
Synthesis of PNVP-Based Copolymers with Tunable Thermosensitivity by Sequential Reversible Addition–Fragmentation Chain Transfer Copolymerization and Ring-Opening Polymerization



Abstract
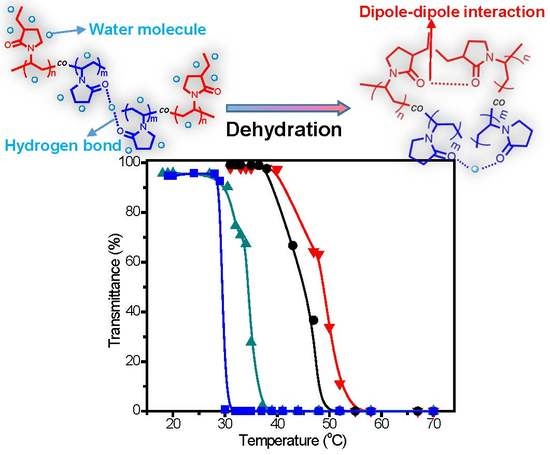
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of 2-Hydroxyethyl 2-Bromopropanoate (HEB)
2.3. Synthesis of S-(1-Methyl-4-hydroxyethyl Acetate) O-Ethyl Xanthate (MHEX)
2.4. Synthesis of 3-Ethyl-1-vinyl-2-pyrrolidone (C2NVP)
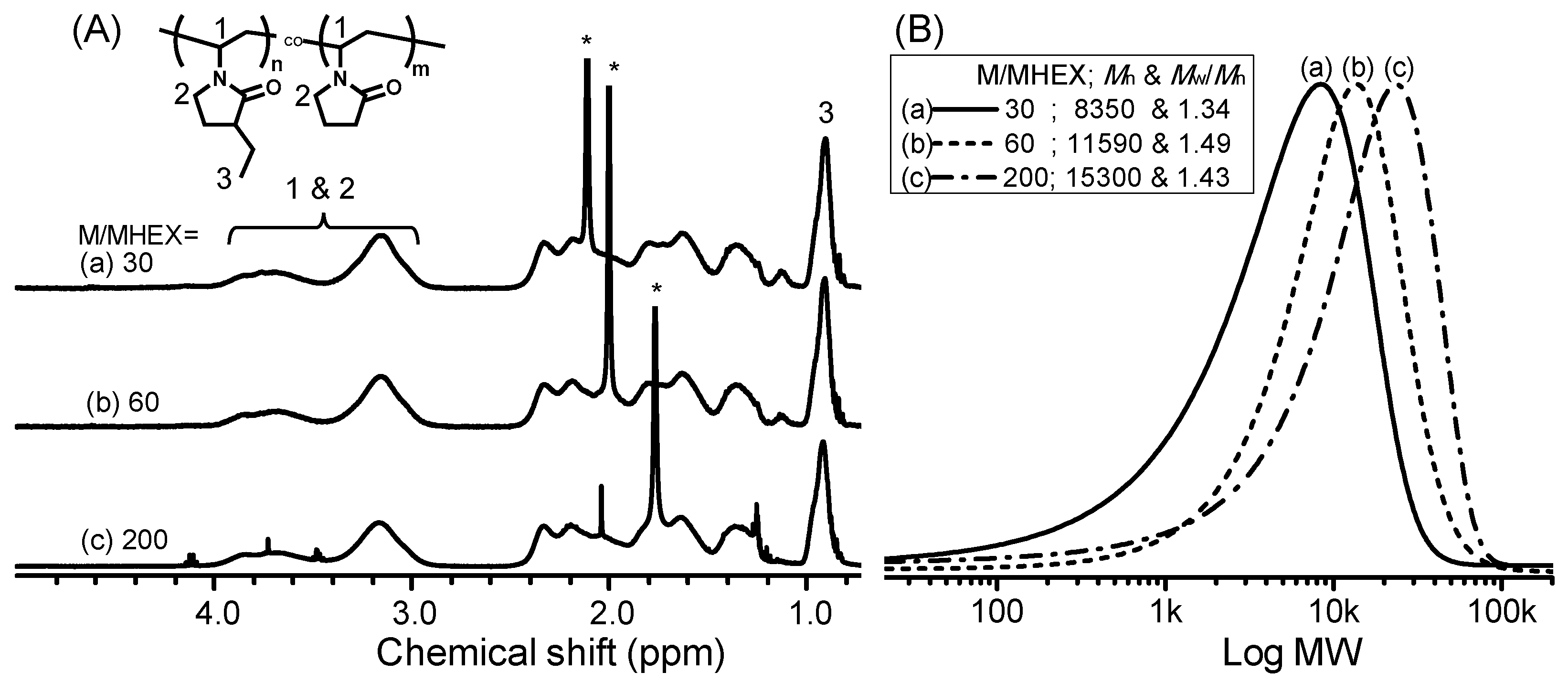
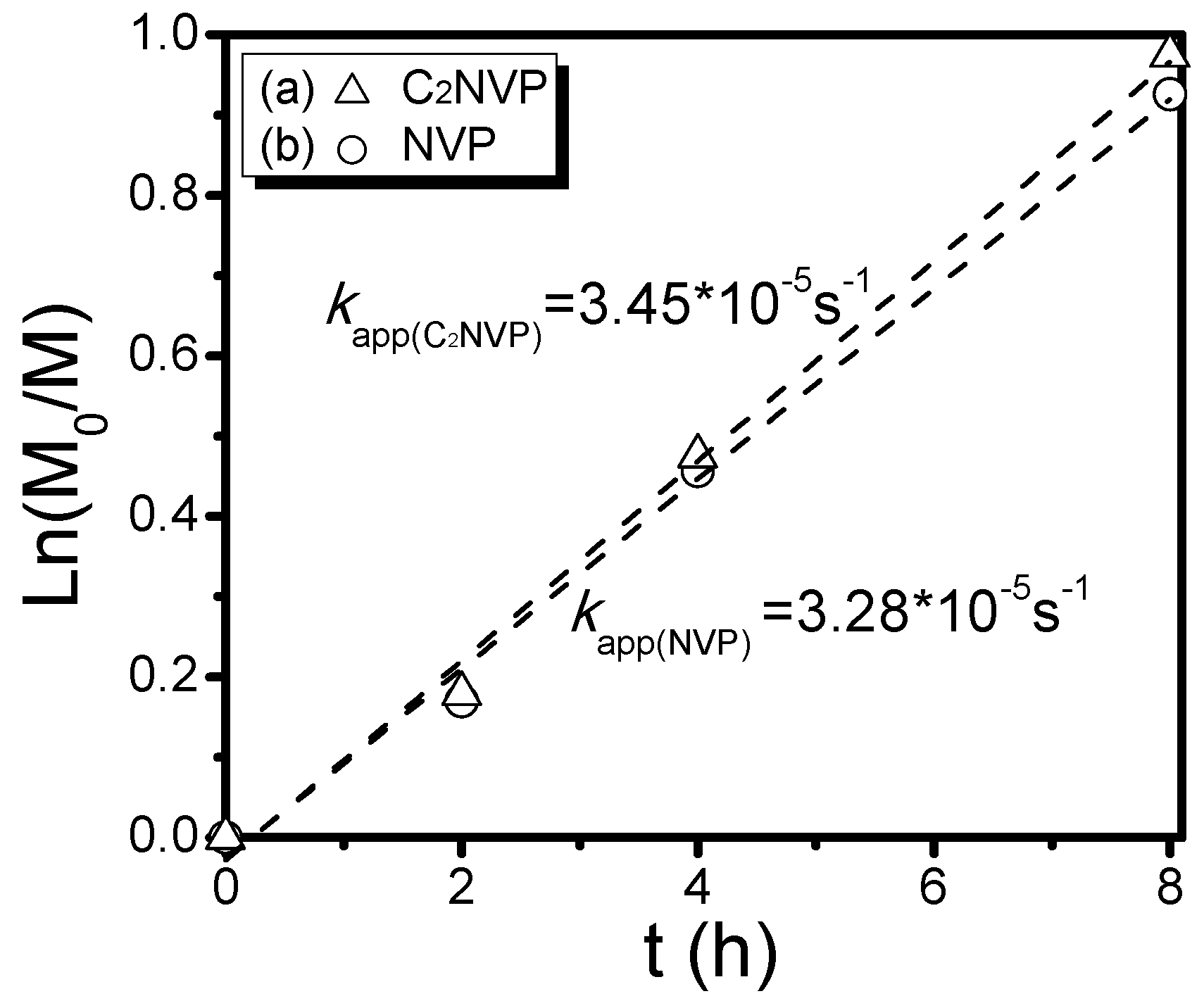
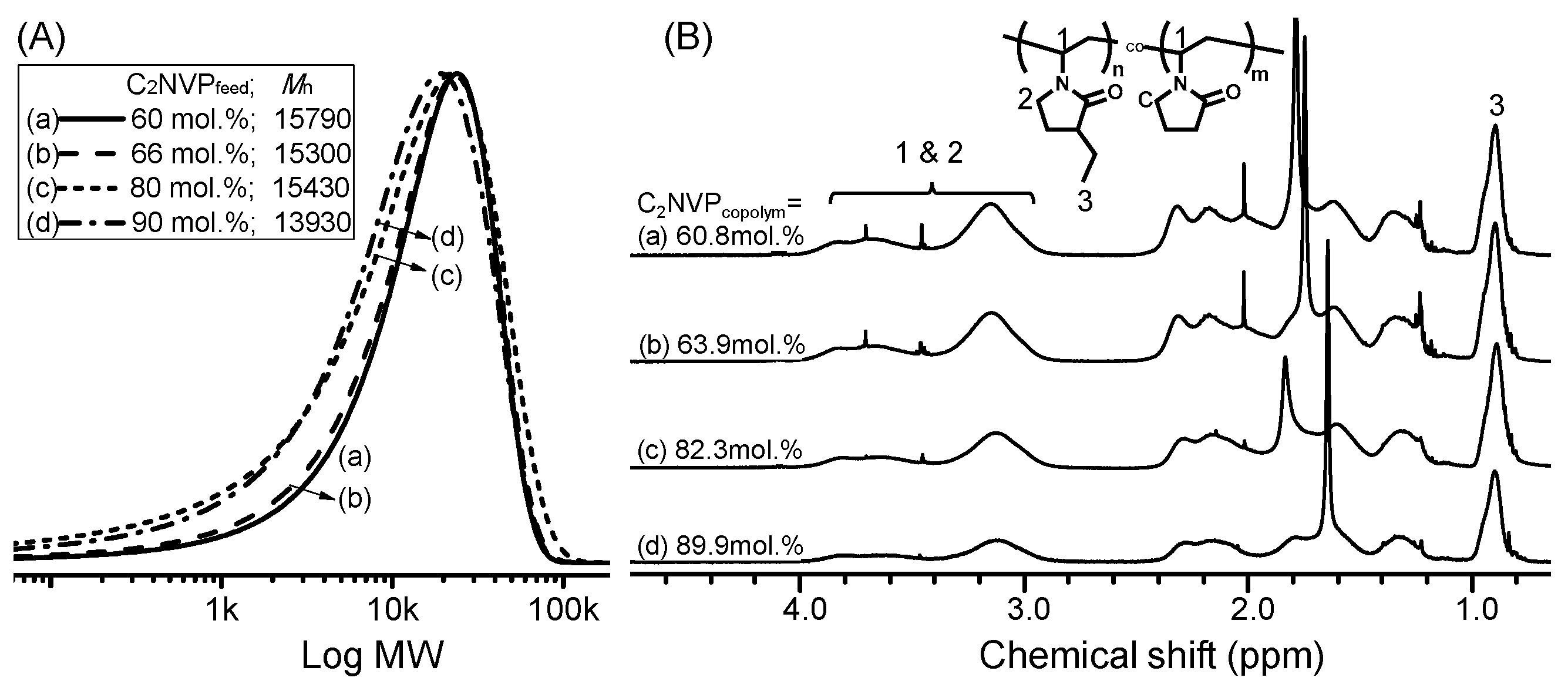
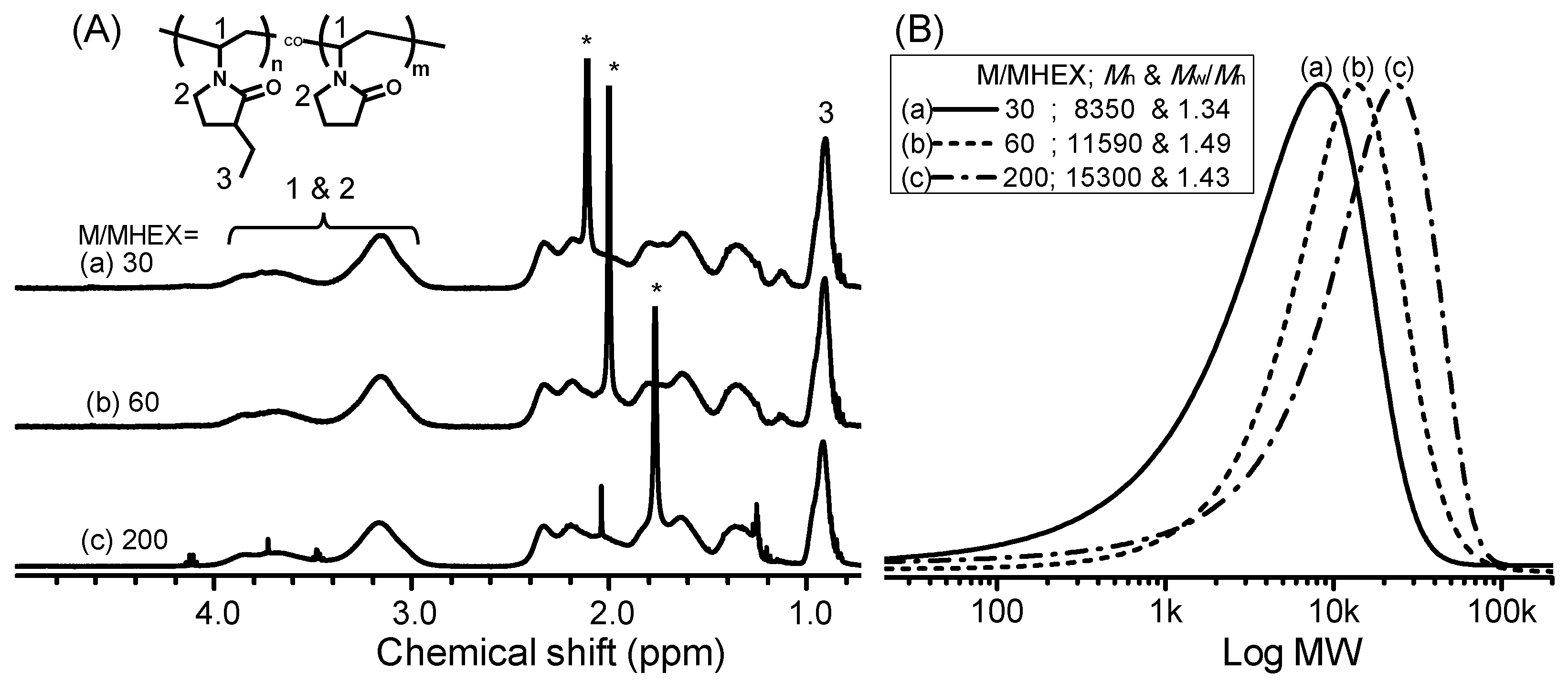
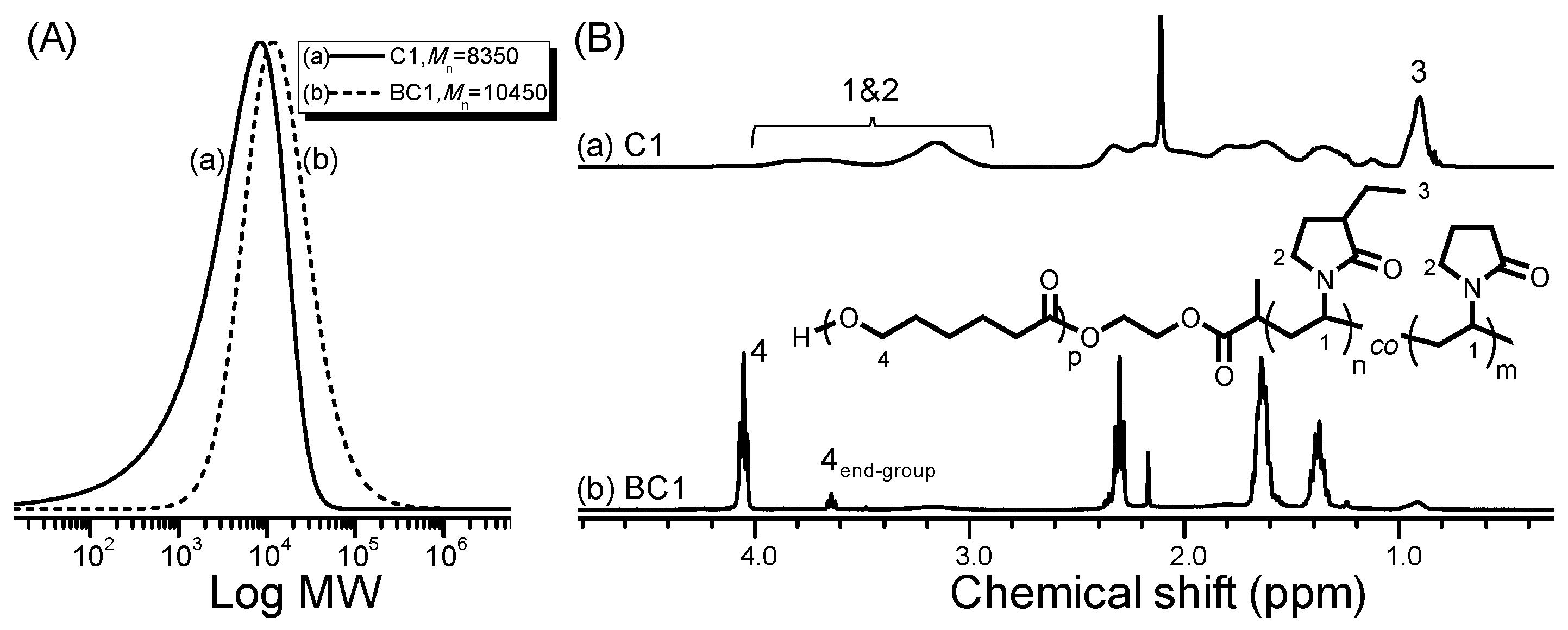
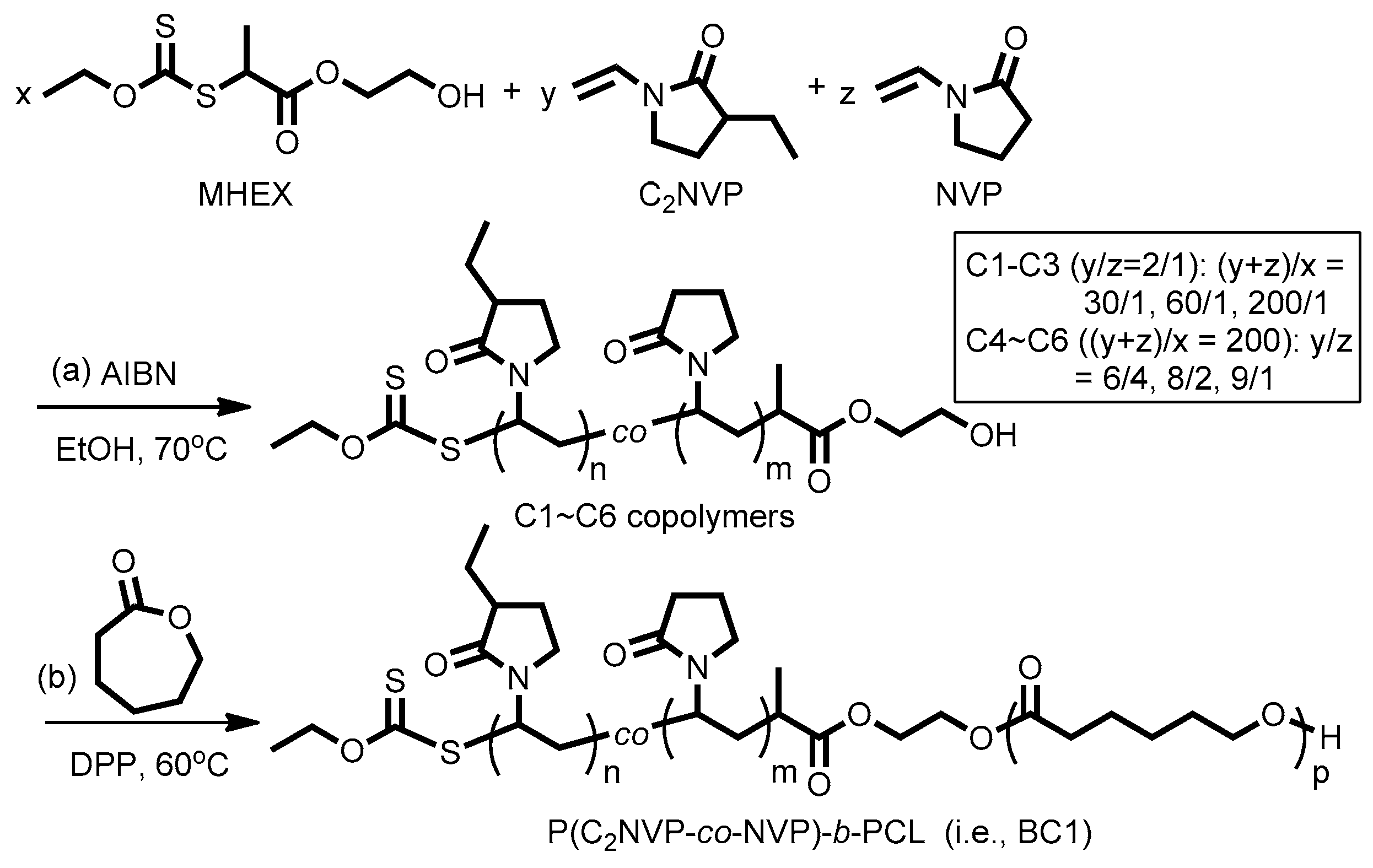
2.5. Synthesis of Poly[(3-ethyl-1-vinyl-2-pyrrolidone)-co-(N-vinyl-2-pyrrolidone)] by RAFT Copolymerization (i.e., Copolymers C1–C6)
2.6. Chain Extension of P(C2NVP-co-NVP)-OH with CL by ROP (i.e., BC1)
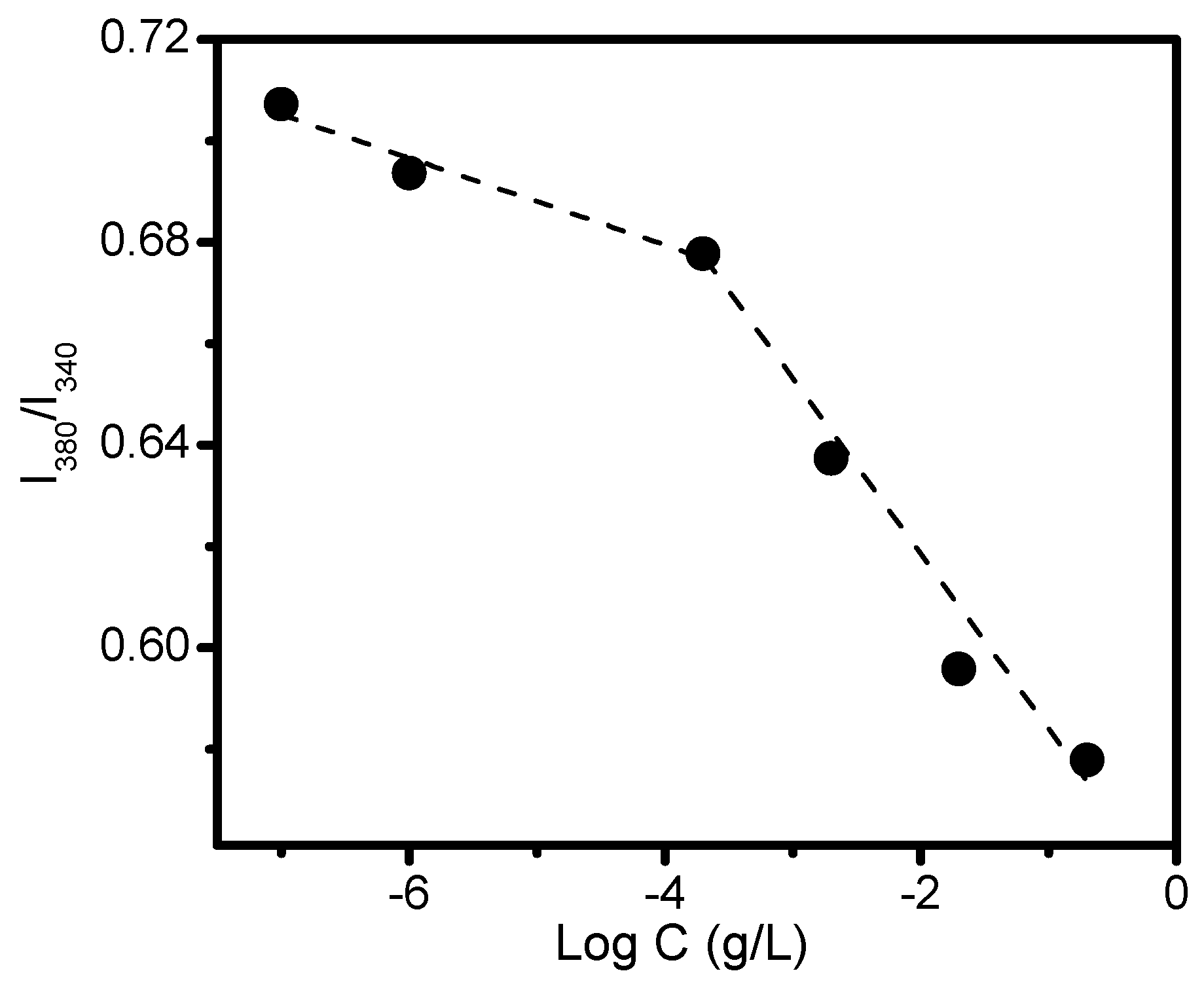
2.7. Characterization
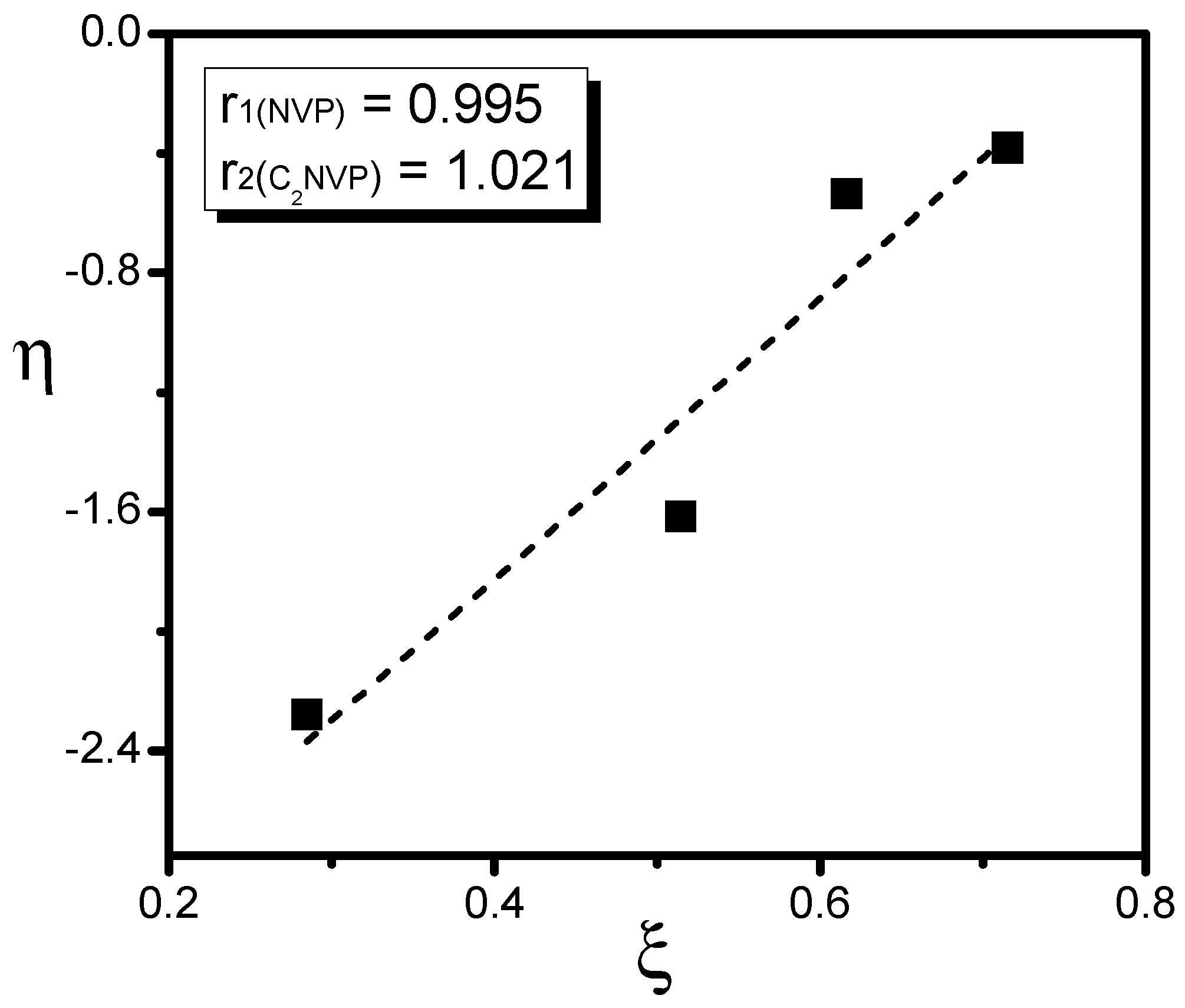
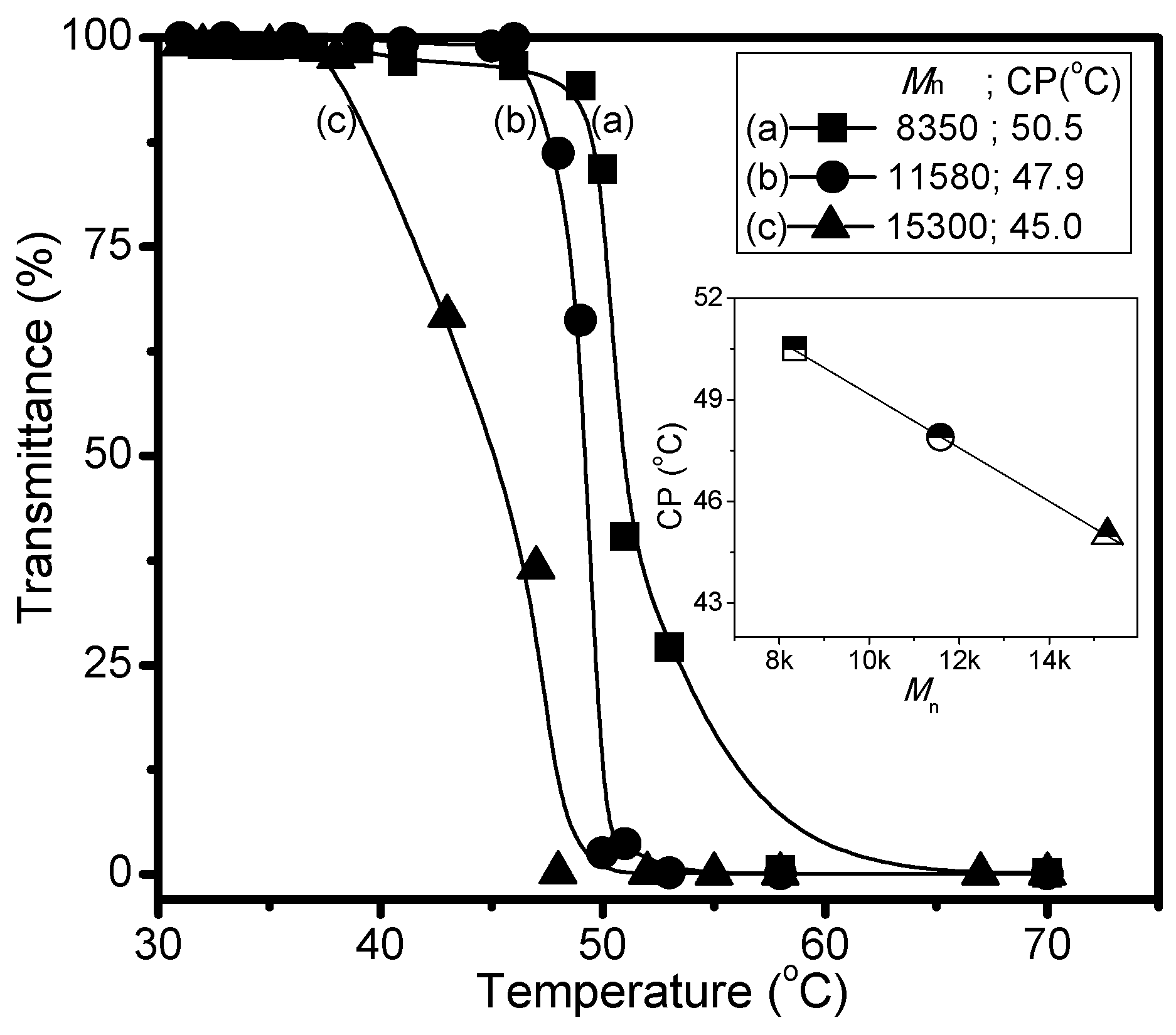
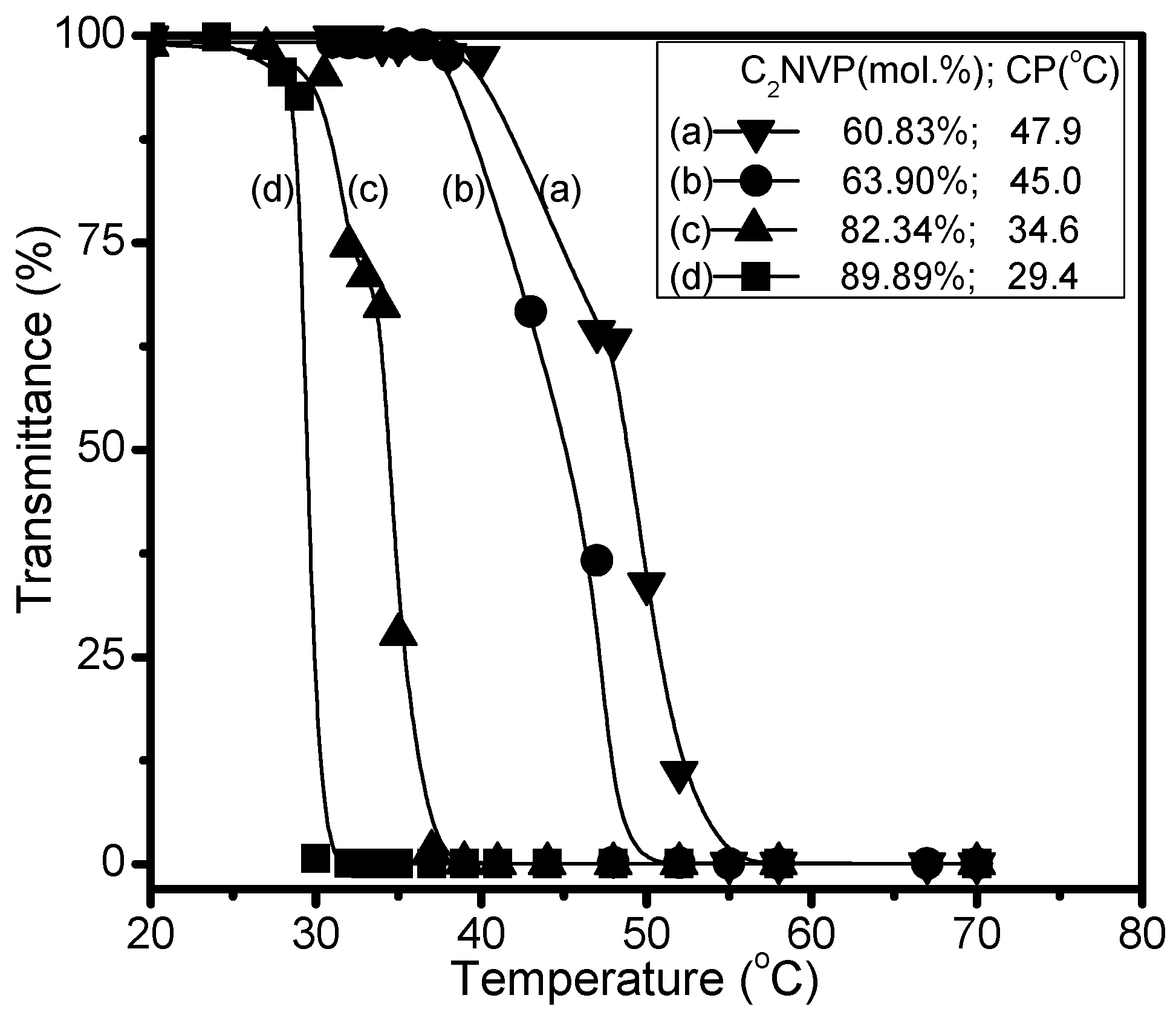
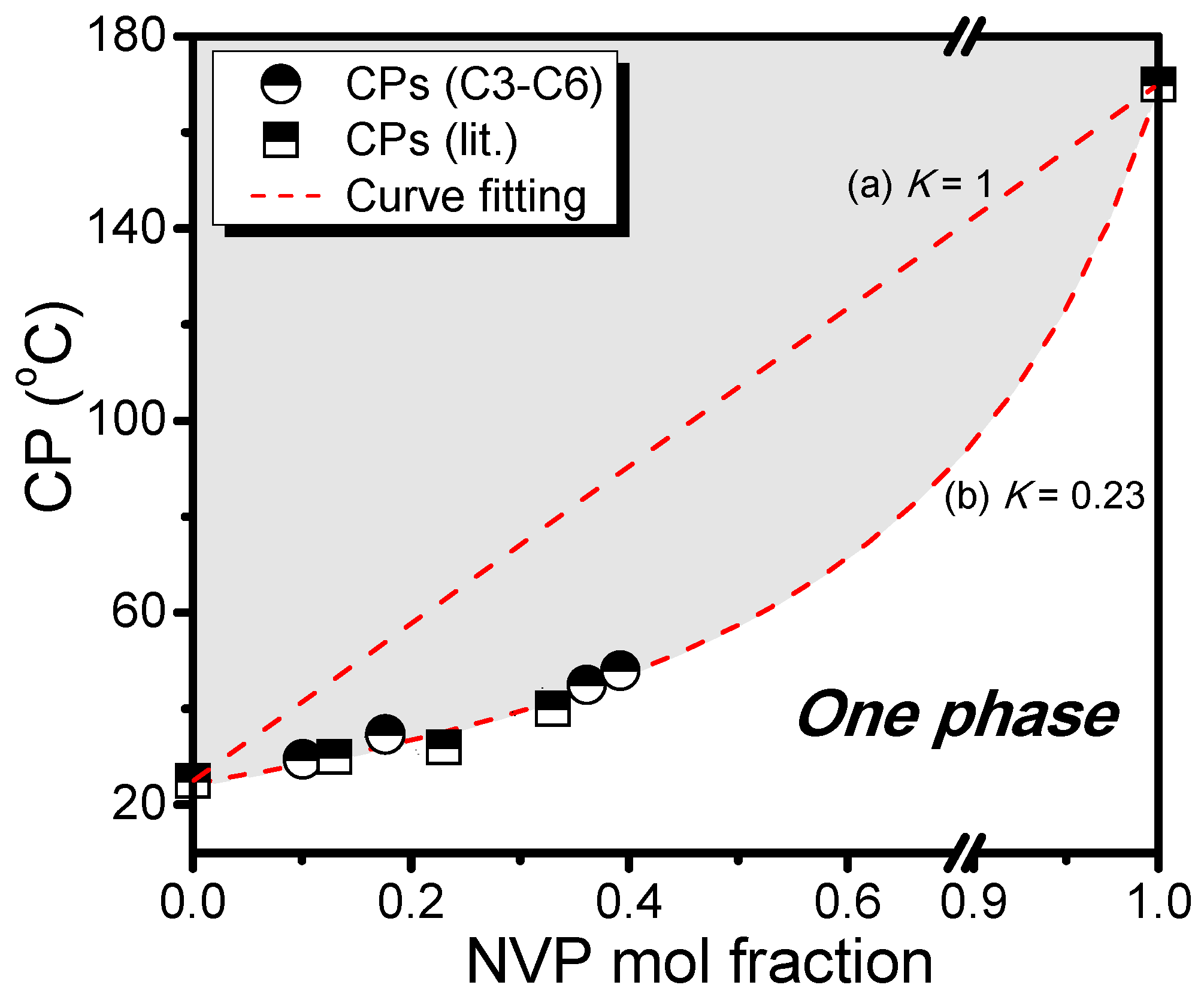
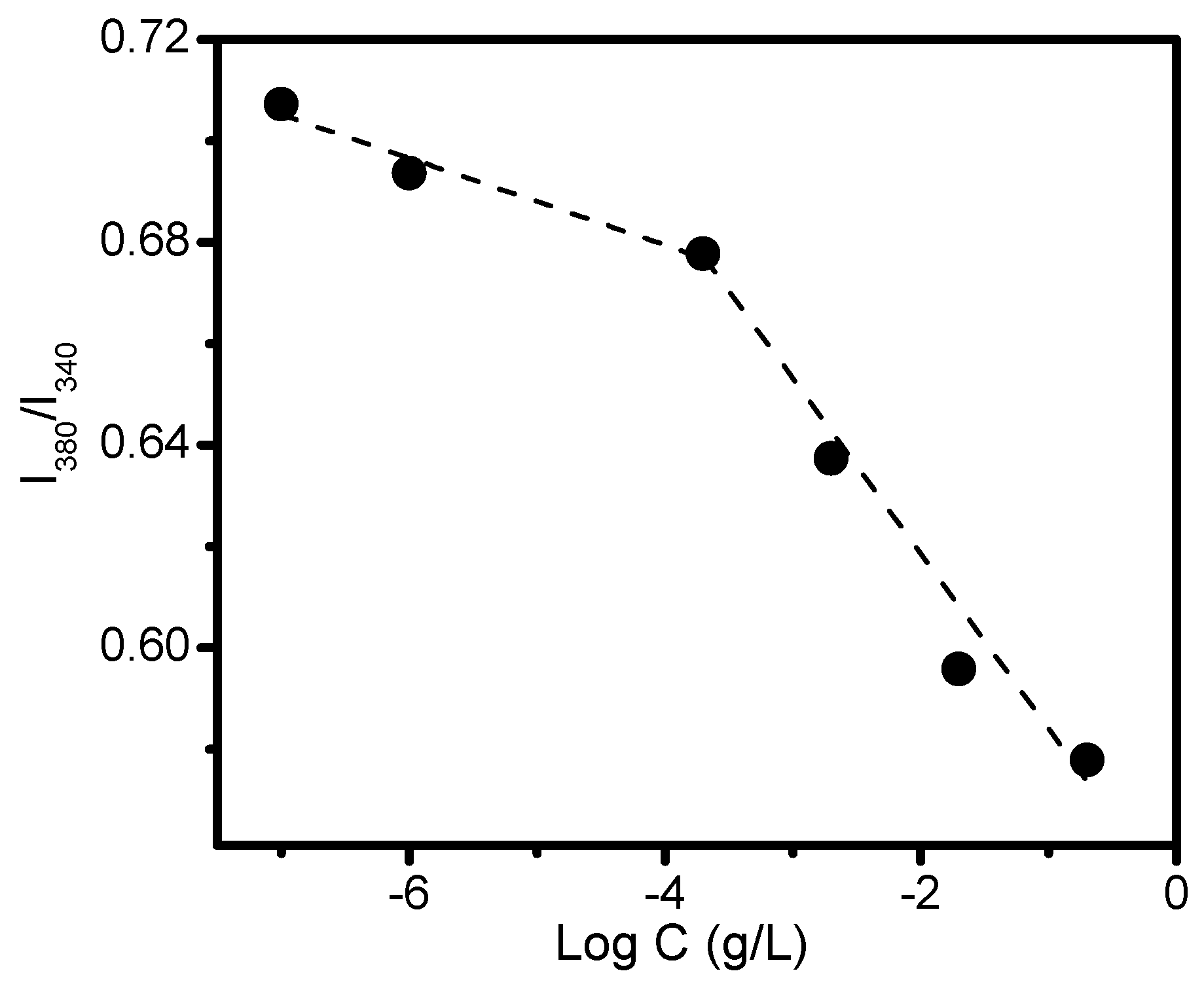
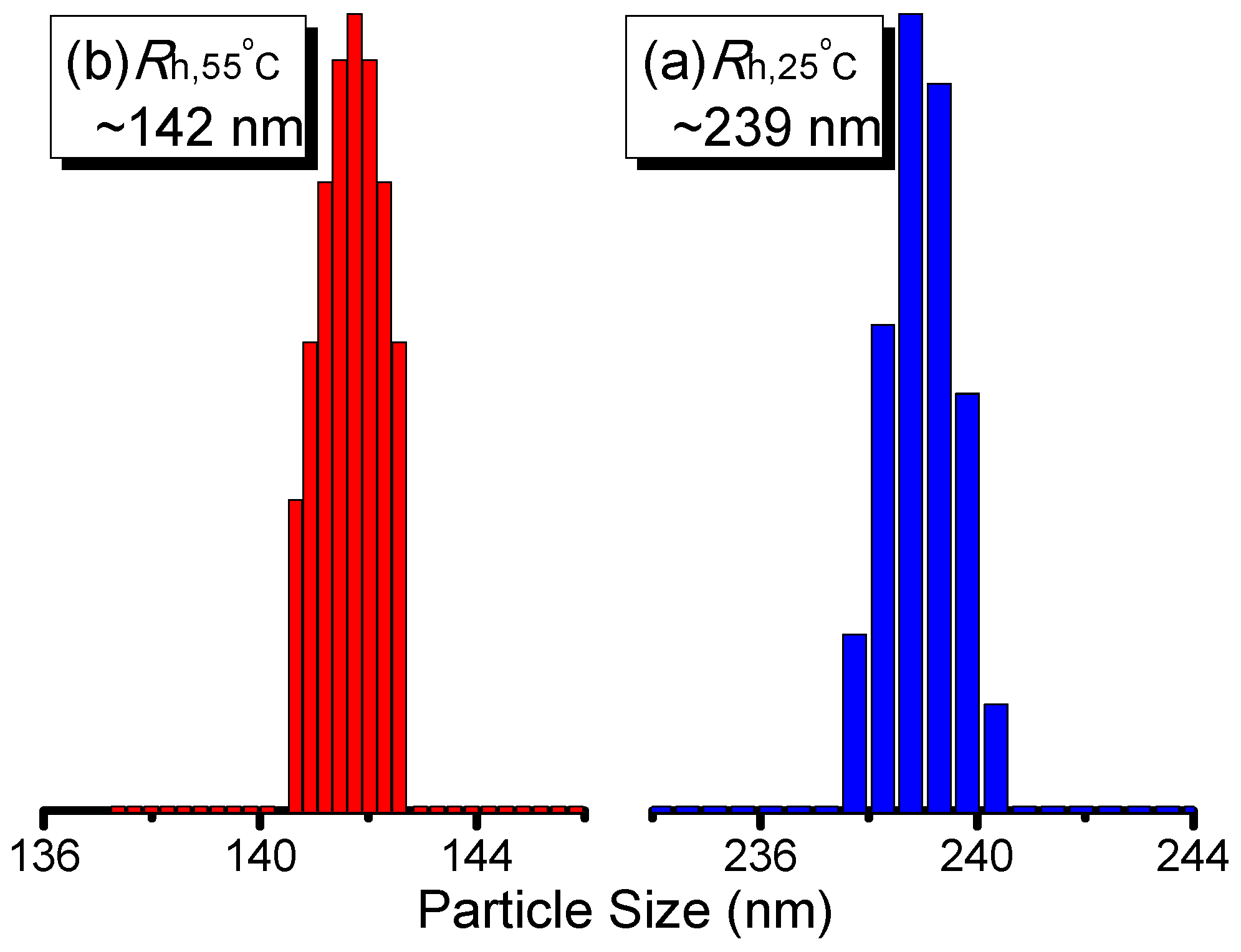
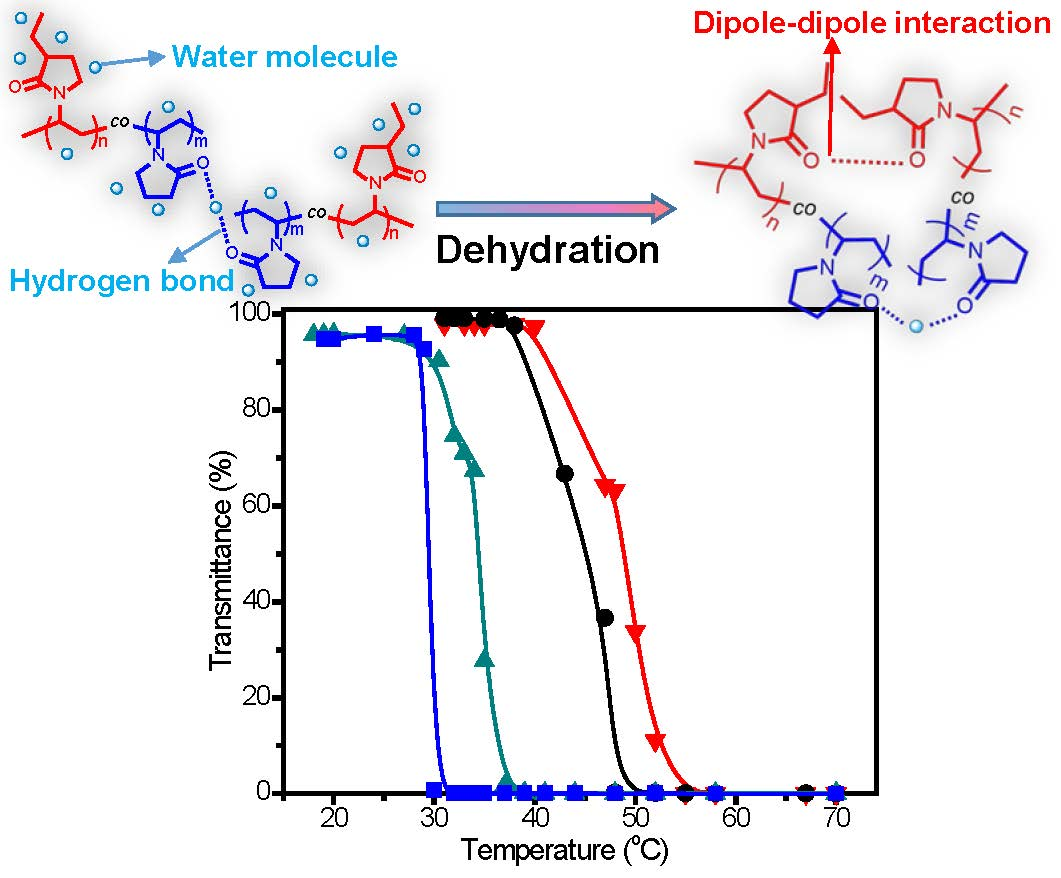
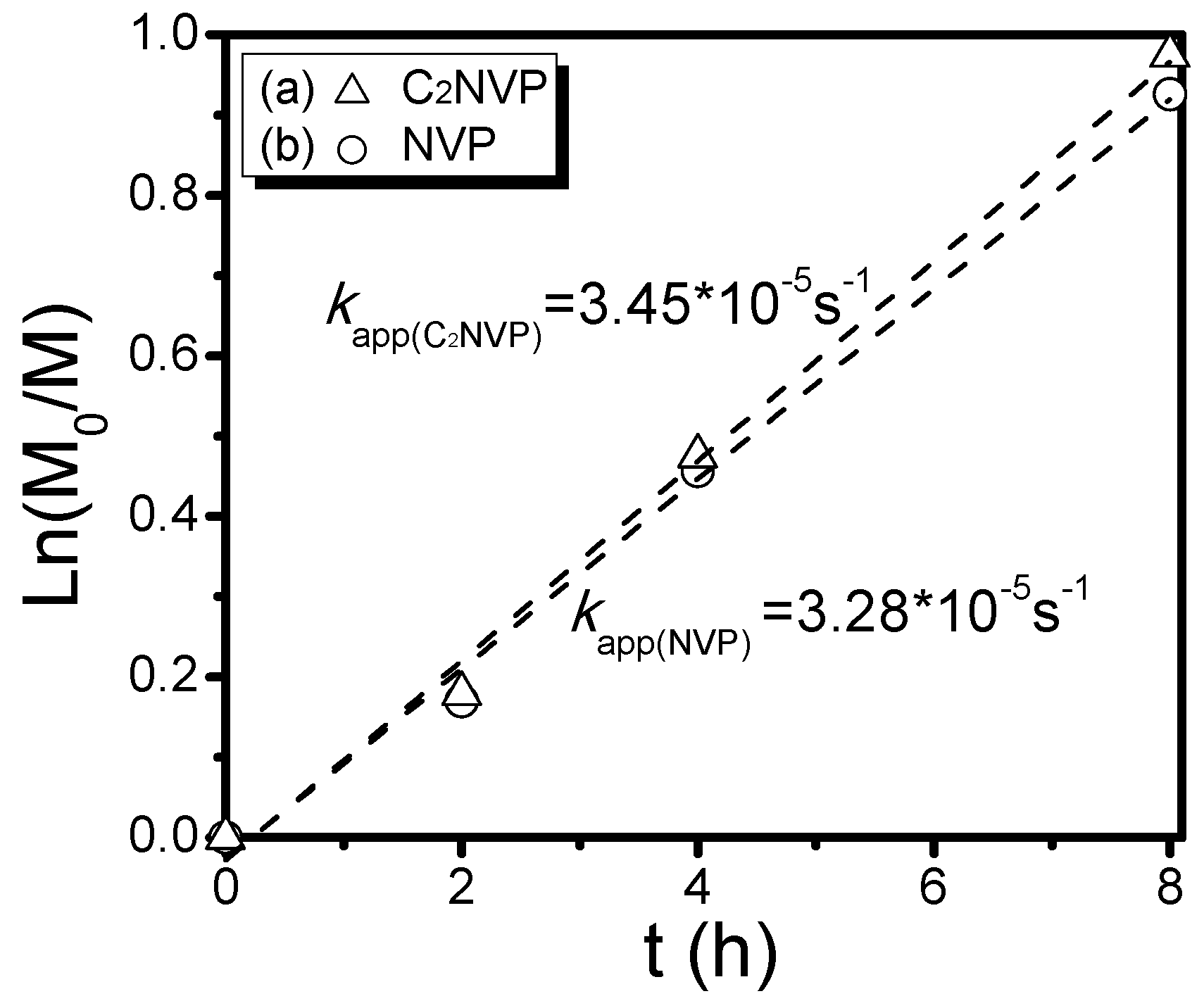
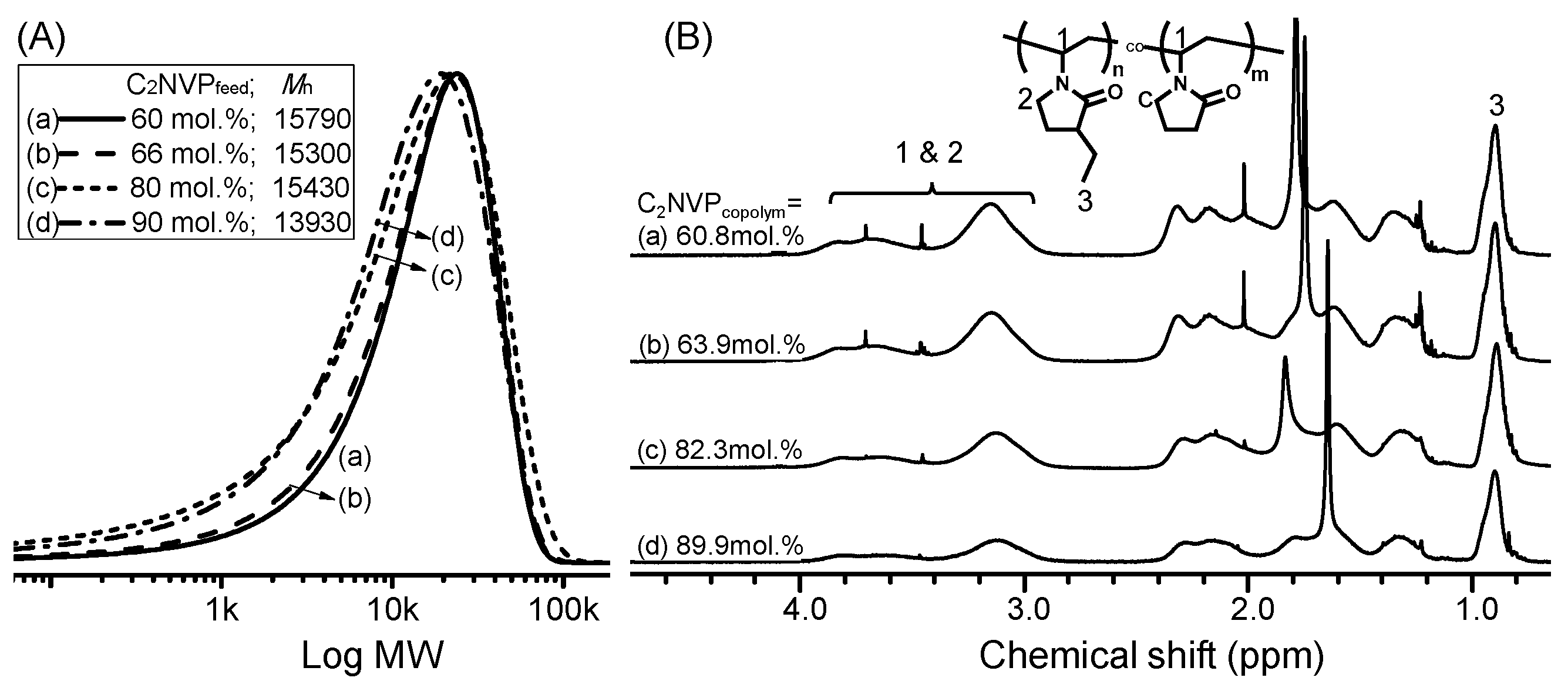
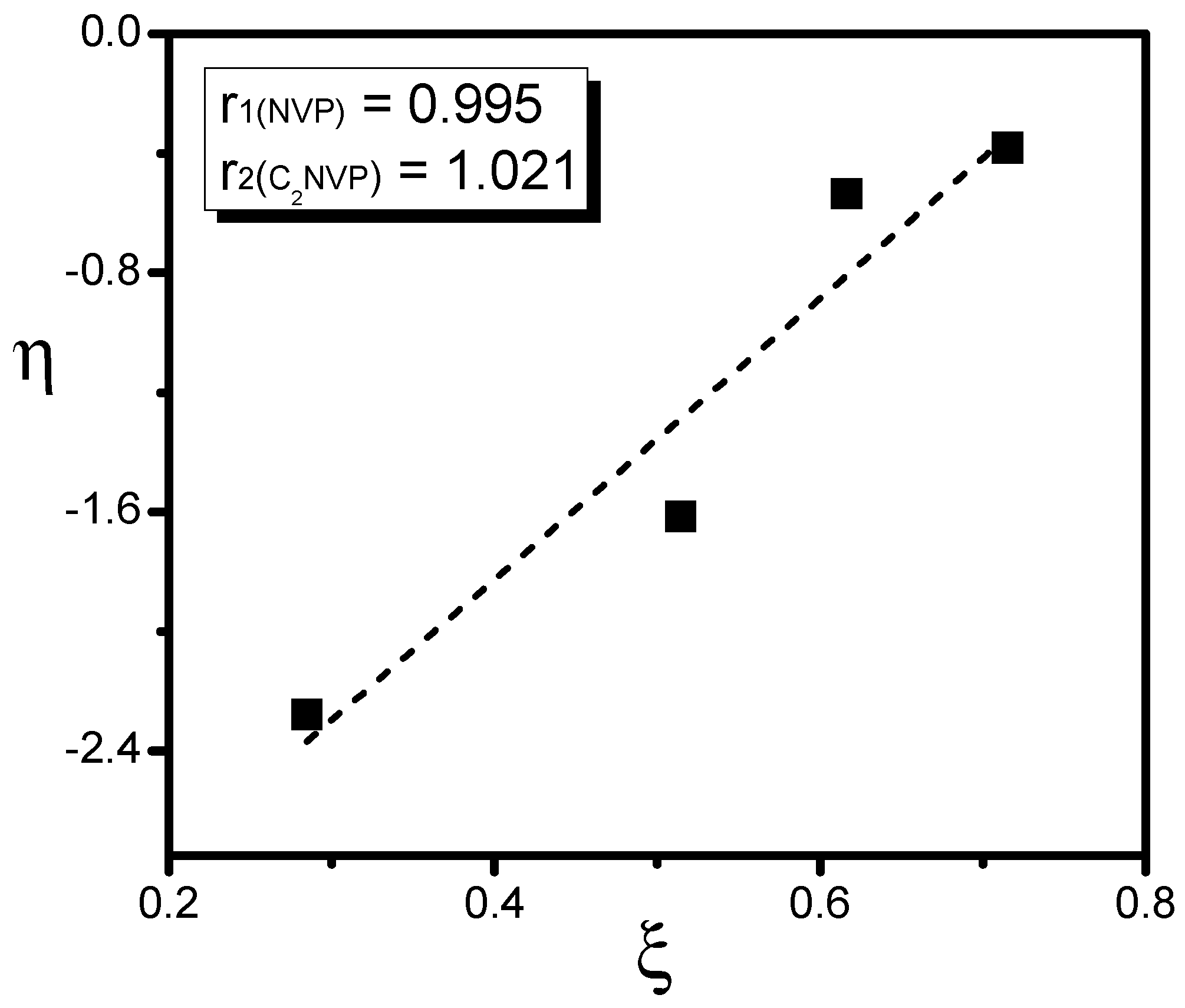
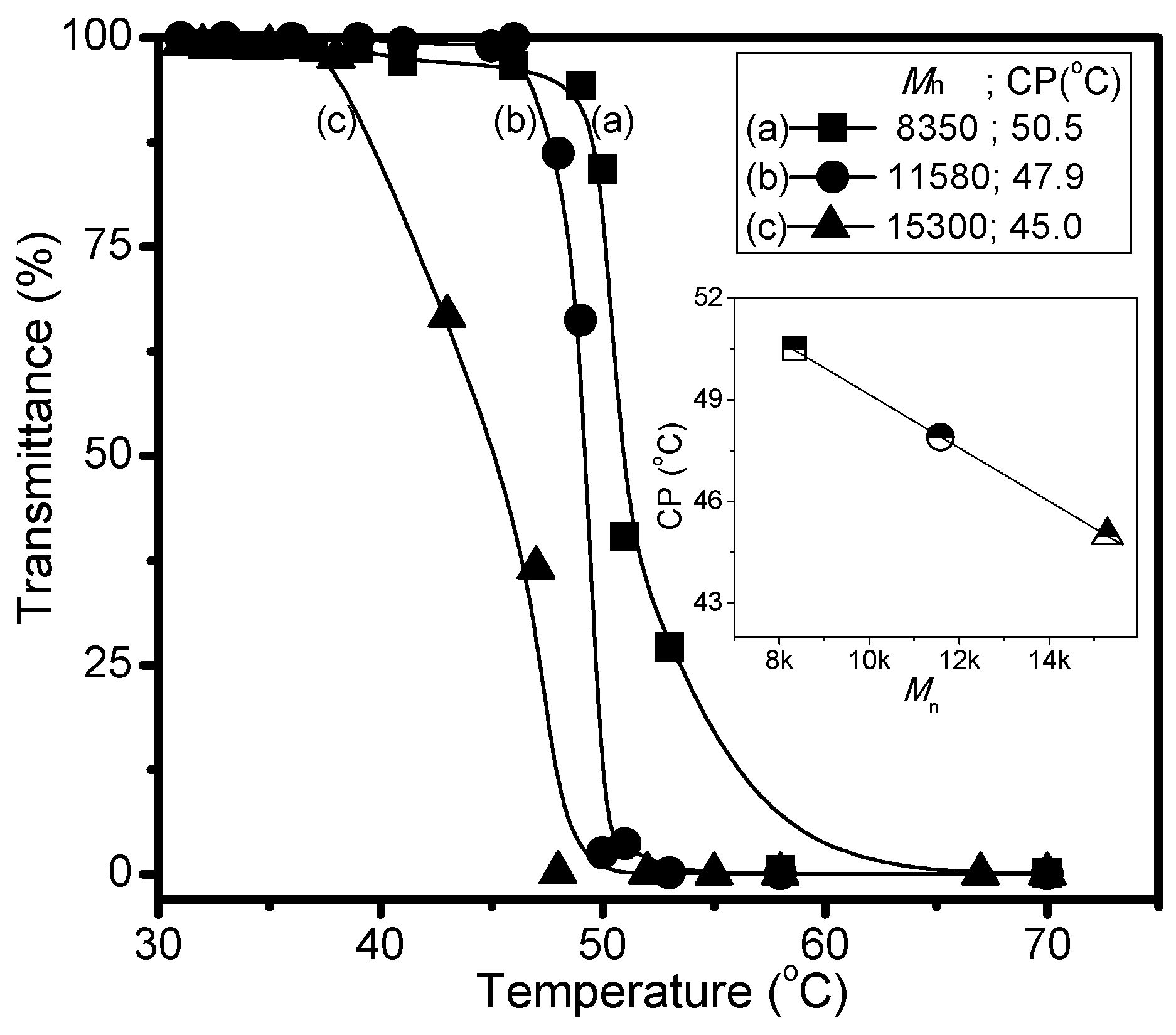
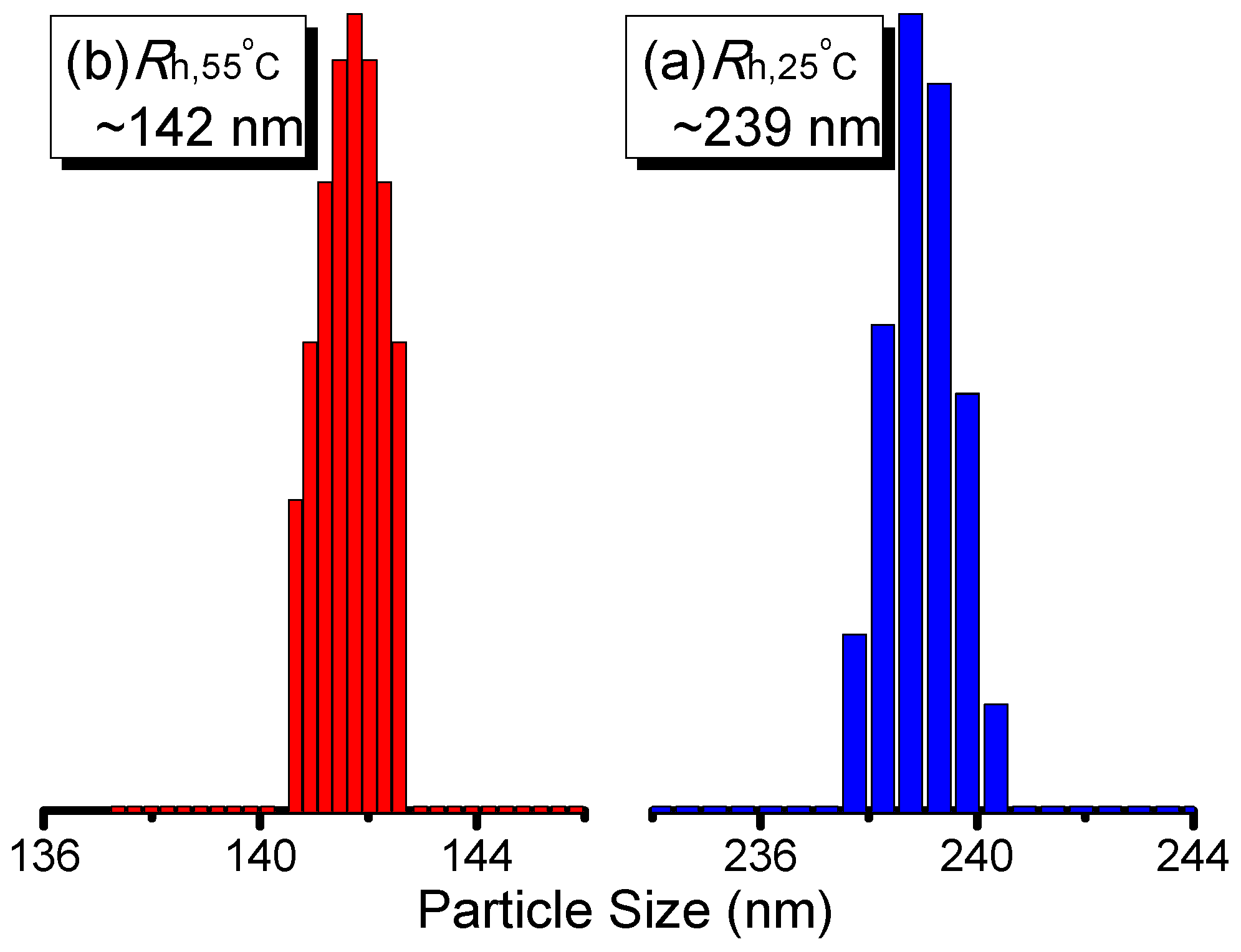
3. Results and Discussion
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stuart, M.A.C.; Huck, W.T.S.; Genzer, J.; Müller, M.; Ober, C.; Stamm, M.; Sukhorukov, G.B.; Szleifer, I.; Tsukruk, V.V.; Urban, M.; et al. Emerging applications of stimuli-responsive polymer materials. Nat. Mater. 2010, 9, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Urban, M.W. Recent advances and challenges in designing stimuli-responsive polymers. Prog. Polym. Sci. 2010, 35, 3–23. [Google Scholar] [CrossRef]
