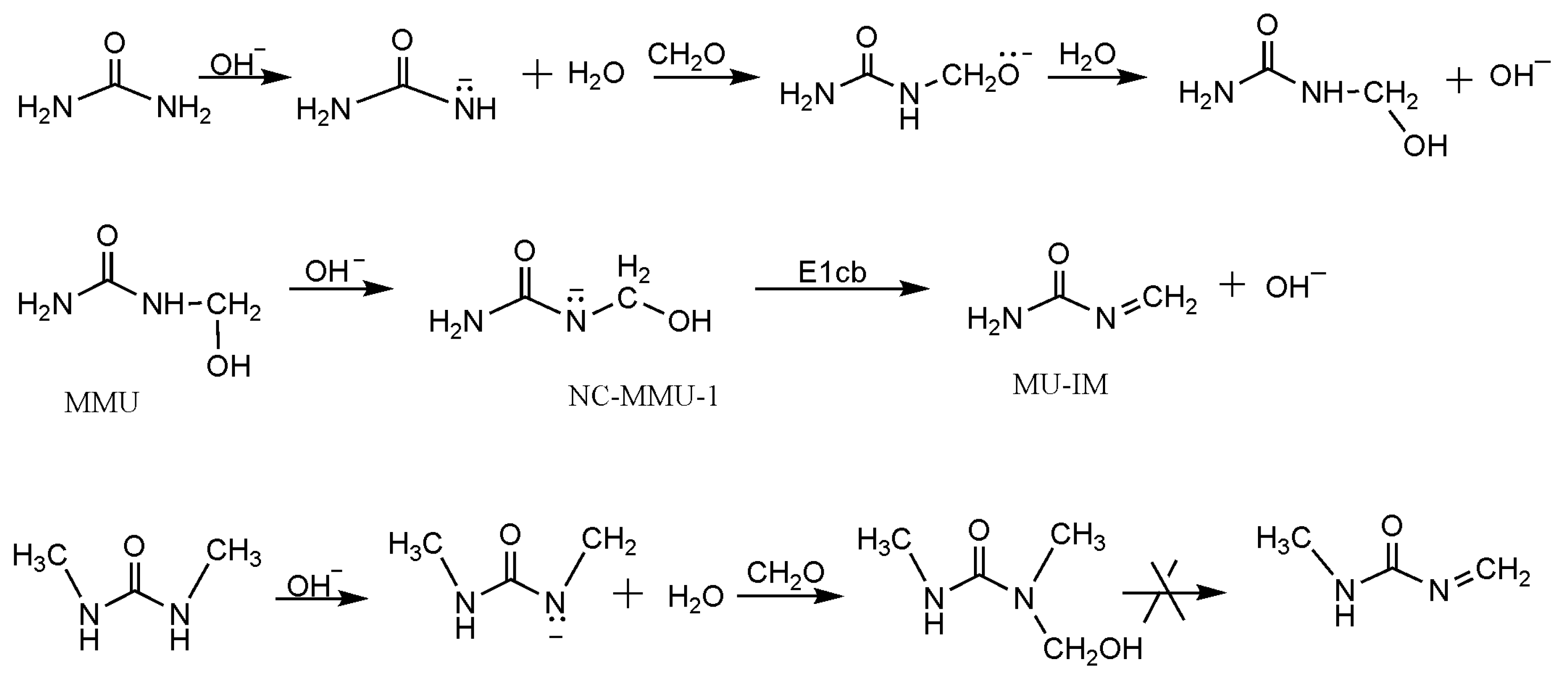
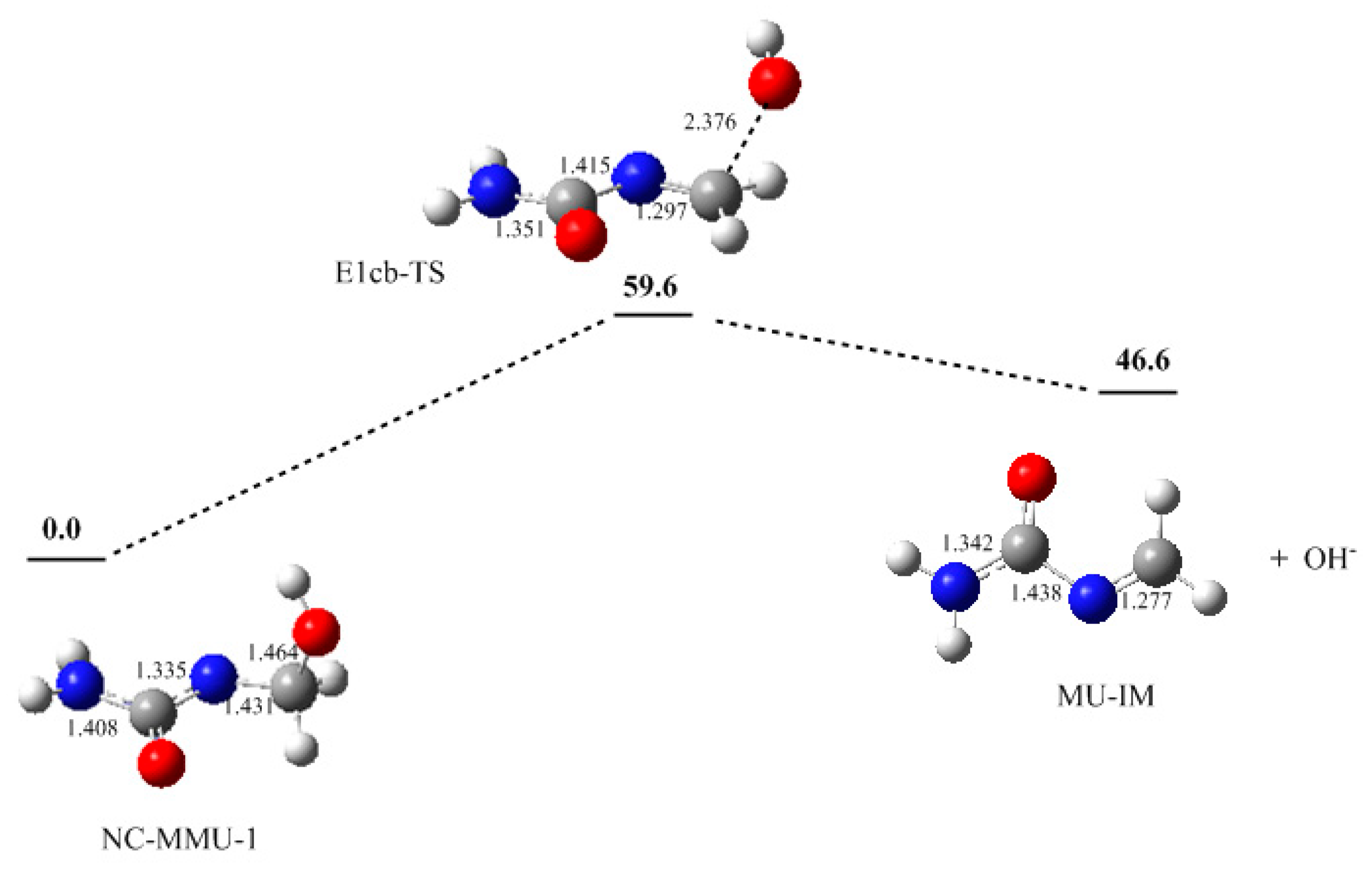
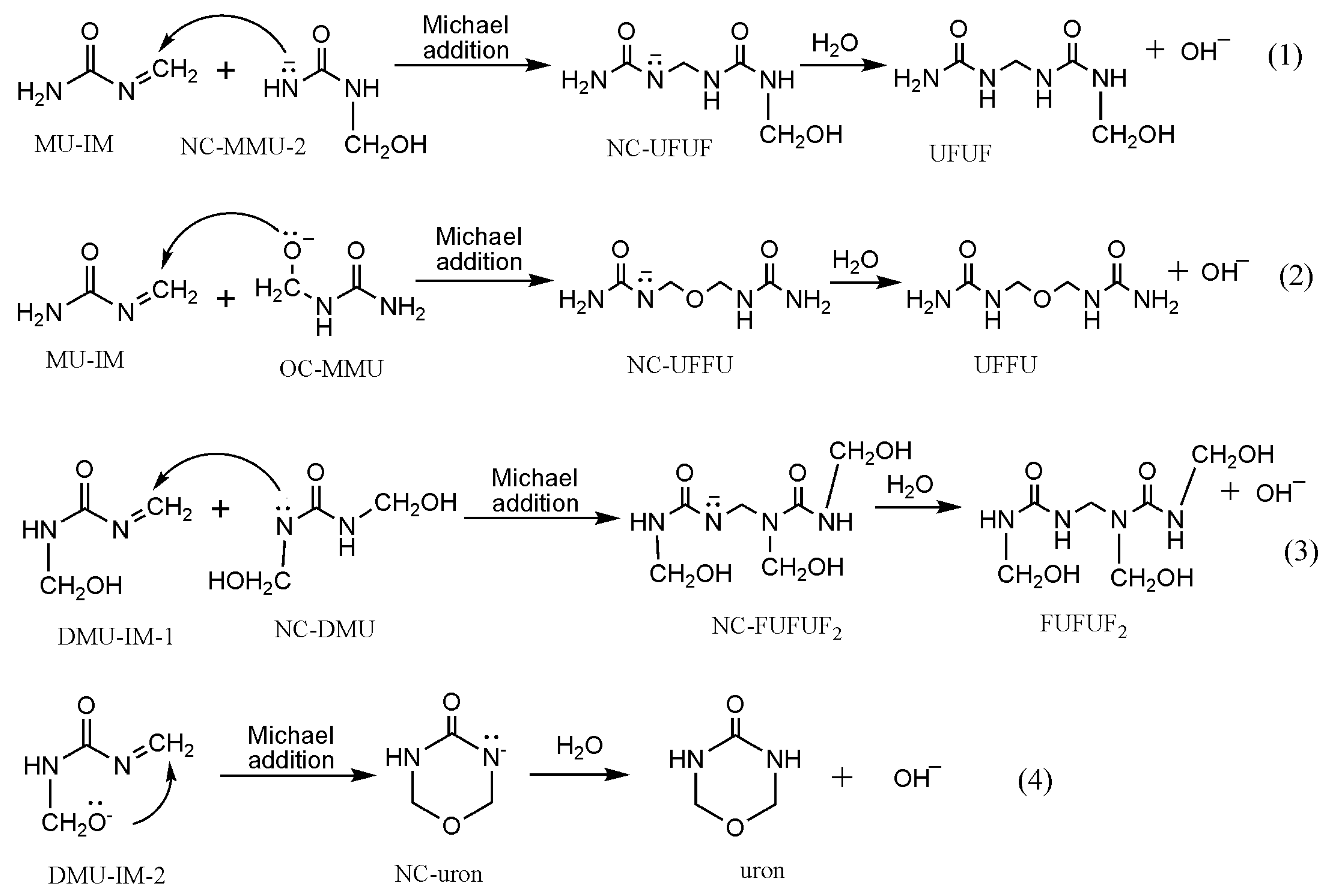
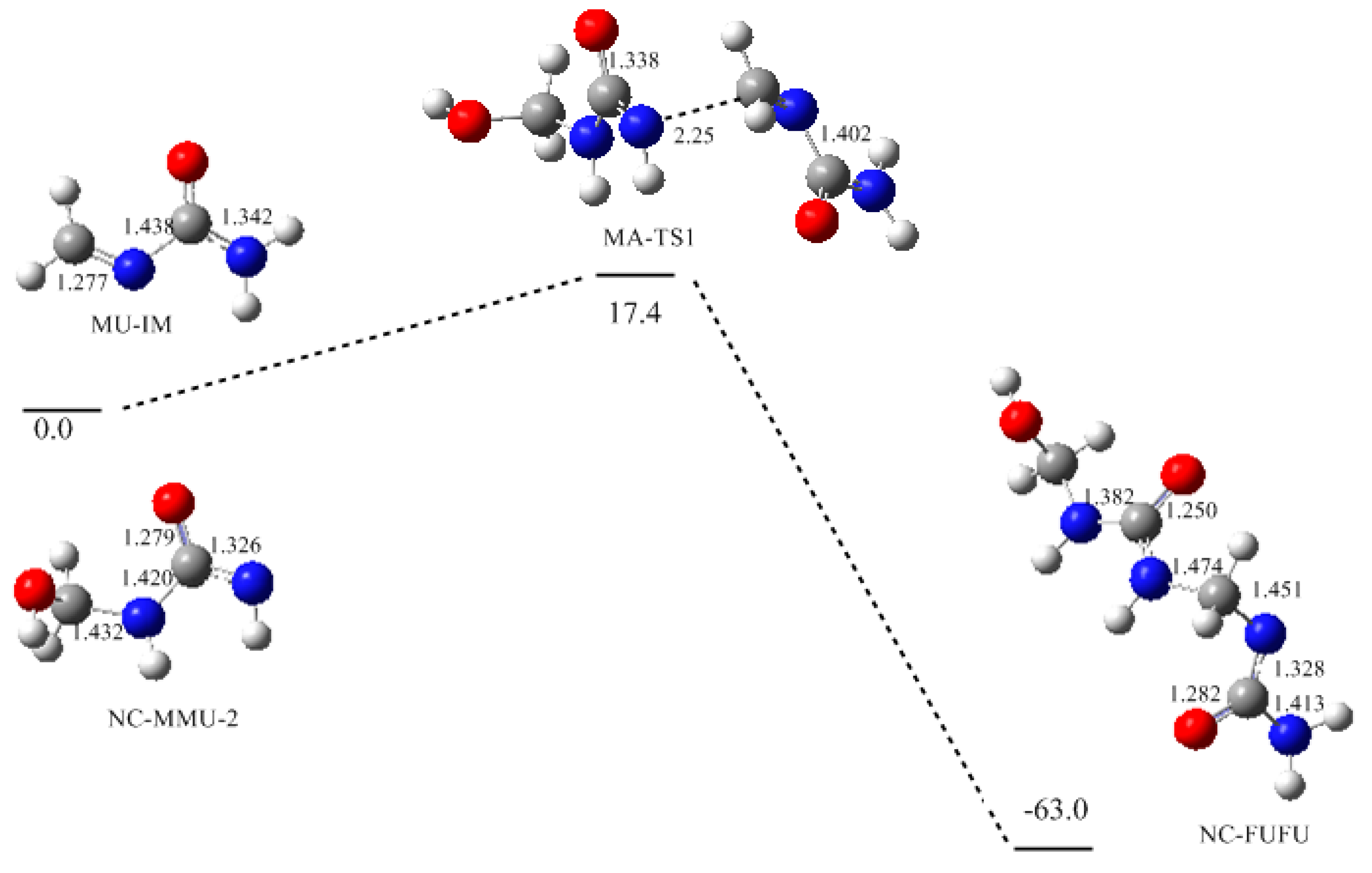
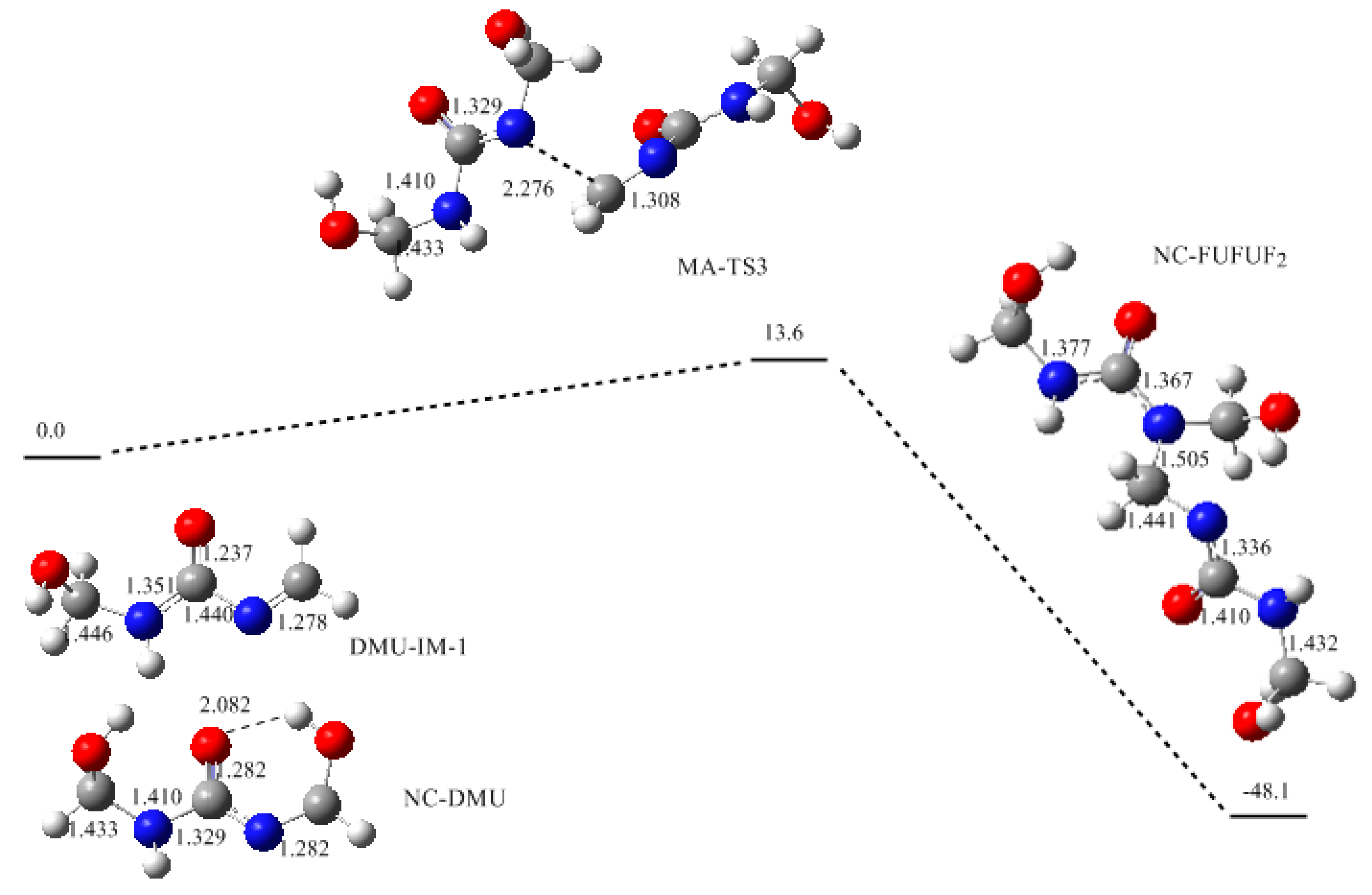
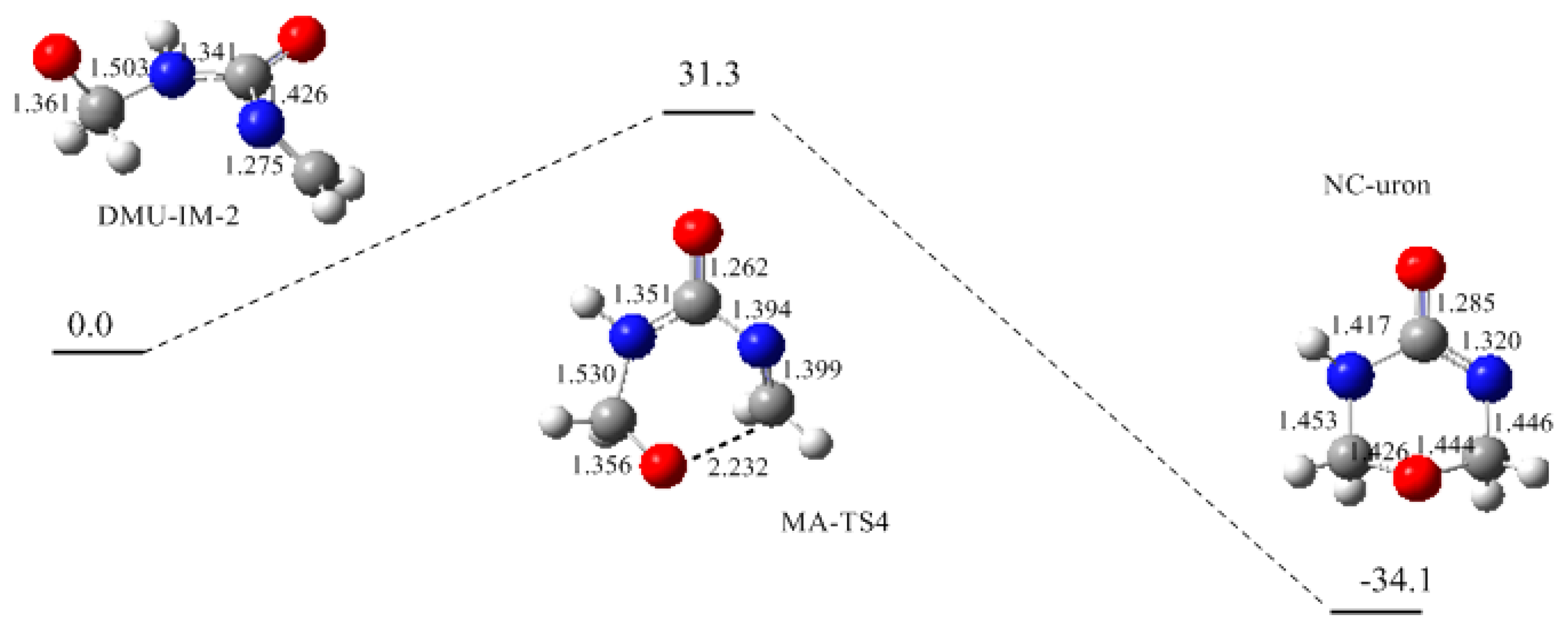
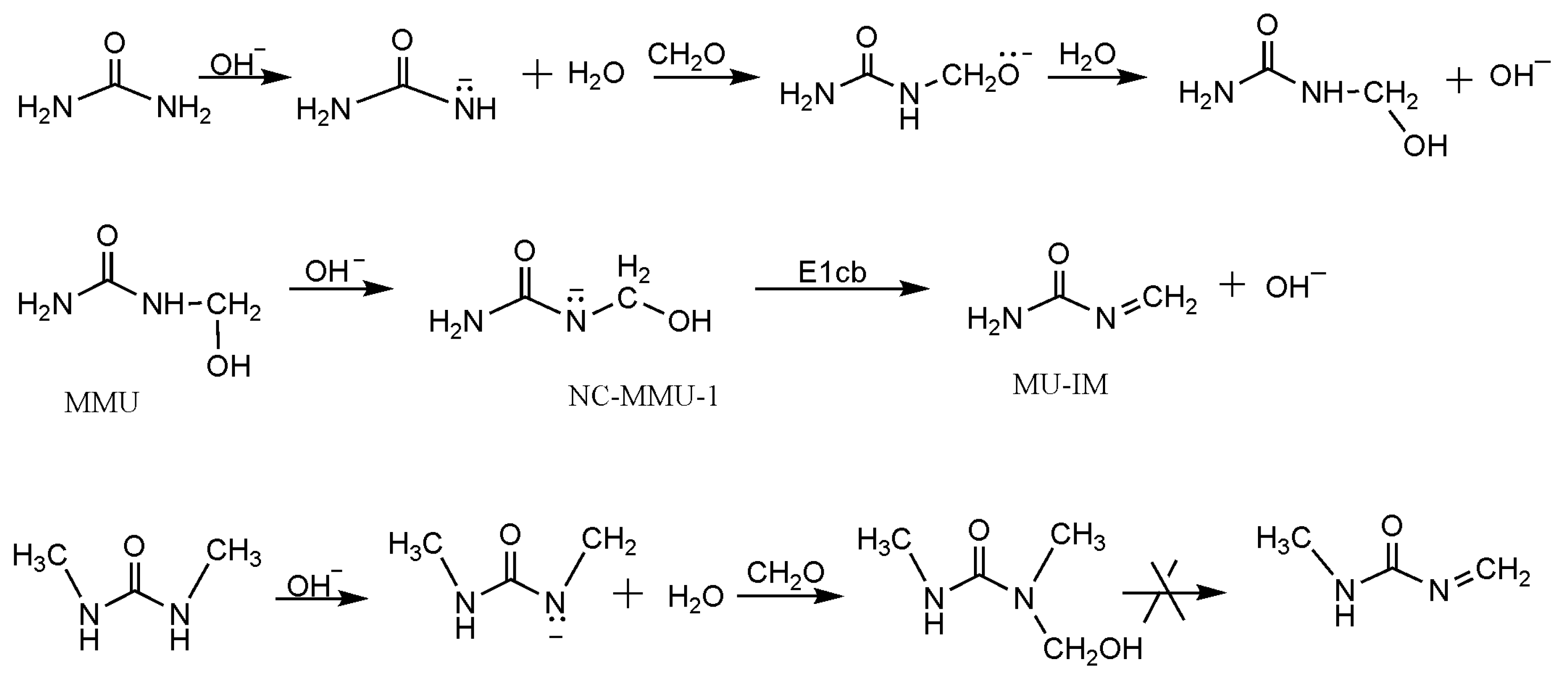
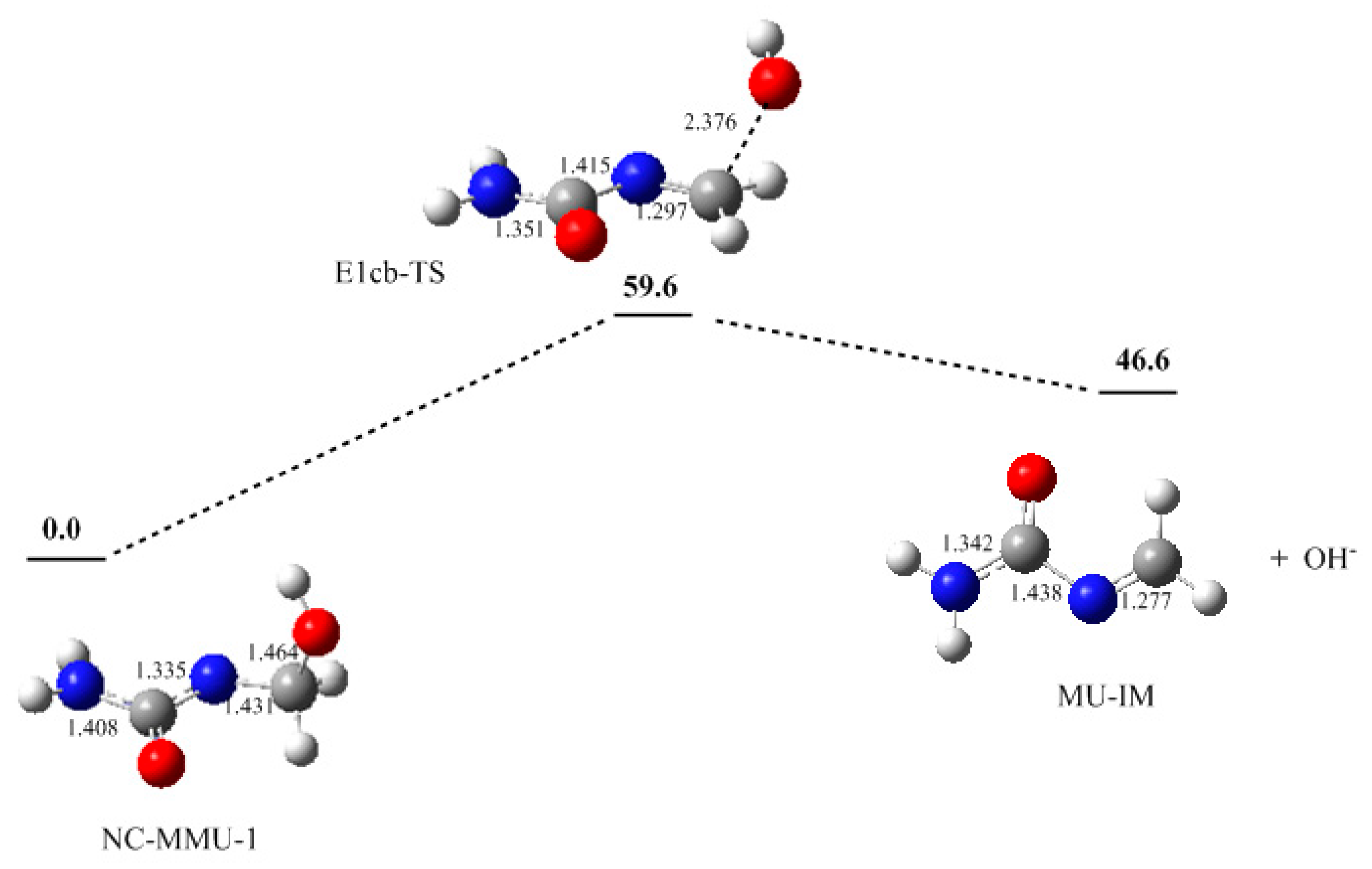
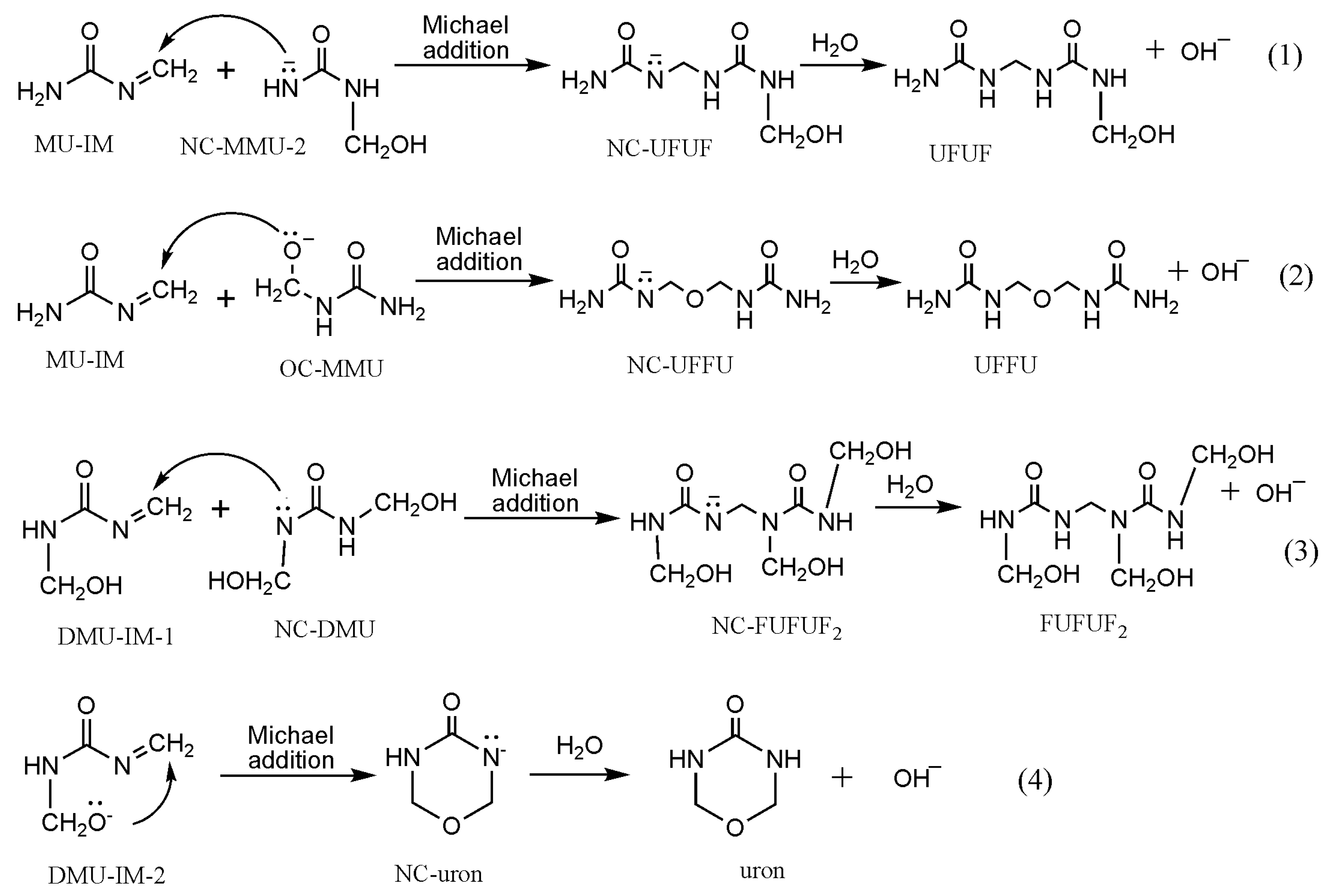
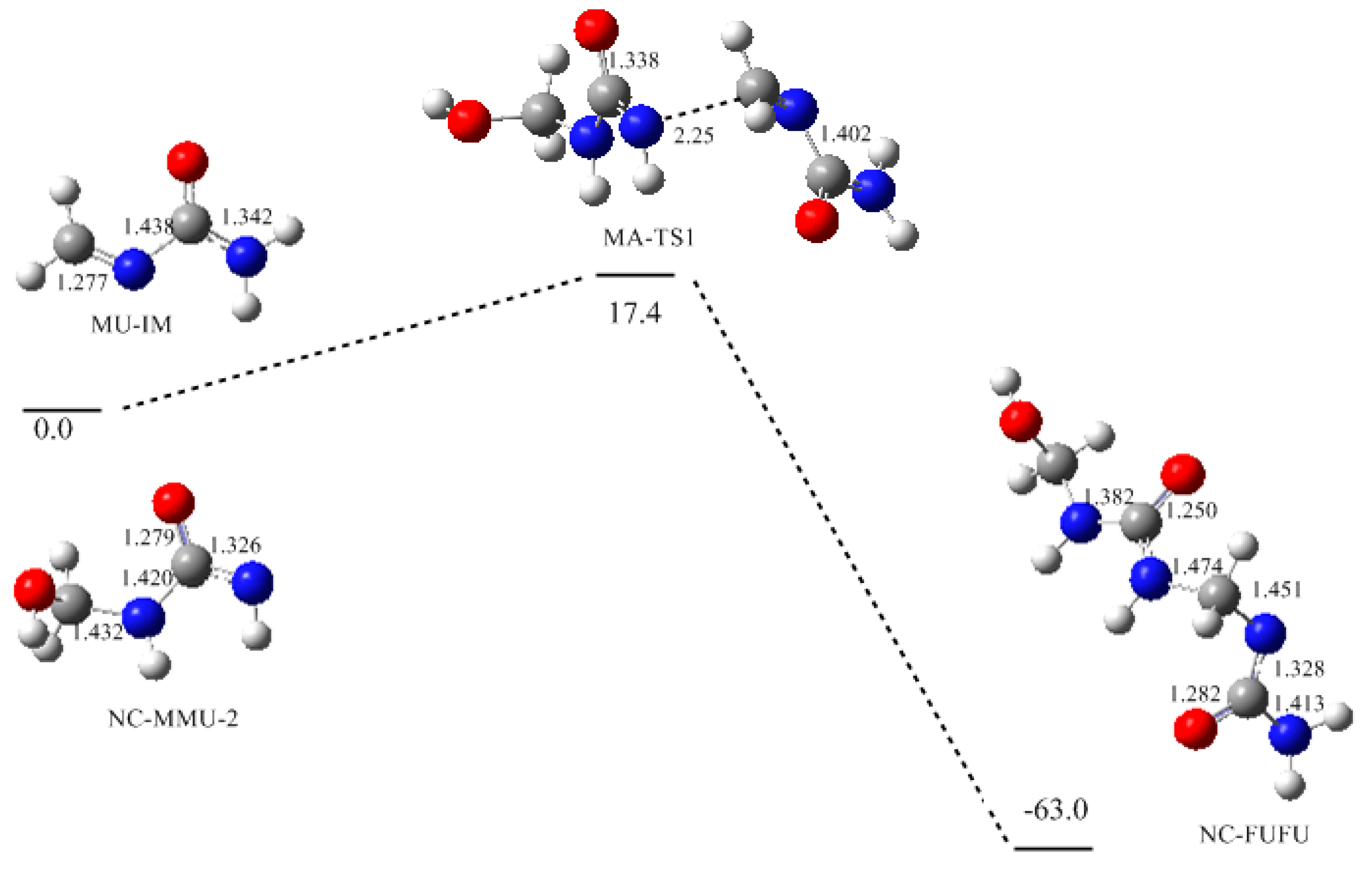
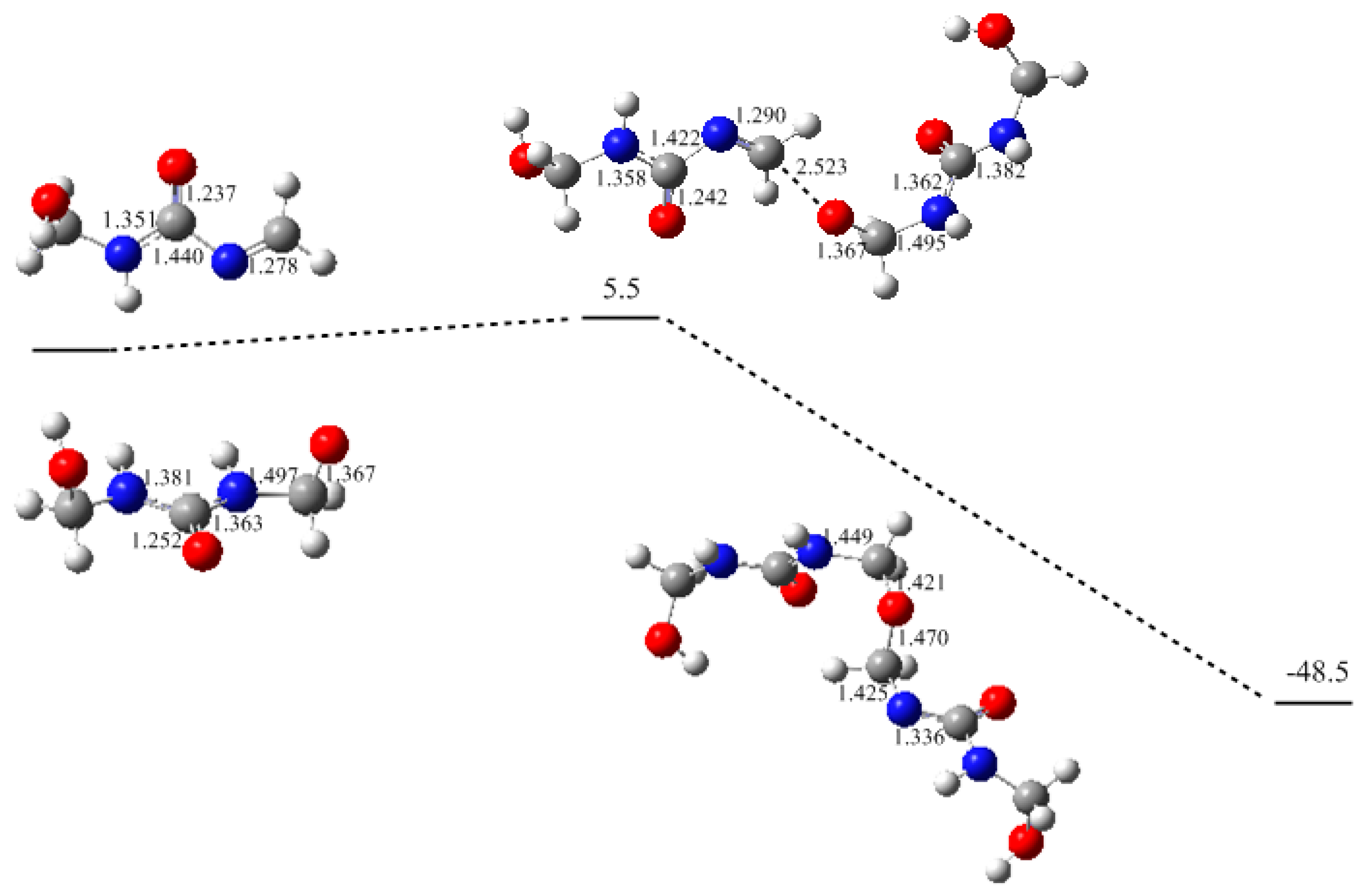
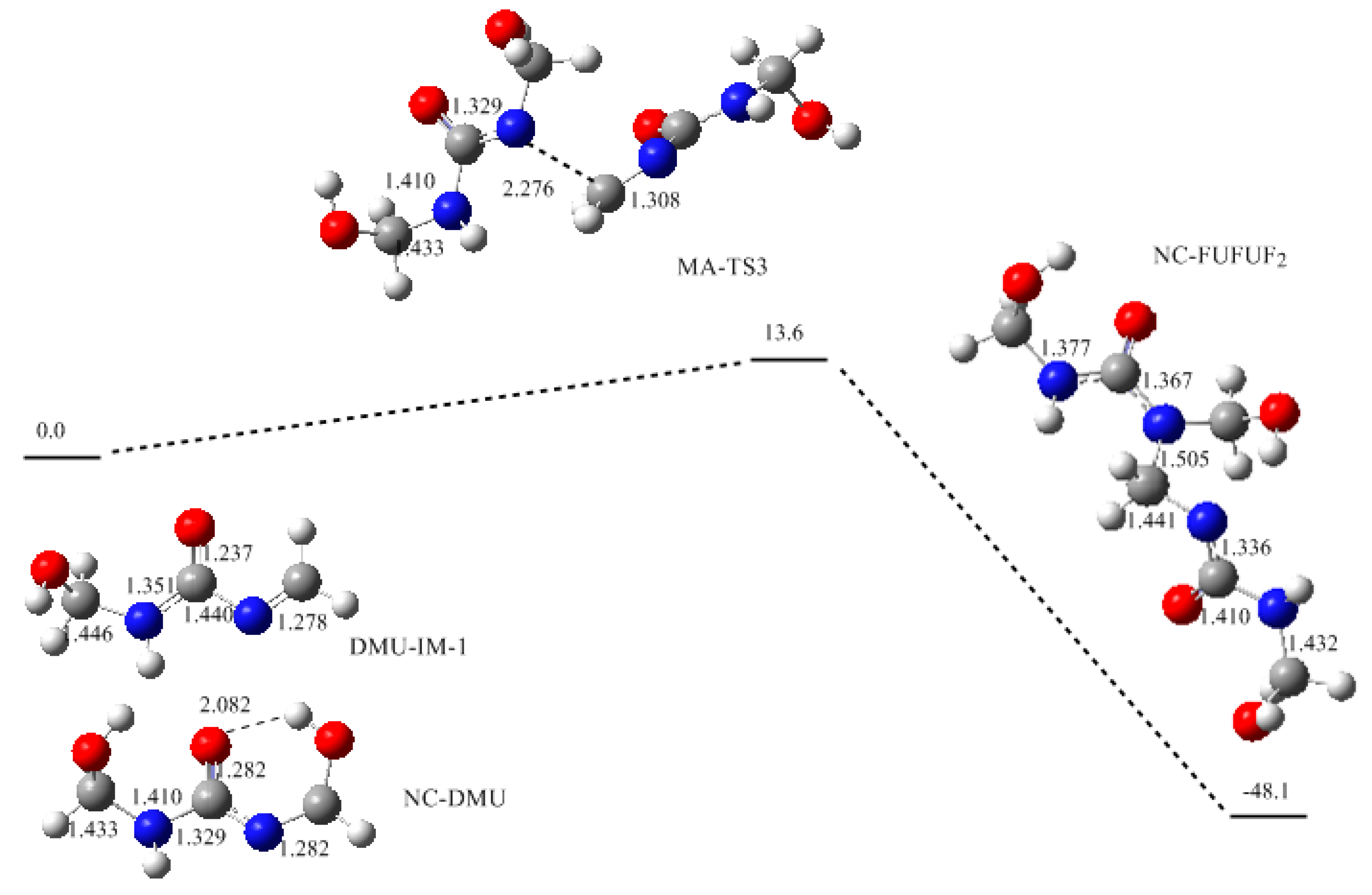
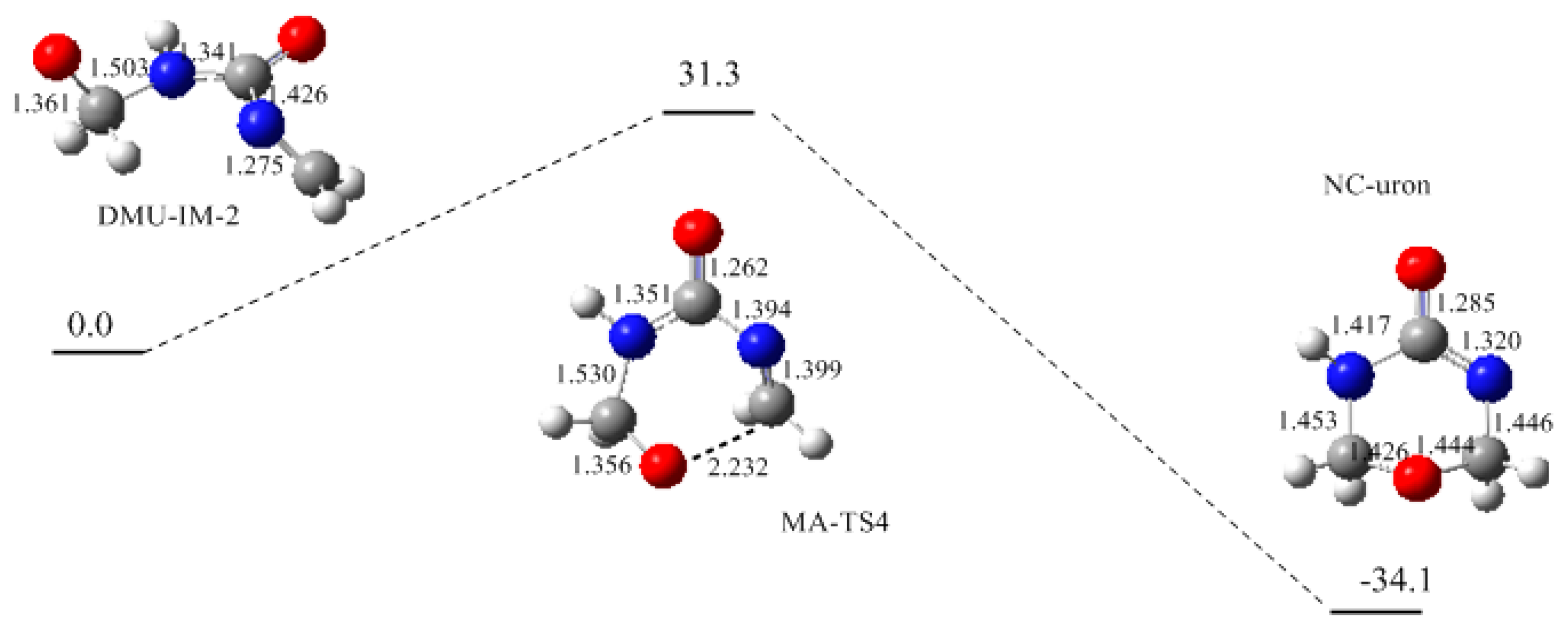
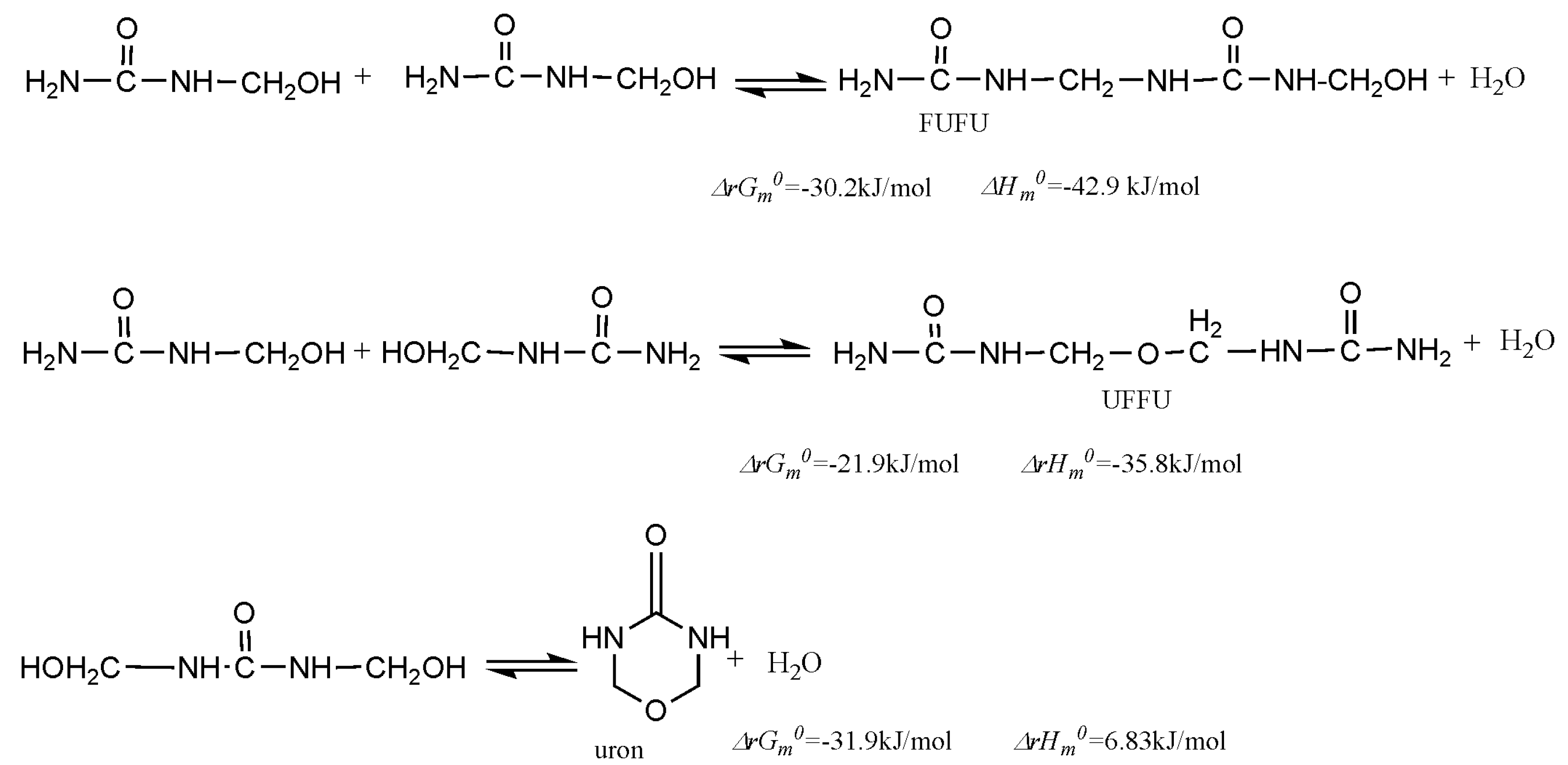
3. Results and Discussion
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dunky, M. Urea–formaldehyde (UF) adhesive resins for wood. Int. J. Adhes. Adhes. 1998, 18, 95–107. [Google Scholar] [CrossRef]
- Dunky, M. Adhesives based on formaldehyde condensation resins. Macromol. Symp. 2004, 217, 417–429. [Google Scholar] [CrossRef]
