
Pathogenetic Mechanisms Underlying Spinocerebellar Ataxia Type 3 Are Altered in Primary Oligodendrocyte Culture


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Mice
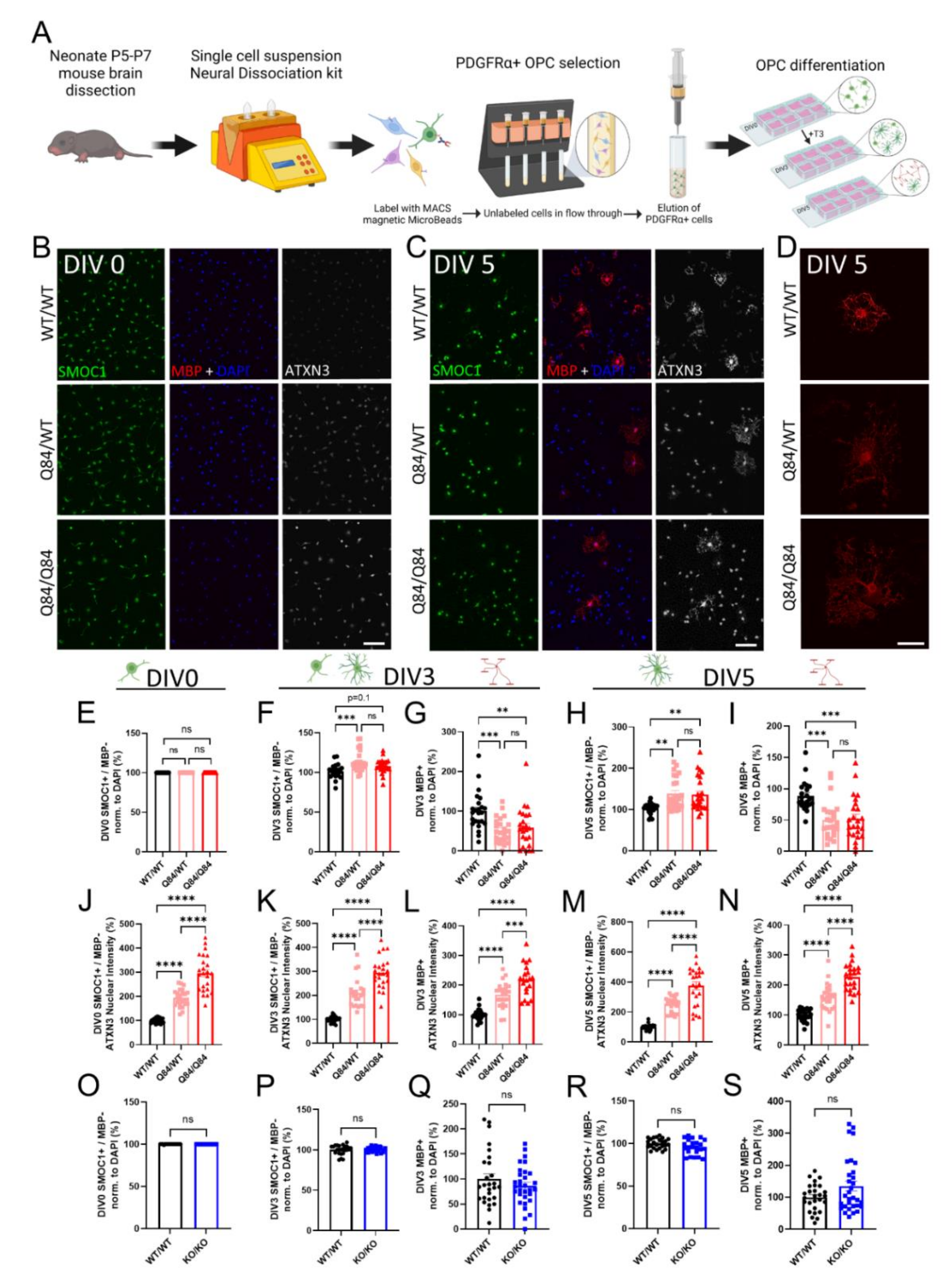
2.2. OPC Isolation and Culture
2.3. Immunocytochemistry
2.4. Image Analysis
2.5. Statistics
3. Results
3.1. Cell Autonomous SCA3 Oligodendrocyte Maturation Is Impaired via a Toxic Gain-of-Function Mechanism
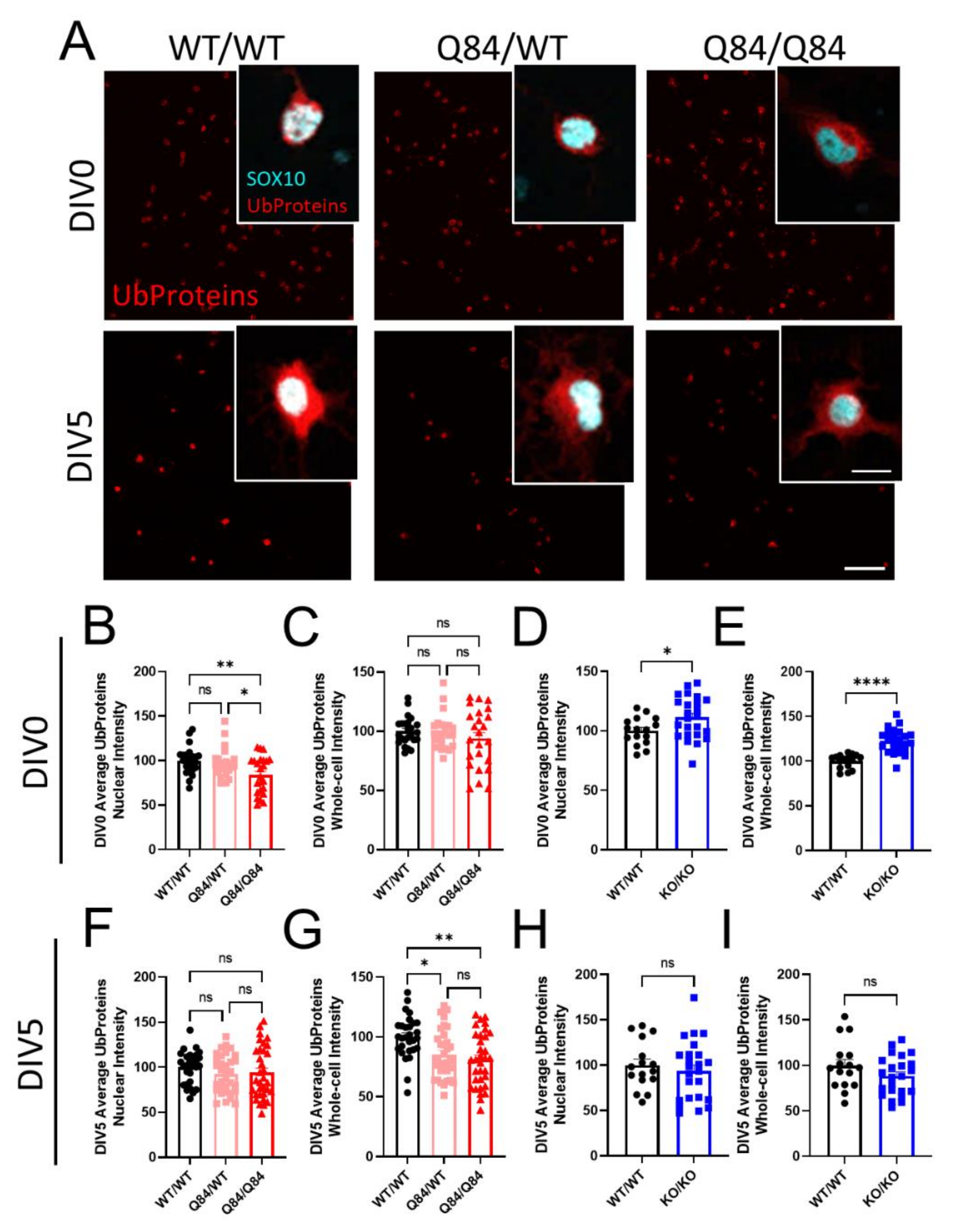
3.2. Protein Ubiquitination Is Dysregulated in Both SCA3 and Atxn3-KO Oligodendrocytes
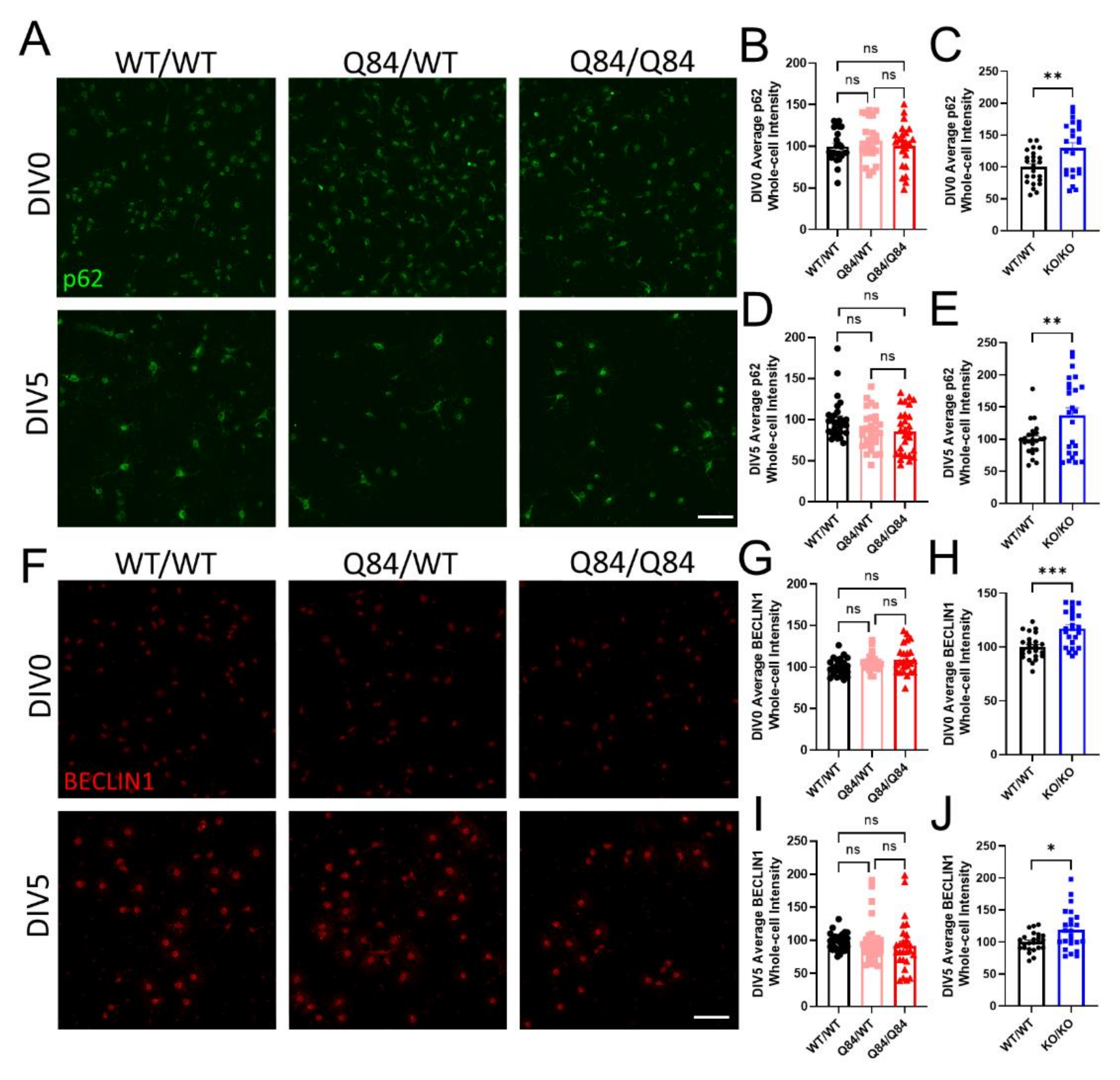
3.3. Autophagy Is Not Affected in SCA3 Oligodendrocytes
3.4. DNA Damage Does Not Play a Role in SCA3 Oligodendrocyte Maturation Impairments
3.5. Histone Methylation Is Impaired in Proliferating and Maturing SCA3 Oligodendrocytes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seidel, K.; Siswanto, S.; Brunt, E.R.P.; Dunnen, W.D.; Korf, H.; Rueb, U. Brain pathology of spinocerebellar ataxias. Acta Neuropathol. 2012, 124, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Rüb, U.; Schöls, L.; Paulson, H.; Auburger, G.; Kermer, P.; Jen, J.C.; Seidel, K.; Korf, H.; Deller, T. Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog. Neurobiol. 2013, 104, 38–66. [Google Scholar] [CrossRef] [PubMed]
- Paulson, H.L.; Shakkottai, V.G.; Clark, H.B.; Orr, H. Polyglutamine spinocerebellar ataxias—From genes to potential treatments. Nat. Rev. Neurosci. 2017, 18, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Paulson, H.L. Dominantly inherited ataxias: Lessons learned from Machado-Joseph disease/spinocerebellar ataxia type 3. Skull Base 2007, 27, 133–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciel, P.; Gaspar, C.; DeStefano, A.L.; Silveira, I.; Coutinho, P.; Radvany, J.; Dawson, D.M.; Sudarsky, L.; Guimarães, J.; E Loureiro, J. Correlation between CAG repeat length and clinical features in Machado-Joseph disease. Am. J. Hum. Genet. 1995, 57, 54–61. [Google Scholar] [PubMed]
- Maruyama, H.; Nakamura, S.; Matsuyama, Z.; Sakai, T.; Doyu, M.; Sobue, G.; Seto, M.; Tsujihata, M.; Oh-I, T.; Nishio, T.; et al. Molecular features of the CAG repeats and clinical manifestation of Machado--Joseph disease. Hum. Mol. Genet. 1995, 4, 807–812. [Google Scholar] [CrossRef]