- Liu, R.; Fraylich, M.; Saunders, B.R. Thermoresponsive copolymers: From fundamental studies to applications. Colloid Polym. Sci. 2009, 287, 627–643. [Google Scholar] [CrossRef]
- Dimitrov, I.; Trzebicka, B.; Müller, A.H.E.; Dworak, A.; Tsvetanov, C.B. Thermosensitive water-soluble copolymers with doubly responsive reversibly interacting entities. Prog. Polym. Sci. 2007, 32, 1275–1343. [Google Scholar] [CrossRef]
- Gil, E.; Hudson, S. Stimuli-reponsive polymers and their bioconjugates. Prog. Polym. Sci. 2004, 29, 1173–1222. [Google Scholar] [CrossRef]
- Katsumoto, Y.; Etoh, Y.; Shimoda, N. Phase diagrams of stereocontrolled poly(N,N-diethylacrylamide) in water. Macromolecules 2010, 43, 3120–3121. [Google Scholar] [CrossRef]
- Maeda, Y.; Sakamoto, J.; Wang, S.-Y.; Mizuno, Y. Lower critical solution temperature behavior of poly(N-(2-ethoxyethyl)acrylamide) as compared with poly(N-isopropylacrylamide). J. Phys. Chem. B 2009, 113, 12456–12461. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, S.; Ritter, H. Access to poly{N-[3-(dimethylamino)propyl](meth)acrylamide} via microwave-assisted synthesis and control of LCST-behavior in water. Macromol. Rapid Commun. 2007, 28, 2080–2083. [Google Scholar] [CrossRef]
- Roth, P.J.; Jochum, F.D.; Forst, F.R.; Zentel, R.; Theato, P. Influence of end groups on the stimulus-responsive behavior of poly[oligo(ethylene glycol) methacrylate] in water. Macromolecules 2010, 43, 4638–4645. [Google Scholar] [CrossRef]
- Maeda, Y.; Yamauchi, H.; Kubota, T. Confocal micro-raman and infrared spectroscopic study on the phase separation of aqueous poly(2-(2-methoxyethoxy)ethyl (meth)acrylate) solutions. Langmuir 2009, 25, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Hoogenboom, R.; Van Assche, G.; Van Mele, B. Demixing and remixing kinetics of poly(2-isopropyl-2-oxazoline) (PIPOZ) aqueous solutions studied by modulated temperature differential scanning calorimetry. Macromolecules 2010, 43, 6853–6860. [Google Scholar] [CrossRef]
