Intricately Regulated: A Cellular Toolbox for Fine-Tuning XBP1 Expression and Activity

Abstract
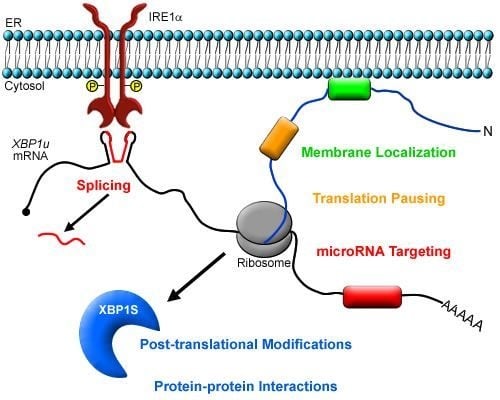
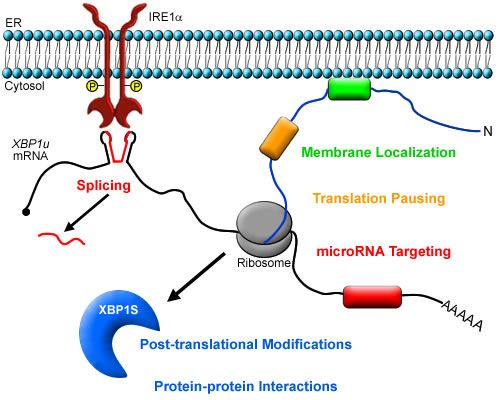
:
1. Introduction
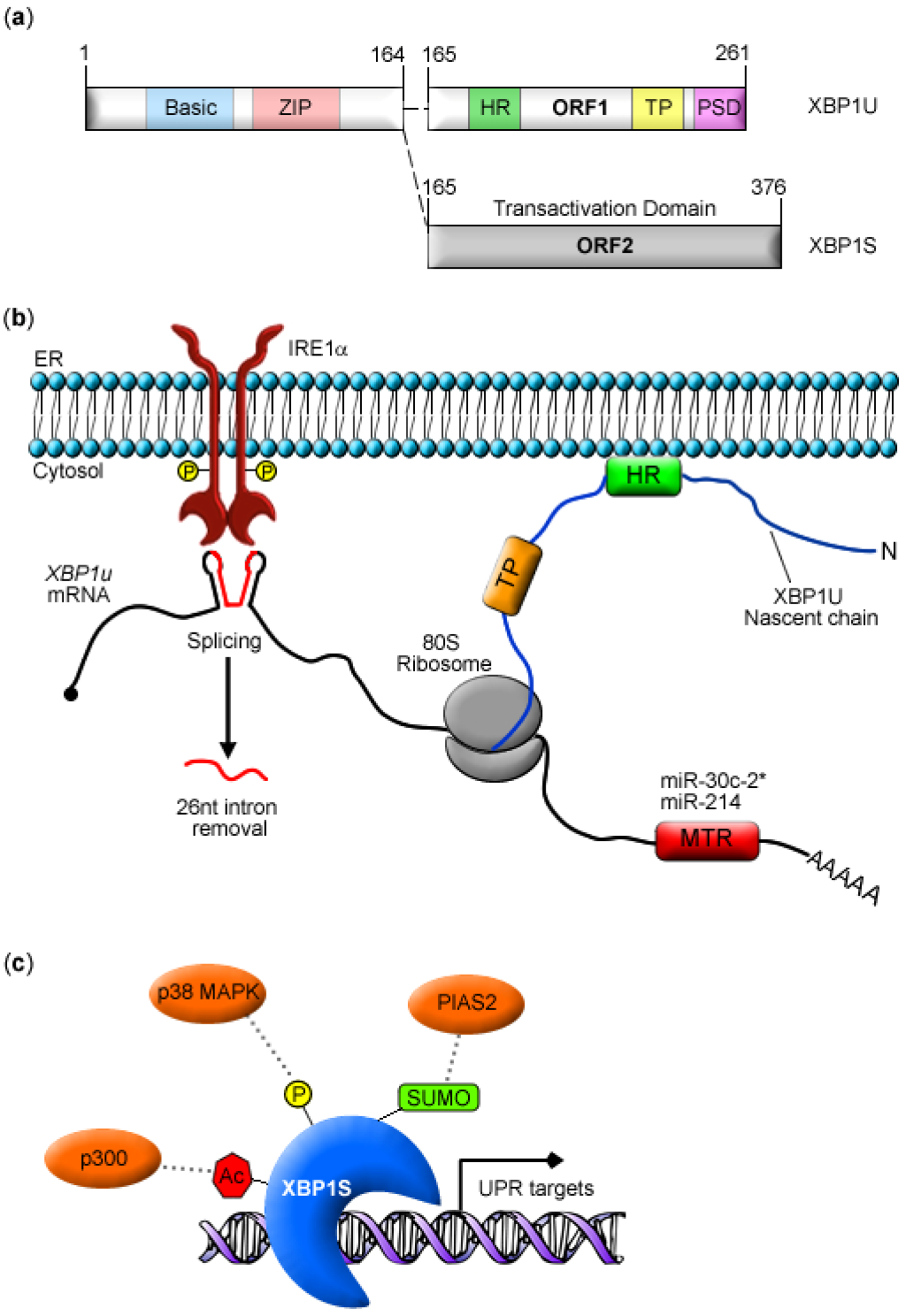
2. XBP1 Biosynthesis and Function
3. Post-transcriptional Modulation of XBP1 Expression
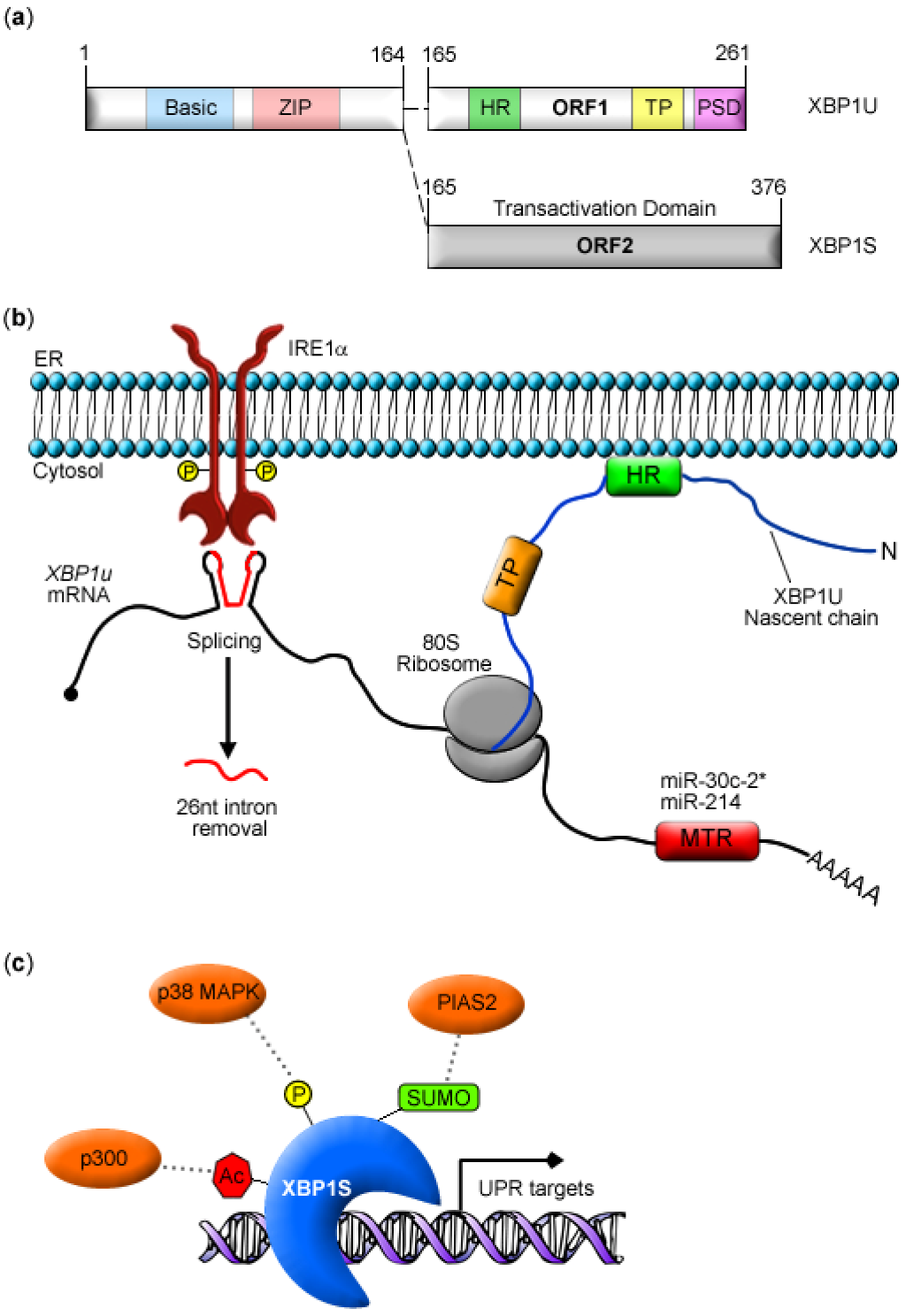
4. Post-translational Modifications of XBP1S
4.1. Phosphorylation
4.2. SUMOylation
4.3. Acetylation and Deacetylation
5. Protein-protein Interactions of XBP1U and XBP1S
5.1. XBP1U Negatively Regulates XBP1S
5.2. p85 Subunit of PI3K Enhances XBP1S Nucleocytoplasmic Shuttling
5.3. XBP1S Directs FOXO1 to Proteasome-Mediated Degradation


{kind=link}
{kind=link}
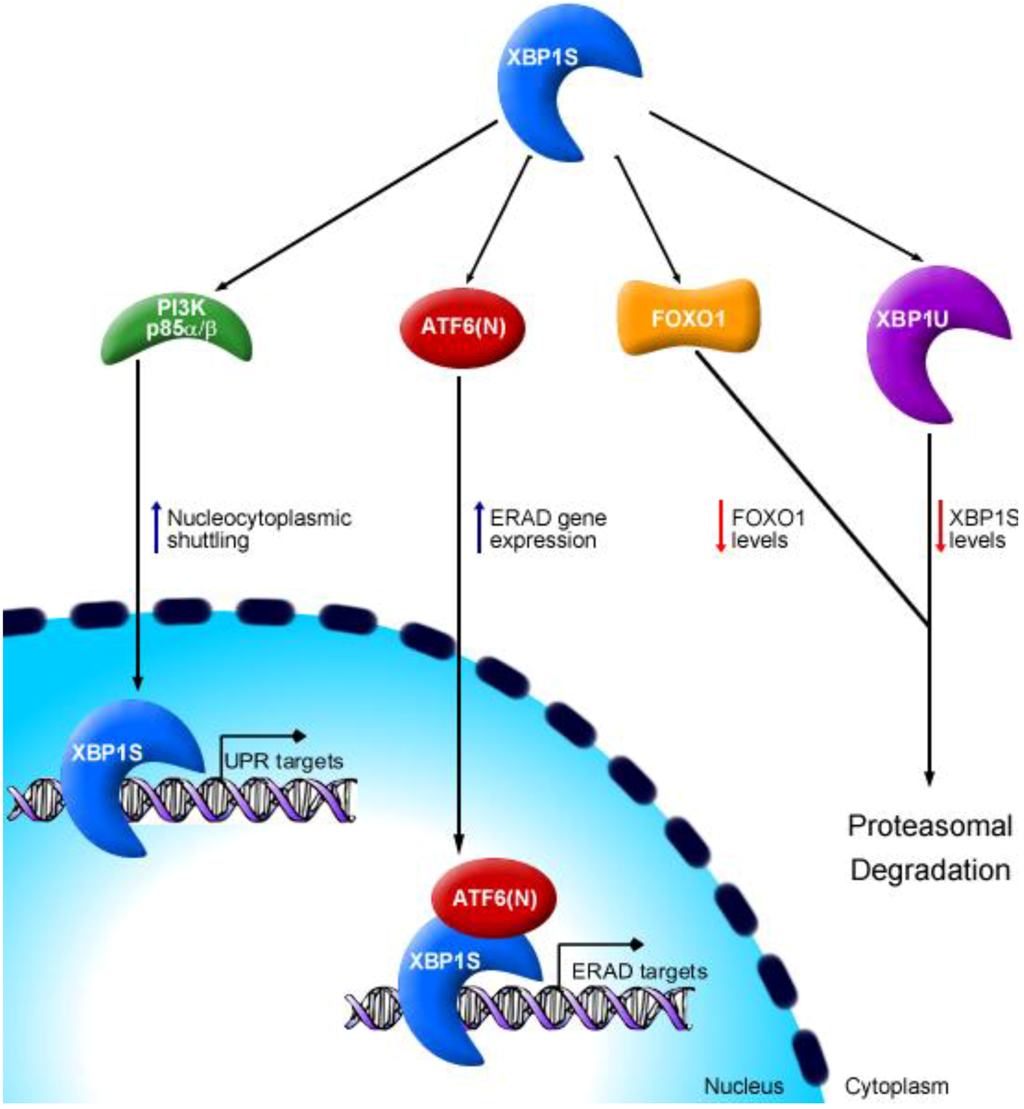{kind=link}
| Cellular Process | Effector | Mechanism | Effect on XBP1 | Model | Reference |
|---|---|---|---|---|---|
| Post-trancription | IRE1 | cytosolic mRNA splicing | cytosolic mRNA splicing/religation/ORF frameshift | pharmacologic-induced ER stress | [11,12,13] |
| HR2 of nascent XBP1U | XBP1u mRNA recruitment | localize XBP1 mRNA at ER membrane within close proximity of IRE1 | in vitro expression of XBP1 | [24] | |
| XBP1u COOH-terminus | translational pausing | stabilize R-RNC complex at ER membrane for efficient targeting and splicing of XBP1u mRNA | pharmacologic-induced ER stress | [25] | |
| miR-30c-2* | inhibits XBP1 | fine-tune XBP1 expression as the UPR proceeds | pharmacologic-induced ER stress | [15] | |
| miR-214 | expression inhibits XBP1 | repress XBP1 expression prior to UPR activation | LPS- and pharmacologic- induced ER stress | [50] | |
| Post-translation | p38 MAPK | phosphorylation | increase XBP1S stability/enhance XBP1S nucleocytoplasmic shuttling | pharmacologic-induced ER stress; ob/ob mice | [17] |
| PIAS2 | SUMOylation | decrease transcriptional activity of XBP1S | in vitro expression of XBP1; pharmacologic-induced ER stress | [16] | |