- Durr, A.; Stevanin, G.; Ms, G.C.; Duyckaerts, C.; Abbas, N.; Bs, O.D.; Chneiweiss, H.; Benomar, A.; Lyon-Caen, O.; Julien, J.; et al. Spinocerebellar ataxia 3 and machado-joseph disease: Clinical, molecular, and neuropathological features. Ann. Neurol. 1996, 39, 490–499. [Google Scholar] [CrossRef]
- Durr, A. Autosomal dominant cerebellar ataxias: Polyglutamine expansions and beyond. Lancet Neurol. 2010, 9, 885–894. [Google Scholar] [CrossRef]
- Gardiner, S.L.; Boogaard, M.W.; Trompet, S.; De Mutsert, R.; Rosendaal, F.R.; Gussekloo, J.; Jukema, J.W.; Roos, R.A.C.; Aziz, N.A. Prevalence of carriers of intermediate and pathological polyglutamine disease–Associated alleles among large population-based cohorts. JAMA Neurol. 2019, 76, 650–656. [Google Scholar] [CrossRef]
- McLoughlin, H.S.; Moore, L.R.; Paulson, H.L. Pathogenesis of SCA3 and implications for other polyglutamine diseases. Neurobiol. Dis. 2019, 134, 104635. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Goto, J.; Hattori, M.; Toyoda, A.; Ishii, K.; Jeong, S.-Y.; Hashida, H.; Masuda, N.; Ogata, K.; Kasai, F.; et al. The genomic structure and expression of MJD, the Machado-Joseph disease gene. J. Hum. Genet. 2001, 46, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Costa Mdo, C.; Paulson, H.L. Toward understanding Machado-Joseph disease. Prog. Neurobiol. 2012, 97, 239–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulson, H.; Perez, M.; Trottier, Y.; Trojanowski, J.; Subramony, S.; Das, S.; Vig, P.; Mandel, J.-L.; Fischbeck, K.; Pittman, R. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron 1997, 19, 333–344. [Google Scholar] [CrossRef] [Green Version]
- Rüb, U.; Brunt, E.R.; Deller, T. New insights into the pathoanatomy of spinocerebellar ataxia type 3 (Machado–Joseph disease). Curr. Opin. Neurol. 2008, 21, 111–116. [Google Scholar] [CrossRef]
- Lukas, C.; Schöls, L.; Bellenberg, B.; Rüb, U.; Przuntek, H.; Schmid, G.; Köster, O.; Suchan, B. Dissociation of grey and white matter reduction in spinocerebellar ataxia type 3 and 6: A voxel-based morphometry study. Neurosci. Lett. 2006, 408, 230–235. [Google Scholar] [CrossRef]
- D’Abreu, A.; Jr, M.F.; Appenzeller, S.; Lopes-Cendes, I.; Cendes, F. Axonal dysfunction in the deep white matter in Machado-Joseph disease. J. Neuroimaging 2009, 19, 9–12. [Google Scholar] [CrossRef]