- Bloksma, M.M.; Bakker, D.J.; Weber, C.; Hoogenboom, R.; Schubert, U.S. The effect of Hofmeister salts on the LCST transition of poly(2-oxazoline)s with varying hydrophilicity. Macromol. Rapid Commun. 2010, 31, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Liang, X.; Gao, L.; Fan, X.; Zhou, Q. Water-soluble triply-responsive homopolymers of N,N-dimethylaminoethyl methacrylate with a terminal azobenzene moiety. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 2564–2570. [Google Scholar] [CrossRef]
- Plamper, F.A.; Ruppel, M.; Schmalz, A.; Borisov, O.; Ballauff, M.; Müller, A.H.E. Tuning the thermoresponsive properties of weak polyelectrolytes: Aqueous solutions of star-shaped and linear poly(N,N-dimethylaminoethyl methacrylate). Macromolecules 2007, 40, 8361–8366. [Google Scholar] [CrossRef]
- Deng, J.J.; Shi, Y.; Jiang, W.D.; Peng, Y.F.; Lu, L.C.; Cai, Y.L. Facile synthesis and thermoresponsive behaviors of a well-defined pyrrolidone based hydrophilic polymer. Macromolecules 2008, 41, 3007–3014. [Google Scholar] [CrossRef]
- Sun, J.; Peng, Y.F.; Chen, Y.; Liu, Y.; Deng, J.J.; Lu, L.C.; Cai, Y.L. Effect of molecular structure on thermoresponsive behaviors of pyrrolidone-based water-soluble polymers. Macromolecules 2010, 43, 4041–4049. [Google Scholar] [CrossRef]
- Yan, N.; Zhang, J.; Yuan, Y.; Chen, G.T.; Dyson, P.J.; Li, Z.C.; Kou, Y. Thermoresponsive polymers based on poly(N-vinylpyrrolidone): Applications in nanoparticle catalysis. Chem. Commun. 2010, 46, 1631–1633. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Zhang, J.G.; Tong, Y.; Yao, S.; Xiao, C.; Li, Z.; Kou, Y. Solubility adjustable nanoparticles stabilized by a novel PVP based family: Synthesis, characterization and catalytic properties. Chem. Commun. 2009, 4423–4425. [Google Scholar] [CrossRef] [PubMed]
- Trellenkamp, T.; Ritter, H. 3-ethylated N-vinyl-2-pyrrolidone with LCST properties in water. Macromol. Rapid Commun. 2009, 30, 1736–1740. [Google Scholar] [CrossRef] [PubMed]