| p300 | acetylation | increase XBP1S stability | pharmacologic-induced ER stress | [22] | |
| SIRT1 | deacetylation | decrease XBP1S stability | pharmacologic-induced ER stress | [22] | |
| Protein interaction | XBP1U | heterodimerization | target XBP1U-XBP1S complex for proteasome-mediated degradation | transient over-expression in vitro | [26] |
| PI3K p85 subunits | heterodimerization | enhance XBP1S nucleocytoplasmic shuttling | pharmacologic-induced ER stress; metabolic induced overload in ob/ob mice | [20,23] | |
| FOXO1 | heterodimerization | direct protein complex to 26S proteasome | in vitro expression; glucose homeostasis in ob/ob mice | [58] | |
| ATF6α( N) | heterodimerization | combinatorially enhance ERAD gene expression | pharmacologic-induced ER stress | [10] |
5.4. ATF6α Interacts with XBP1S to Optimize ERAD
6. Concluding Remarks
Acknowledgements
Conflict of Interest
References
- Walter, P.; Ron, D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Scheuner, D.; Song, B.; McEwen, E.; Liu, C.; Laybutt, R.; Gillespie, P.; Saunders, T.; Bonner-Weir, S.; Kaufman, R.J. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell 2001, 7, 1165–1176. [Google Scholar] [CrossRef]
- Shi, Y.; Vattem, K.M.; Sood, R.; An, J.; Liang, J.; Stramm, L.; Wek, R.C. Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol. Cell. Biol. 1998, 18, 7499–7509. [Google Scholar]
- Adachi, Y.; Yamamoto, K.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct. Funct. 2008, 33, 75–89. [Google Scholar] [CrossRef]
- Haze, K.; Okada, T.; Yoshida, H.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. Identification of the g13 (camp-response-element-binding protein-related protein) gene product related to activating transcription factor 6 as a transcriptional activator of the mammalian unfolded protein response. Biochem. J. 2001, 355, 19–28. [Google Scholar] [CrossRef]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar]