- Guimaraes, R.P.; D’Abreu, A.; Yasuda, C.L.; Franca, M.C., Jr.; Silva, B.H.; Cappabianco, F.A.; Bergo, F.P.; Lopes-Cendes, I.T.; Cendes, F. A multimodal evaluation of microstructural white matter damage in spinocerebellar ataxia type 3. Mov. Disord. 2013, 28, 1125–1132. [Google Scholar] [CrossRef]
- Kang, J.-S.; Klein, J.C.; Baudrexel, S.; Deichmann, R.; Nolte, D.; Hilker, R. White matter damage is related to ataxia severity in SCA3. J. Neurol. 2013, 261, 291–299. [Google Scholar] [CrossRef]
- Rezende, T.J.R.; De Paiva, J.L.R.; Martinez, A.R.M.; Lopes-Cendes, I.; Pedroso, J.L.; Barsottini, O.G.P.; Cendes, F.; França, M.C., Jr. Structural signature of SCA3: From presymptomatic to late disease stages. Ann. Neurol. 2018, 84, 401–408. [Google Scholar] [CrossRef]
- Schuster, K.H.; Zalon, A.J.; Zhang, H.; DiFranco, D.M.; Stec, N.R.; Haque, Z.; Blumenstein, K.G.; Pierce, A.M.; Guan, Y.; Paulson, H.L.; et al. Impaired oligodendrocyte maturation is an early feature in SCA3 disease pathogenesis. J. Neurosci. 2022, 42, 1604–1617. [Google Scholar] [CrossRef] [PubMed]
- Philips, T.; Bento-Abreu, A.; Nonneman, A.; Haeck, W.; Staats, K.; Geelen, V.; Hersmus, N.; Küsters, B.; Van Den Bosch, L.; Van Damme, P.; et al. Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis. Brain 2013, 136 (Pt 2), 471–482. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Misra Sen, J.; Gorospe, M.; et al. Senolytic therapy alleviates Abeta-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Errea, O.; Rodriguez-Oroz, M.C. Oligodendrocytes, a new player in the etiology of Parkinson’s disease. Mov. Disord. 2021, 36, 83. [Google Scholar] [CrossRef] [PubMed]
- Ettle, B.; Schlachetzki, J.C.M.; Winkler, J. Oligodendroglia and myelin in neurodegenerative diseases: More than just bystanders? Mol. Neurobiol. 2016, 53, 3046–3062. [Google Scholar] [CrossRef] [Green Version]
- Bardile, C.F.; Garcia-Miralles, M.; Caron, N.S.; Rayan, N.A.; Langley, S.R.; Harmston, N.; Rondelli, A.M.; Teo, R.T.Y.; Waltl, S.; Anderson, L.M.; et al. Intrinsic mutant HTT-mediated defects in oligodendroglia cause myelination deficits and behavioral abnormalities in Huntington disease. Proc. Natl. Acad. Sci. USA 2019, 116, 9622–9627. [Google Scholar] [CrossRef] [Green Version]