- Li, T.; Du, G.; Guo, X.; Liang, J.; Wang, H.; Xie, X. Competitive Formation of the Methylene and Methylene ether Bridges in the Urea–formaldehyde Reaction in Alkaline Solution: A Combined Experimental and Theoretical Study. Wood Sci. Technol. 2015, 49, 475–493. [Google Scholar] [CrossRef]
- Fan, D.; Chang, J.; Li, J.; Mao, A.; Zhang, L. 13C-NMR study on the structure of phenol-urea–formaldehyde resins prepared by methylolureas and phenol. J. Appl. Polym. Sci. 2009, 112, 2195–2202. [Google Scholar] [CrossRef]
- Du, G.; Lei, H.; Pizzi, A.; Pasch, H. Synthesis-structure-performance relationship of cocondensed phenol-urea–formaldehyde resins by MALDI-TOF and 13C-NMR. J. Appl. Polym. Sci. 2008, 110, 1182–1194. [Google Scholar] [CrossRef]
- Fan, D.; Li, J.; Chang, J.; Gou, J.; Jiang, J. Chemical structure and curing behavior of phenol-urea–formaldehyde cocondensed resins of high urea content. J. Adhes. Sci. Technol. 2009, 23, 1787–1797. [Google Scholar] [CrossRef]
- Tomita, B.; Ohyama, M.; Hse, C.Y.; Tomita, B.; Ohyama, M.; Hse, C.Y. Synthesis of phenol-urea–formaldehyde cocondehsed resins from UF-concentrate and phenol. Holzforschung 1994, 48, 522–526. [Google Scholar] [CrossRef]
- Tohmura, S.; Inoue, A.; Sahari, S.H. Influence of the melamine content in melamine-urea–formaldehyde resins on formaldehyde emission and cured resin structure. J. Wood Sci. 2001, 47, 451–457. [Google Scholar] [CrossRef]
- No, B.Y.; Kim, M.G. Curing of low level melamine-modified urea–formaldehyde particleboard binder resins studied with dynamic mechanical analysis (DMA). J. Appl. Polym. Sci. 2005, 97, 377–389. [Google Scholar]