- Wu, J.; Rutkowski, D.T.; Dubois, M.; Swathirajan, J.; Saunders, T.; Wang, J.; Song, B.; Yau, G.D.; Kaufman, R.J. ATF6α optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev. Cell 2007, 13, 351–364. [Google Scholar] [CrossRef]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6α and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002, 415, 92–96. [Google Scholar] [CrossRef]
- Shen, X.; Ellis, R.; Lee, K.; Liu, C.-Y.; Yang, K.; Solomon, A.; Yoshida, H.; Morimoto, R.; Kurnit, D.M.; Mori, K.; et al. Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell 2001, 107, 893–903. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef]
- Byrd, A.E.; Aragon, I.V.; Brewer, J.W. MicroRNA-30c-2* limits expression of proadaptive factor XBP1 in the unfolded protein response. J. Cell Biol. 2012, 196, 689–698. [Google Scholar] [CrossRef]
- Chen, H.; Qi, L. SUMO modification regulates the transcriptional activity of XBP1. Biochem. J. 2011, 429, 95–102. [Google Scholar] [CrossRef]
- Lee, J.; Sun, C.; Zhou, Y.; Lee, J.; Gokalp, D.; Herrema, H.; Park, S.W.; Davis, R.J.; Ozcan, U. p38 MAPK-mediated regulation of Xbp1s is crucial for glucose homeostasis. Nat. Med. 2011, 17, 1251–1260. [Google Scholar] [CrossRef]
- Majumder, M.; Huang, C.; Snider, M.D.; Komar, A.A.; Tanaka, J.; Kaufman, R.J.; Krokowski, D.; Hatzoglou, M. A novel feedback loop regulates the response to endoplasmic reticulum stress via the cooperation of cytoplasmic splicing and mRNA translation. Mol. Cell. Biol. 2012, 32, 992–1003. [Google Scholar] [CrossRef]
- Navon, A.; Gatushkin, A.; Zelcbuch, L.; Shteingart, S.; Farago, M.; Hadar, R.; Tirosh, B. Direct proteasome binding and subsequent degradation of unspliced XBP-1 prevent its intracellular aggregation. FEBS Letters 2010, 584, 67–73. [Google Scholar] [CrossRef]