- Kenigsbuch, M.; Bost, P.; Halevi, S.; Chang, Y.; Chen, S.; Ma, Q.; Hajbi, R.; Schwikowski, B.; Bodenmiller, B.; Fu, H.; et al. A shared disease-associated oligodendrocyte signature among multiple CNS pathologies. Nat. Neurosci. 2022, 25, 876–886. [Google Scholar] [CrossRef]
- Kang, S.H.; Li, Y.; Fukaya, M.; Lorenzini, I.; Cleveland, D.; Ostrow, L.; Rothstein, J.D.; E Bergles, D. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat. Neurosci. 2013, 16, 571–579. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Wei, W.; Wang, G.; Gaertig, M.A.; Feng, Y.; Wang, W.; Li, X.-J.; Li, S. Mutant huntingtin downregulates myelin regulatory factor-mediated myelin gene expression and affects mature oligodendrocytes. Neuron 2015, 85, 1212–1226. [Google Scholar] [CrossRef] [Green Version]
- Osipovitch, M.; Martinez, A.A.; Mariani, J.N.; Cornwell, A.; Dhaliwal, S.; Zou, L.; Chandler-Militello, D.; Wang, S.; Li, X.; Benraiss, S.-J.; et al. Human ESC-derived chimeric mouse models of Huntington’s disease reveal cell-intrinsic defects in glial progenitor cell differentiation. Cell Stem Cell 2018, 24, 107–122.e7. [Google Scholar] [CrossRef] [Green Version]
- Benraiss, A.; Wang, S.; Herrlinger, S.; Li, X.; Chandler-Militello, D.; Mauceri, J.; Burm, H.B.; Toner, M.; Osipovitch, M.; Xu, Q.J. Human glia can both induce and rescue aspects of disease phenotype in Huntington disease. Nat. Commun. 2016, 7, 11758. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.R.; Rajpal, G.; Dillingham, I.T.; Qutob, M.; Blumenstein, K.G.; Gattis, D.; Hung, G.; Kordasiewicz, H.B.; Paulson, H.L.; McLoughlin, H.S. Evaluation of antisense oligonucleotides targeting ATXN3 in SCA3 mouse models. Mol. Ther. Nucleic Acids 2017, 7, 200–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLoughlin, H.S.; Moore, L.R.; Chopra, R.; Komlo, R.; McKenzie, M.; Blumenstein, K.G.; Zhao, H.; Kordasiewicz, H.B.; Shakkottai, V.G.; Paulson, H.L. Oligonucleotide therapy mitigates disease in spinocerebellar ataxia type 3 mice. Ann. Neurol. 2018, 84, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Reina, C.P.; Nabet, B.Y.; Young, P.D.; Pittman, R.N. Basal and stress-induced Hsp70 are modulated by ataxin-3. Cell Stress Chaperon 2012, 17, 729–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, T.R.; Kang, I.H.; Wheeler, D.B.; A Lindquist, R.; Papallo, A.; Sabatini, D.M.; Golland, P.; E Carpenter, A. CellProfiler Analyst: Data exploration and analysis software for complex image-based screens. BMC Bioinform. 2008, 9, 482. [Google Scholar] [CrossRef] [Green Version]