- Du, F.-S.; Huang, X.-N.; Chen, G.-T.; Lin, S.-S.; Liang, D.; Li, Z.-C. Aqueous solution properties of the acid-labile thermoresponsive poly(meth)acrylamides with pendent cyclic orthoester groups. Macromolecules 2010, 43, 2474–2483. [Google Scholar] [CrossRef]
- Lambermont-Thijs, H.M.L.; Hoogenboom, R.; Fustin, C.-A.; Bomal-D’Haese, C.; Gohy, J.-F.; Schubert, U.S. Solubility behavior of amphiphilic block and random copolymers based on 2-ethyl-2-oxazoline and 2-nonyl-2-oxazoline in binary water-ethanol mixtures. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 515–522. [Google Scholar] [CrossRef]
- Hoogenboom, R.; Thijs, H.M.L.; Wouters, D.; Hoeppener, S.; Schubert, U.S. Tuning solution polymer properties by binary water–ethanolsolvent mixtures. Soft Matter 2008, 4, 103–107. [Google Scholar] [CrossRef]
- Huang, X.; Du, F.; Ju, R.; Li, Z. Novel acid-labile, thermoresponsive poly(methacrylamide)s with pendentortho ester moieties. Macromol. Rapid Commun. 2007, 28, 597–603. [Google Scholar] [CrossRef]
- Pakuro, N.; Yakimansky, A.; Chibirova, F.; Arest-Yakubovich, A. Thermo- and pH-sensitivity of aqueous poly(N-vinylpyrrolidone) solutions in the presence of organic acids. Polymer 2009, 50, 148–153. [Google Scholar] [CrossRef]
- Braunecker, W.A.; Matyjaszewski, K. Controlled/living radical polymerization: Features, developments, and perspectives. Prog. Polym. Sci. 2007, 32, 93–146. [Google Scholar] [CrossRef]
- Yokozawa, T.; Ogawa, M.; Sekino, A.; Sugi, R.; Yokoyama, A. Chain-growth polycondensation for well-defined aramide. Synthesis of unprecedented block copolymer containing aramide with low polydispersity. J. Am. Chem. Soc. 2002, 124, 15158–15159. [Google Scholar] [CrossRef] [PubMed]