- Park, S.W.; Zhou, Y.; Lee, J.; Lu, A.; Sun, C.; Chung, J.; Ueki, K.; Ozcan, U. The regulatory subunits of PI3K, p85α and p85β, interact with XBP-1 and increase its nuclear translocation. Nat. Med. 2010, 16, 429–437. [Google Scholar] [CrossRef]
- Tirosh, B.; Iwakoshi, N.N.; Glimcher, L.H.; Ploegh, H.L. Rapid turnover of unspliced Xbp-1 as a factor that modulates the unfolded protein response. J. Biol. Chem. 2006, 281, 5852–5860. [Google Scholar]
- Wang, F.-M.; Chen, Y.-J.; Ouyang, H.-J. Regulation of unfolded protein response modulator XBP1s by acetylation and deacetylation. Biochem. J. 2011, 433, 245–252. [Google Scholar] [CrossRef]
- Winnay, J.N.; Boucher, J.; Mori, M.A.; Ueki, K.; Kahn, C.R. A regulatory subunit of phosphoinositide 3-kinase increases the nuclear accumulation of X-box-binding protein-1 to modulate the unfolded protein response. Nat. Med. 2010, 16, 438–445. [Google Scholar] [CrossRef]
- Yanagitani, K.; Imagawa, Y.; Iwawaki, T.; Hosoda, A.; Saito, M.; Kimata, Y.; Kohno, K. Cotranslational targeting of XBP1 protein to the membrane promotes cytoplasmic splicing of its own mRNA. Mol. Cell 2009, 34, 191–200. [Google Scholar] [CrossRef]
- Yanagitani, K.; Kimata, Y.; Kadokura, H.; Kohno, K. Translational pausing ensures membrane targeting and cytoplasmic splicing of XBP1u mRNA. Science 2011, 331, 586–589. [Google Scholar] [CrossRef]
- Yoshida, H.; Oku, M.; Suzuki, M.; Mori, K. pXBP1(u) encoded in XBP1 pre-mRNA negatively regulates unfolded protein response activator pXBP1(s) in mammalian ER stress response. J. Cell. Biol. 2006, 172, 565–575. [Google Scholar] [CrossRef]
- Liou, H.C.; Boothby, M.R.; Finn, P.W.; Davidon, R.; Nabavi, N.; Zeleznik-Le, N.J.; Ting, J.P.; Glimcher, L.H. A new member of the leucine zipper class of proteins that binds to the HLA DR α promoter. Science 1990, 247, 1581–1584. [Google Scholar]