- Schuster, K.H.; Zalon, A.J.; DiFranco, D.M.; Putka, A.F.; Stec, N.; Jarrah, S.; Naeem, A.; Haque, Z.; Zhang, H.; Guan, Y.; et al. ASOs are an effective treatment for disease-associated oligodendrocyte signatures in premanifest and symptomatic SCA3 mice. bioRxiv 2022. [Google Scholar] [CrossRef]
- Dugas, J.C.; Tai, Y.C.; Speed, T.P.; Ngai, J.; Barres, B.A. Functional genomic analysis of oligodendrocyte differentiation. J. Neurosci. 2006, 26, 10967–10983. [Google Scholar] [CrossRef]
- Warrington, A.E.; Pfeiffer, S.E. Proliferation and differentiation of O4+ oligodendrocytes in postnatal rat cerebellum: Analysis in unfixed tissue slices using anti-glycolipid antibodies. J. Neurosci. Res. 1992, 33, 338–353. [Google Scholar] [CrossRef]
- Spitzer, S.O.; Sitnikov, S.; Kamen, Y.; Evans, K.A.; Kronenberg-Versteeg, D.; Dietmann, S.; de Faria, O.; Agathou, S.; Káradóttir, R.T. Oligodendrocyte progenitor cells become regionally diverse and heterogeneous with age. Neuron 2019, 101, 459–471.e5. [Google Scholar] [CrossRef] [Green Version]
- Hughes, E.G.; Stockton, M.E. Premyelinating oligodendrocytes: Mechanisms underlying cell survival and integration. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Richter-Landsberg, C. The cytoskeleton in oligodendrocytes. Microtubule dynamics in health and disease. J. Mol. Neurosci. 2008, 35, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Barateiro, A.; Fernandes, A. Temporal oligodendrocyte lineage progression: In vitro models of proliferation, differentiation and myelination. Biochim. Biophys. Acta 2014, 1843, 1917–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadelmann, C.; Timmler, S.; Barrantes-Freer, A.; Simons, M. Myelin in the central nervous system: Structure, function, and pathology. Physiol. Rev. 2019, 99, 1381–1431. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin–proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashkenazi, A.; Bento, C.F.; Ricketts, T.; Vicinanza, M.; Siddiqi, F.; Pavel, M.; Squitieri, F.; Hardenberg, M.; Imarisio, S.; Menzies, F.M.; et al. Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature 2017, 545, 108–111. [Google Scholar] [CrossRef] [Green Version]