- Sugi, R.; Yokoyama, A.; Furuyama, T.; Uchiyama, M.; Yokozawa, T. Inductive effect-assisted chain-growth polycondensation. Synthetic development from para- to meta-substituted aromatic polyamides with low polydispersities. J. Am. Chem. Soc. 2005, 127, 10172–10173. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-F.; Chen, M.-J.; Lin, C.-H.; Chiang, Y.-W. Synthesis of well-defined poly(N-H benzamide-co-N-octyl benzamide)s and the study of their blends with nylon 6. Polymers 2017, 9, 172. [Google Scholar] [CrossRef]
- Huang, C.-F.; Yokoyama, A.; Yokozawa, T. Synthesis of polybenzamide-b-polystyrene block copolymer via combination of chain-growth condensation polymerization and atom transfer radical polymerization. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 2948–2954. [Google Scholar] [CrossRef]
- Mecerreyes, D.; Jerome, R.; Dubois, P. Novel macromolecular architectures based on aliphatic polyesters: Relevance of the ”coordination-insertion” ring-opening polymerization. Adv. Polym. Sci. 1999, 147, 1–59. [Google Scholar]
- Hadjichristidis, N.; Pitsikalis, M.; Pispas, S.; Iatrou, H. Polymers with complex architecture by living anionic polymerization. Chem. Rev. 2001, 101, 3747–3792. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Goseki, R.; Ishizone, T.; Hirao, A. Synthesis of well-controlled graft polymers by living anionic polymerization towards exact graft polymers. Polym. Chem. 2014, 5, 5523–5534. [Google Scholar] [CrossRef]
- Sawamoto, M. Modern cationic vinyl polymerization. Prog. Polym. Sci. 1991, 16, 111–172. [Google Scholar] [CrossRef]
- Bielawski, C.W.; Grubbs, R.H. Living ring-opening metathesis polymerization. Prog. Polym. Sci. 2007, 32, 1–29. [Google Scholar] [CrossRef]
- Huang, C.F.; Aimi, J.; Lai, K.Y. Synthesis of novel μ-star copolymers with poly(N-octyl benzamide) and poly(ε-caprolactone) miktoarms through chain-growth condensation polymerization, styrenics-assisted atom transfer radical coupling, and ring-opening polymerization. Macromol. Rapid Commun. 2017, 38, 1600607. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-F.; Ohta, Y.; Yokoyama, A.; Yokozawa, T. Efficient low-temperature atom transfer radical coupling and its application to synthesis of well-defined symmetrical polybenzamides. Macromolecules 2011, 44, 4140–4148. [Google Scholar] [CrossRef]