- Glimcher, L.H. XBP1: The last two decades. Ann. Rheum. Dis. 2010, 69 (Suppl. 1), i67–i71. [Google Scholar] [CrossRef]
- Reimold, A.M.; Iwakoshi, N.N.; Manis, J.; Vallabhajosyula, P.; Szomolanyi-Tsuda, E.; Gravallese, E.M.; Friend, D.; Grusby, M.J.; Alt, F.; Glimcher, L.H. Plasma cell differentiation requires the transcription factor XBP-1. Nature 2001, 412, 300–307. [Google Scholar] [CrossRef]
- Lee, A.H.; Chu, G.C.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J. 2005, 24, 4368–4380. [Google Scholar] [CrossRef]
- Lee, A.-H.; Heidtman, K.; Hotamisligil, G.S.; Glimcher, L.H. Dual and opposing roles of the unfolded protein response regulated by IRE1α and XBP1 in proinsulin processing and insulin secretion. Proc. Natl. Acad. Sci. USA 2011, 108, 8885–8890. [Google Scholar] [CrossRef]
- Lee, A.-H.; Scapa, E.F.; Cohen, D.E.; Glimcher, L.H. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 2008, 320, 1492–1496. [Google Scholar] [CrossRef]
- Reimold, A.M.; Etkin, A.; Clauss, I.; Perkins, A.; Friend, D.S.; Zhang, J.; Horton, H.F.; Scott, A.; Orkin, S.H.; Byrne, M.C.; et al. An essential role in liver development for transcription factor XBP-1. Genes Dev. 2000, 14, 152–157. [Google Scholar]
- Iwakoshi, N.N.; Pypaert, M.; Glimcher, L.H. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J. Exp. Med. 2007, 204, 2267–2275. [Google Scholar] [CrossRef]
- Martinon, F.; Chen, X.; Lee, A.-H.L.; Glimcher, L.H. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 2010, 11, 411–418. [Google Scholar] [CrossRef]
- Kaser, A.; Lee, A.-H.; Franke, A.; Glickman, J.N.; Zeissig, S.; Tilg, H.; Nieuwenhuis, E.E.S.; Higgins, D.E.; Schreiber, S.; Glimcher, L.H.; et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008, 134, 743–756. [Google Scholar] [CrossRef]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef]