- Nascimento-Ferreira, I.; Ferreira, T.; Sousa-Ferreira, L.; Auregan, G.; Onofre, I.; Alves, S.; Dufour, N.; Gould, V.F.C.; Koeppen, A.; Déglon, N.; et al. Overexpression of the autophagic beclin-1 protein clears mutant ataxin-3 and alleviates Machado–Joseph disease. Brain 2011, 134, 1400–1415. [Google Scholar] [CrossRef]
- Herzog, L.K.; Kevei, E.; Marchante, R.; Böttcher, C.; Bindesbøll, C.; Lystad, A.H.; Pfeiffer, A.; Gierisch, M.E.; Salomons, F.A.; Simonsen, A.; et al. The Machado–Joseph disease deubiquitylase ataxin-3 interacts with LC3C/GABARAP and promotes autophagy. Aging Cell 2019, 19, e13051. [Google Scholar] [CrossRef]
- Tse, K.-H.; Herrup, K. DNA damage in the oligodendrocyte lineage and its role in brain aging. Mech. Ageing Dev. 2016, 161, 37–50. [Google Scholar] [CrossRef] [Green Version]
- French, H.M.; Reid, M.; Mamontov, P.; Simmons, R.A.; Grinspan, J.B. Oxidative stress disrupts oligodendrocyte maturation. J. Neurosci. Res. 2009, 87, 3076–3087. [Google Scholar] [CrossRef] [Green Version]
- Gao, R.; Liu, Y.; Silva-Fernandes, A.; Fang, X.; Paulucci-Holthauzen, A.; Chatterjee, A.; Zhang, H.L.; Matsuura, T.; Choudhary, S.; Ashizawa, T.; et al. Inactivation of PNKP by mutant ATXN3 triggers apoptosis by activating the DNA damage-response pathway in SCA3. PLoS Genet. 2015, 11, e1004834. [Google Scholar] [CrossRef] [Green Version]
- Gao, R.; Chakraborty, A.; Geater, C.; Pradhan, S.; Gordon, K.L.; Snowden, J.; Yuan, S.; Dickey, A.; Choudhary, S.; Ashizawa, T.; et al. Mutant huntingtin impairs PNKP and ATXN3, disrupting DNA repair and transcription. eLife 2019, 8, e42988. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, T.; Suart, C.E.; Hung, C.L.K.; Graham, K.J.; Bazan, C.A.B.; Truant, R. DNA damage repair in Huntington’s disease and other neurodegenerative diseases. Neurotherapeutics 2019, 16, 948–956. [Google Scholar] [CrossRef] [PubMed]
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. ATM Phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001, 276, 42462–42467. [Google Scholar] [CrossRef] [Green Version]