- Hawker, C.J.; Bosman, A.W.; Harth, E. New polymer synthesis by nitroxide mediated living radical polymerizations. Chem. Rev. 2001, 101, 3661–3688. [Google Scholar] [CrossRef] [PubMed]
- Chiefari, J.; Chong, Y.K.; Ercole, F.; Krstina, J.; Jeffery, J.; Le, T.P.T.; Mayadunne, R.T.A.; Meijs, G.F.; Moad, C.L.; Moad, G.; et al. Living free-radical polymerization by reversible addition-fragmentation chain transfer: The RAFT process. Macromolecules 1998, 31, 5559–5562. [Google Scholar] [CrossRef]
- Kamigaito, M.; Ando, T.; Sawamoto, M. Metal-catalyzed living radical polymerization. Chem. Rev. 2001, 101, 3689–3745. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.S.; Matyjaszewski, K. Controlled living radical polymerization-atom-transfer radical polymerization in the presence of transition-metal complexes. J. Am. Chem. Soc. 1995, 117, 5614–5615. [Google Scholar] [CrossRef]
- Yusa, S.; Yamago, S.; Sugahara, M.; Morikawa, S.; Yamamoto, T.; Morishima, Y. Thermo-responsive diblock copolymers of poly(N-isopropylacrylamide) and poly(N-vinyl-2-pyrroridone) synthesized via organotellurium-mediated controlled radical polymerization (TERP). Macromolecules 2007, 40, 5907–5915. [Google Scholar] [CrossRef]
- Yamago, S.; Kayahara, E.; Kotani, M.; Ray, B.; Kwak, Y.; Goto, A.; Fukuda, T. Highly controlled living radical polymerization through dual activation of organobismuthines. Angew. Chem. Int. Ed. 2007, 46, 1304–1306. [Google Scholar] [CrossRef] [PubMed]
- Yamago, S.; Ray, B.; Iida, K.; Yoshida, J.; Tada, T.; Yoshizawa, K.; Kwak, Y.; Goto, A.; Fukuda, T. Highly versatile organostibine mediators for living radical polymerization. J. Am. Chem. Soc. 2004, 126, 13908–13909. [Google Scholar] [CrossRef] [PubMed]