- Cox, J.S.; Shamu, C.E.; Walter, P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 1993, 73, 1197–1206. [Google Scholar] [CrossRef]
- Mori, K.; Ma, W.; Gething, M.J.; Sambrook, J. A transmembrane protein with a cdc2+/cdc28-related kinase activity is required for signaling from the ER to the nucleus. Cell 1993, 74, 743–756. [Google Scholar] [CrossRef]
- Sidrauski, C.; Walter, P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 1997, 90, 1031–1039. [Google Scholar] [CrossRef]
- Tirasophon, W.; Welihinda, A.A.; Kaufman, R.J. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998, 12, 1812–1824. [Google Scholar] [CrossRef]
- Wang, X.Z.; Harding, H.P.; Zhang, Y.; Jolicoeur, E.M.; Kuroda, M.; Ron, D. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J. 1998, 17, 5708–5717. [Google Scholar] [CrossRef]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef]
- Shaffer, A.L.; Shapiro-Shelef, M.; Iwakoshi, N.N.; Lee, A.H.; Qian, S.B.; Zhao, H.; Yu, X.; Yang, L.; Tan, B.K.; Rosenwald, A.; et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 2004, 21, 81–93. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Brewer, J.W.; Rab, A.; Crossman, D.K.; Bartoszewska, S.; Kapoor, N.; Fuller, C.; Collawn, J.F.; Bebok, Z. The unfolded protein response (UPR)-activated transcription factor X-box-binding protein 1 (XBP1) induces microRNA-346 expression that targets the human antigen peptide transporter 1 (TAP1) mRNA and governs immune regulatory genes. J. Biol. Chem. 2011, 286, 41862–41870. [Google Scholar]
- Iwakoshi, N.N.; Lee, A.H.; Vallabhajosyula, P.; Otipoby, K.L.; Rajewsky, K.; Glimcher, L.H. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat. Immunol. 2003, 4, 321–329. [Google Scholar]
- Back, S.H.; Lee, K.; Vink, E.; Kaufman, R.J. Cytoplasmic IRE1α-mediated XBP1 mRNA splicing in the absence of nuclear processing and endoplasmic reticulum stress. J. Biol. Chem. 2006, 281, 18691–18706. [Google Scholar] [CrossRef]