- Sher, F.; Rößler, R.; Brouwer, N.; Balasubramaniyan, V.; Boddeke, E.; Copray, S. Differentiation of neural stem cells into oligodendrocytes: Involvement of the polycomb group protein Ezh2. Stem Cells 2008, 26, 2875–2883. [Google Scholar] [CrossRef] [PubMed]
- Berry, K.; Wang, J.; Lu, Q.R. Epigenetic regulation of oligodendrocyte myelination in developmental disorders and neurodegenerative diseases. F1000Research 2020, 9, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Magri, L.; Zhang, F.; Marsh, N.O.; Albrecht, S.; Huynh, J.L.; Kaur, J.; Kuhlmann, T.; Zhang, W.; Slesinger, P.A.; et al. Chromatin landscape defined by repressive histone methylation during oligodendrocyte differentiation. J. Neurosci. 2015, 35, 352–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramani, B.; Panwar, B.; Moore, L.R.; Wang, B.; Huang, R.; Guan, Y.; Paulson, H.L. Comparison of spinocerebellar ataxia type 3 mouse models identifies early gain-of-function, cell-autonomous transcriptional changes in oligodendrocytes. Hum. Mol. Genet. 2017, 26, 3362–3374. [Google Scholar] [CrossRef] [Green Version]
- Costa, M.D.C.; Radzwion, M.; McLoughlin, H.S.; Ashraf, N.S.; Fischer, S.; Shakkottai, V.G.; Maciel, P.; Paulson, H.L.; Öz, G. In vivo molecular signatures of cerebellar pathology in spinocerebellar ataxia type 3. Mov. Disord. 2020, 35, 1774–1786. [Google Scholar] [CrossRef]
- Haas, E.; Incebacak, R.D.; Hentrich, T.; Huridou, C.; Schmidt, T.; Casadei, N.; Maringer, Y.; Bahl, C.; Zimmermann, F.; Mills, J.D.; et al. A Novel SCA3 knock-in mouse model mimics the human SCA3 disease phenotype including neuropathological, behavioral, and transcriptional abnormalities especially in oligodendrocytes. Mol. Neurobiol. 2021, 59, 495–522. [Google Scholar] [CrossRef]
- Rodrigues, A.-J.; Costa, M.D.C.; Silva, T.-L.; Ferreira, D.; Bajanca, F.; Logarinho, E.; Maciel, P. Absence of ataxin-3 leads to cytoskeletal disorganization and increased cell death. Biochim. Biophys. Acta 2010, 1803, 1154–1163. [Google Scholar] [CrossRef] [Green Version]