- Gaynor, S.G.; Wang, J.S.; Matyjaszewski, K. Controlled radical polymerization by degenerative transfer-effect of the structure of the transfer agent. Macromolecules 1995, 28, 8051–8056. [Google Scholar] [CrossRef]
- Teodorescu, M.; Matyjaszewski, K. Atom transfer radical polymerization of (meth)acrylamides. Macromolecules 1999, 32, 4826–4831. [Google Scholar] [CrossRef]
- Matyjaszewski, K. Transition metal catalysis in controlled radical polymerization: Atom transfer radical polymerization. Chem. Eur. J. 1999, 5, 3095–3102. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Patten, T.E.; Xia, J.H. Controlled/”living” radical polymerization. Kinetics of the homogeneous atom transfer radical polymerization of styrene. J. Am. Chem. Soc. 1997, 119, 674–680. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Jo, S.M.; Paik, H.J.; Shipp, D.A. An investigation into the CuX/2,2'-bipyridine (X = Br or Cl) mediated atom transfer radical polymerization of acrylonitrile. Macromolecules 1999, 32, 6431–6438. [Google Scholar] [CrossRef]
- Jakubowski, W.; Min, K.; Matyjaszewski, K. Activators regenerated by electron transfer for atom transfer radical polymerization of styrene. Macromolecules 2006, 39, 39–45. [Google Scholar] [CrossRef]
- Jakubowski, W.; Matyjaszewski, K. Activators regenerated by electron transfer for atom-transfer radical polymerization of (meth)acrylates and related block copolymers. Angew. Chem. Int. Ed. 2006, 45, 4482–4486. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.T.; Wang, C.H.; Zhang, J.G.; Wang, Y.; Zhang, R.; Du, F.S.; Yan, N.; Kou, Y.A.; Li, Z.C. Toward functionalization of thermoresponsive poly(N-vinyl-2-pyrrolidone). Macromolecules 2010, 43, 9972–9981. [Google Scholar] [CrossRef]
- Huang, C.-F.; Chang, F.-C. Comparison of hydrogen bonding interaction between PMMA/PMAA blends and PMMA-co-PMAA copolymers. Polymer 2003, 44, 2965–2974. [Google Scholar] [CrossRef]