- Uemura, A.; Oku, M.; Mori, K.; Yoshida, H. Unconventional splicing of XBP1 mRNA occurs in the cytoplasm during the mammalian unfolded protein response. J. Cell Sci. 2009, 122, 2877–2886. [Google Scholar] [CrossRef]
- Stephens, S.B.; Dodd, R.D.; Brewer, J.W.; Lager, P.J.; Keene, J.D.; Nicchitta, C.V. Stable ribosome binding to the endoplasmic reticulum enables compartment-specific regulation of mRNA translation. Mol. Biol. Cell 2005, 16, 5819–5831. [Google Scholar] [CrossRef]
- Duan, Q.; Wang, X.; Gong, W.; Ni, L.; Chen, C.; He, X.; Chen, F.; Yang, L.; Wang, P.; Wang, D.W. ER stress negatively modulates the expression of the mir-199a/214 cluster to regulate tumor survival and progression in human hepatocellular cancer. PLoS One 2012, 7, e31518. [Google Scholar]
- Bartel, D.P. MicroRNAs: target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Behrman, S.; Acosta-Alvear, D.; Walter, P. A CHOP-regulated microRNA controls rhodopsin expression. J. Cell Biol. 2011, 192, 919–927. [Google Scholar] [CrossRef]
- Deng, J.; Lu, P.D.; Zhang, Y.; Scheuner, D.; Kaufman, R.J.; Sonenberg, N.; Harding, H.P.; Ron, D. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol. Cell. Biol. 2004, 24, 10161–10168. [Google Scholar] [CrossRef]
- Jiang, H.Y.; Wek, S.A.; McGrath, B.C.; Scheuner, D.; Kaufman, R.J.; Cavener, D.R.; Wek, R.C. Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-kappaB in response to diverse cellular stresses. Mol. Cell. Biol. 2003, 23, 5651–5663. [Google Scholar] [CrossRef]
- Romero-Ramirez, L.; Cao, H.; Nelson, D.; Hammond, E.; Lee, A.-H.; Yoshida, H.; Mori, K.; Glimcher, L.H.; Denko, N.C.; Giaccia, A.J.; et al. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 2004, 64, 5943–5947. [Google Scholar] [CrossRef]
- Hay, R.T. SUMO: A history of modification. Mol. Cell 2005, 18, 1–12. [Google Scholar] [CrossRef]