- Kandelbauer, A.; Despres, A.; Pizzi, A.; Taudes, I. Testing by fourier transform infrared species variation during melamine–urea–formaldehyde resin preparation. J. Appl. Polym. Sci. 2007, 106, 2192–2197. [Google Scholar] [CrossRef]
- Despres, A.; Pizzi, A.; Pasch, H.; Kandelbauer, A. Comparative 13C-NMR and matrix-assisted laser desorption/ionization time-of-flight analyses of species variation and structure maintenance during melamine–urea–formaldehyde resin preparation. J. Appl. Polym. Sci. 2007, 106, 1106–1128. [Google Scholar] [CrossRef]
- Li, T.; Cao, M.; Liang, J.; Xie, X.; Du, G. Theoretical Confirmation of the Quinone Methide Hypothesis for the Condensation Reactions in Phenol-Formaldehyde Resin Synthesis. Polymers 2017, 9, 45. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Miehlich, B.; Savin, A.; Stoll, H. Results obtained with the correlation-energy density functionals of Becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-consistent molecular orbital methods. 9. Extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 724. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-consistent molecular orbital methods. 12. Further extensions of Gaussian-type basis sets for use in molecular-orbital studies of organic-molecules. J. Chem. Phys. 1972, 56, 2257. [Google Scholar]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W. Efficient diffuse function-augmented basis-sets for anion calculations. III. The 3–21+G basis set for 1st-row elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods. 25. Supplementary functions for Gaussian Basis Sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Miertus, S.; Tomasi, J. Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes. Chem. Phys. 1982, 65, 239–245. [Google Scholar] [CrossRef]