- Liu, H.Y.; Zhu, X.X. Lower critical solution temperatures of N-substituted acrylamide copolymers in aqueous solutions. Polymer 1999, 40, 6985–6990. [Google Scholar] [CrossRef]
- Popkov, Y.M.; Nakhmanovich, B.I.; Chibirova, F.K.; Bune, E.V.; Arest-Yakubovich, A.A. Copolymerization in N-vinylcaprolactam-N-vinylpyrrolidone and N,N-diethylacrylamide-N,N-dimethylacrylamide systems: The effect of composition and spatial structure of copolymers on their thermal sensitivity. Polym. Sci. Ser. B 2007, 49, 155–158. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
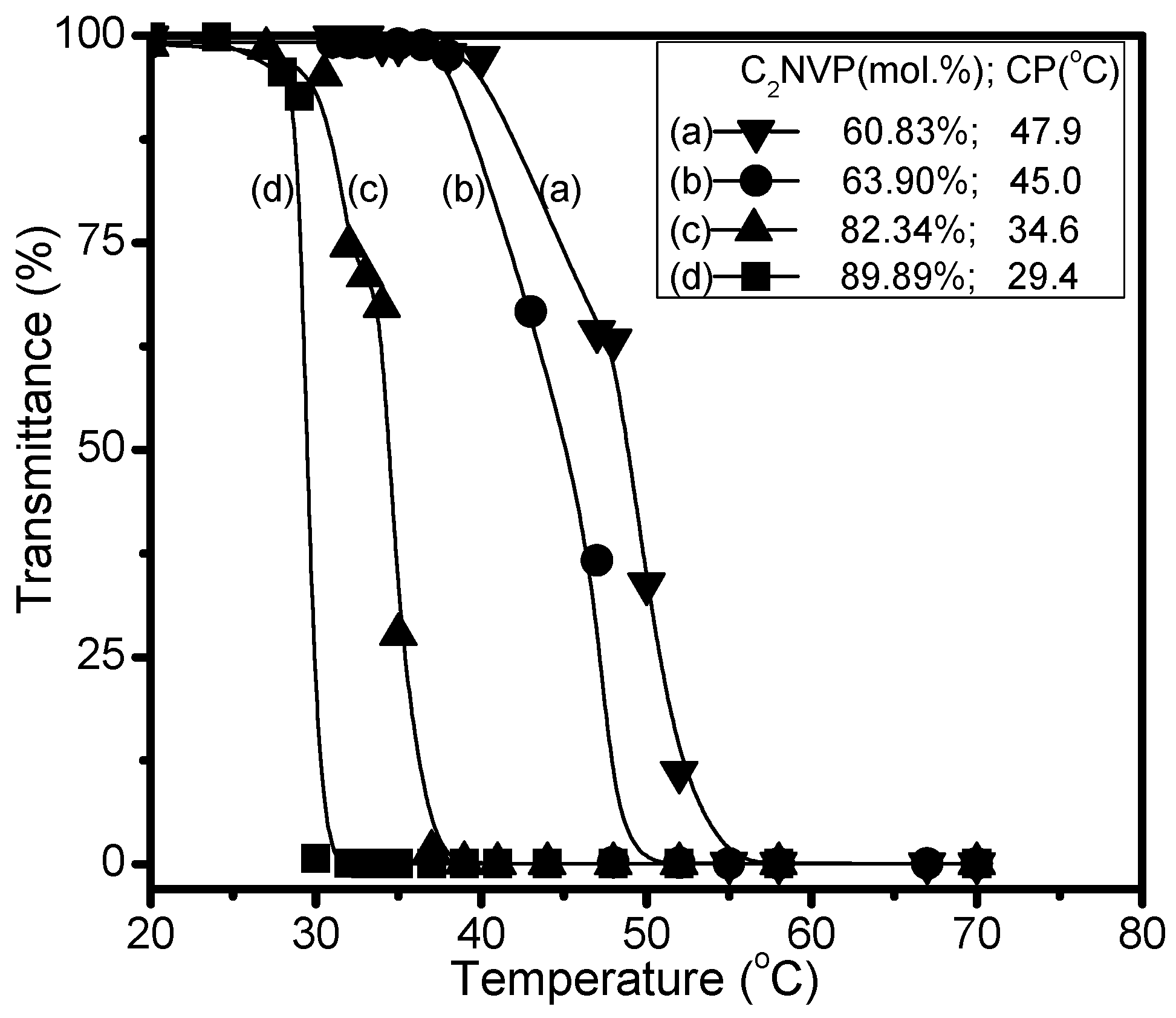
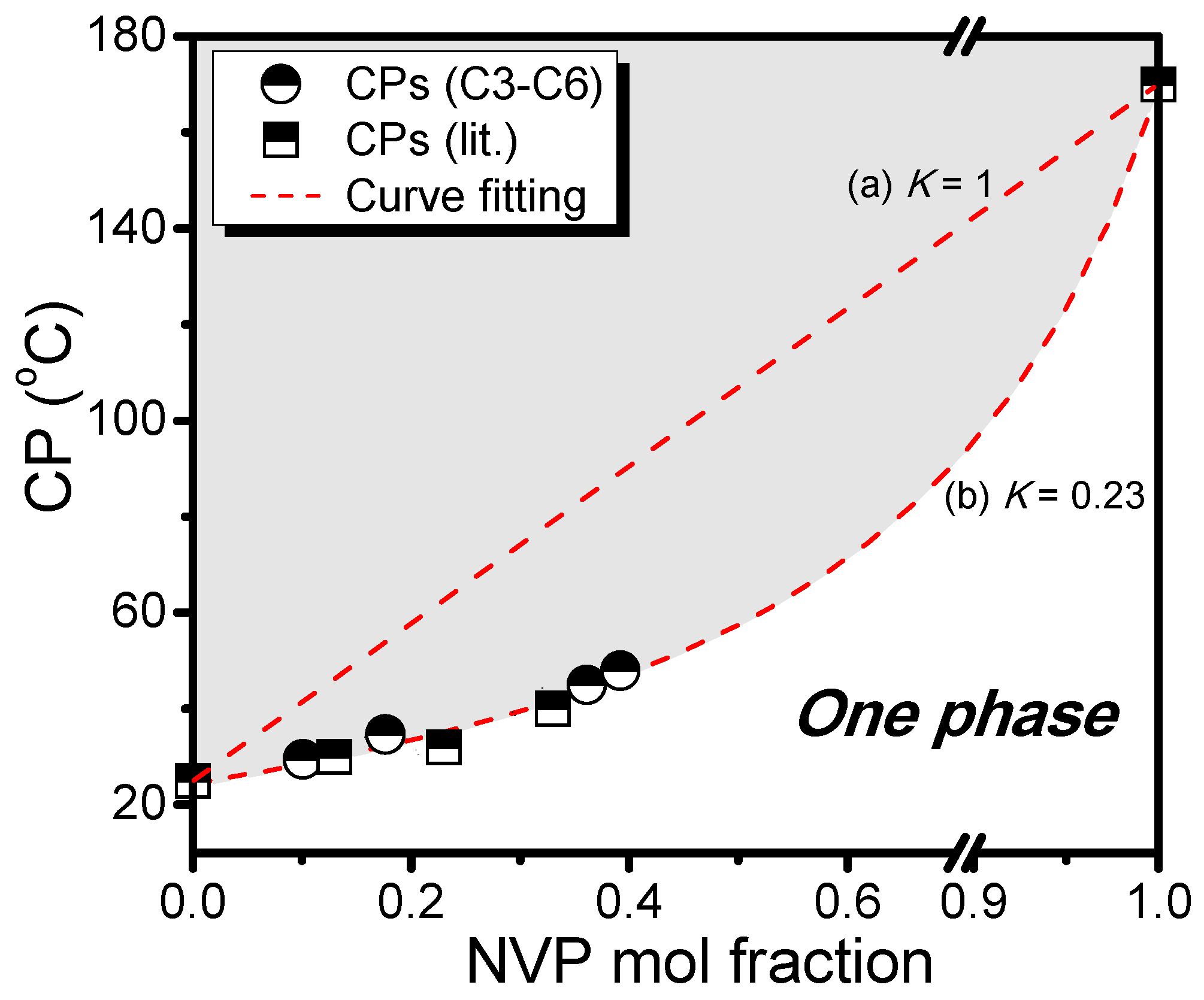
| No. | [M]/[I] a | C2NVPfeed (mol %) | C2NVPcopolym b (mol %) | Mn c | Mw/Mn c | CP (°C) |
|---|---|---|---|---|---|---|
| C1 | 30 | 66.7 | 64.0 | 8350 | 1.34 | 50.5 |
| C2 | 60 | 66.7 | 64.6 | 11,580 | 1.42 | 47.9 |
| C3 | 200 | 66.7 | 63.9 | 15,300 | 1.49 | 45.0 |
| C4 | 200 | 60.0 | 60.8 | 15,790 | 1.49 | 47.9 |
| C5 | 200 | 80.0 | 82.3 | 15,430 | 1.48 | 34.6 |
| C6 | 200 | 90.0 | 89.9 | 13,930 | 1.43 | 29.4 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.-S.; Chen, J.-K.; Chen, T.; Huang, C.-F. Synthesis of PNVP-Based Copolymers with Tunable Thermosensitivity by Sequential Reversible Addition–Fragmentation Chain Transfer Copolymerization and Ring-Opening Polymerization. Polymers 2017, 9, 231. https://doi.org/10.3390/polym9060231
Huang Y-S, Chen J-K, Chen T, Huang C-F. Synthesis of PNVP-Based Copolymers with Tunable Thermosensitivity by Sequential Reversible Addition–Fragmentation Chain Transfer Copolymerization and Ring-Opening Polymerization. Polymers. 2017; 9(6):231. https://doi.org/10.3390/polym9060231
Chicago/Turabian StyleHuang, Yi-Shen, Jem-Kun Chen, Tao Chen, and Chih-Feng Huang. 2017. "Synthesis of PNVP-Based Copolymers with Tunable Thermosensitivity by Sequential Reversible Addition–Fragmentation Chain Transfer Copolymerization and Ring-Opening Polymerization" Polymers 9, no. 6: 231. https://doi.org/10.3390/polym9060231