- Lee, A.-H.; Iwakoshi, N.N.; Anderson, K.C.; Glimcher, L.H. Proteasome inhibitors disrupt the unfolded response in myeloma cells. Proc. Natl. Acad. Sci. 2003, 100, 9946–9951. [Google Scholar]
- Zhou, Y.; Lee, J.; Reno, C.M.; Sun, C.; Park, S.W.; Chung, J.; Lee, J.; Fisher, S.J.; White, M.F.; Biddinger, S.B.; et al. Regulation of glucose homeostasis through a XBP-1-FoxO1 interaction. Nat. Med. 2011, 17, 356–365. [Google Scholar] [CrossRef]
- Accili, D.; Arden, K.C. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell 2004, 117, 421–426. [Google Scholar] [CrossRef]
- Gross, D.N.; van den Heuvel, A.P.J.; Birnbaum, M.J. The role of FoxO in the regulation of metabolism. Oncogene 2008, 27, 2320–2336. [Google Scholar] [CrossRef]
- Yoshida, H.; Uemura, A.; Mori, K. pXBP1(u), a negative regulator of the unfolded protein response activator pXBP1(s), targets ATF6 but not ATF4 in proteasome-mediated degradation. Cell Struct. Funct. 2009, 34, 1–10. [Google Scholar] [CrossRef]
- Newman, J.R.S.; Keating, A.E. Comprehensive identification of human bZIP interactions with coiled-coil arrays. Science 2003, 300, 2097–2101. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef]
- Koong, A.C.; Chauhan, V.; Romero-Ramirez, L. Targeting XBP-1 as a novel anti-cancer strategy. Cancer Biol. Ther. 2006, 5, 756–759. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Byrd, A.E.; Brewer, J.W. Intricately Regulated: A Cellular Toolbox for Fine-Tuning XBP1 Expression and Activity. Cells 2012, 1, 738-753. https://doi.org/10.3390/cells1040738
Byrd AE, Brewer JW. Intricately Regulated: A Cellular Toolbox for Fine-Tuning XBP1 Expression and Activity. Cells. 2012; 1(4):738-753. https://doi.org/10.3390/cells1040738
Chicago/Turabian StyleByrd, Andrew E., and Joseph W. Brewer. 2012. "Intricately Regulated: A Cellular Toolbox for Fine-Tuning XBP1 Expression and Activity" Cells 1, no. 4: 738-753. https://doi.org/10.3390/cells1040738