- Miertus, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilization of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Mennucci, B. Ab initio study of ionic solutions by a polarizable continuum dielectric model. Chem. Phys. Lett. 1998, 286, 253–260. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B. Gaussian 2003, Revision B.03; Gaussian, Inc.: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Steinhof, O.; Scherrb, G.; Hasse, H. Investigation of the reaction of 1,3-dimethylurea with formaldehyde by quantitative on-line NMR spectroscopy: A model for the urea–formaldehyde system. Magn. Reson. Chem. 2016, 54, 457–476. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Li, T.; Liang, J.; Du, G. The influence of pH on the melamine-dimethylurea–formaldehyde co-condensations: A quantitative 13C-NMR study. Polymers 2017, 9, 109. [Google Scholar] [CrossRef]
- Little, R.D.; Masjedizadeh, M.R.; Wallquist, O.; McLoughlin, J.I. The Intramolecular Michael Reaction. Org. React. 1995, 47, 315–552. [Google Scholar]
- Mather, B.; Viswanathan, K.; Miller, K.; Long, T. Michael addition reactions in macromolecular design for emerging technologies. Prog. Polym. Sci. 2006, 31, 487–531. [Google Scholar] [CrossRef]
- Li, T.; Liang, J.; Cao, M.; Guo, X.; Xie, X.; Du, G. Re-elucidation of the acid-catalyzed urea–formaldehyde reactions: A theoretical and 13C-NMR study. J. Appl. Polym. Sci. 2016, 133, 44339–44356. [Google Scholar] [CrossRef]
- Liang, J.; Li, T.; Cao, M.; Du, G. Urea–formaldehyde Resin Structure Formation under Alkaline Condition: A Quantitative 13C-NMR Study. J. Adhes. Sci. Technol. 2016, in press. [Google Scholar]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, T.; Cao, M.; Liang, J.; Xie, X.; Du, G. New Mechanism Proposed for the Base-Catalyzed Urea–Formaldehyde Condensation Reactions: A Theoretical Study. Polymers 2017, 9, 203. https://doi.org/10.3390/polym9060203
Li T, Cao M, Liang J, Xie X, Du G. New Mechanism Proposed for the Base-Catalyzed Urea–Formaldehyde Condensation Reactions: A Theoretical Study. Polymers. 2017; 9(6):203. https://doi.org/10.3390/polym9060203
Chicago/Turabian StyleLi, Taohong, Ming Cao, Jiankun Liang, Xiaoguang Xie, and Guanben Du. 2017. "New Mechanism Proposed for the Base-Catalyzed Urea–Formaldehyde Condensation Reactions: A Theoretical Study" Polymers 9, no. 6: 203. https://doi.org/10.3390/polym9060203
APA StyleLi, T., Cao, M., Liang, J., Xie, X., & Du, G. (2017). New Mechanism Proposed for the Base-Catalyzed Urea–Formaldehyde Condensation Reactions: A Theoretical Study. Polymers, 9(6), 203. https://doi.org/10.3390/polym9060203