- Wiatr, K.; Piasecki, P.; Marczak, L.; Wojciechowski, P.; Kurkowiak, M.; Ploski, R.; Rydzanicz, M.; Handschuh, L.; Jungverdorben, J.; Brustle, O.; et al. Altered levels of proteins and phosphoproteins, in the absence of early causative transcriptional changes, shape the molecular pathogenesis in the brain of young presymptomatic Ki91 SCA3/MJD mouse. Mol. Neurobiol. 2019, 56, 8168–8202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seiberlich, V.; Bauer, N.G.; Schwarz, L.; Ffrench-Constant, C.; Goldbaum, O.; Richter-Landsberg, C. Downregulation of the microtubule associated protein Tau impairs process outgrowth and myelin basic protein mRNA transport in oligodendrocytes. Glia 2015, 63, 1621–1635. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, M.-M.; McAlear, T.S.; Nguyen, H.; Oses-Prieto, J.A.; Valenzuela, A.; Shi, R.D.; Perrino, J.J.; Huang, T.-T.; Burlingame, A.L.; Bechstedt, S.; et al. The Golgi outpost protein TPPP nucleates microtubules and is critical for myelination. Cell 2019, 179, 132–146.e14. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Pan, L.; Tsai, L.-H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, A.; Saha, S.; Chakraborty, A.; Silva-Fernandes, A.; Mandal, S.M.; Carvalho, A.; Liu, Y.; Pandita, R.K.; Hegde, M.; Hegde, P.M.; et al. The role of the mammalian DNA end-processing enzyme polynucleotide kinase 3’-phosphatase in spinocerebellar ataxia type 3 pathogenesis. PLoS Genet. 2015, 11, e1004749. [Google Scholar] [CrossRef] [Green Version]
- Kuo, L.J.; Yang, L.-X. Gamma-H2AX—A novel biomarker for DNA double-strand breaks. In Vivo 2008, 22, 305–309. [Google Scholar]
- Thorburne, S.K.; Juurlink, B.H.J. Low glutathione and high iron govern the susceptibility of oligodendroglial precursors to oxidative stress. J. Neurochem. 2002, 67, 1014–1022. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Host | Dilution | Company | Catalog # |
|---|---|---|---|---|
| ATXN3 (1H9) | Mouse IgG1 | 1:500 | Millipore | MAB5360 |
| SMOC1 | Rabbit | 1:100 | ThermoFisher | PA5-31392 |
| MBP | Rat | 1:1000 | Abcam | Ab7349 |
| SQSTM1/p62 | Mouse IgG2a | 1:200 | Abnova | 89-015-843 |
| BECLIN1 | Rabbit | 1:100 | Abcam | Ab207612 |
| SOX10 | Mouse IgG1 | 1:100 | Santa Cruz Biotechnology | sc-365692 |
| SOX10 | Rabbit | 1:500 | Cell Signaling | 893565 |
| Ubiquitinated Proteins | Mouse IgM | 1:500 | Millipore Sigma | 04-262 |
| Ki-67 | Rat | 1:300 | ThermoFisher | 14-5698-82 |
| H3K27me3 | Rabbit | 1:500 | Millipore | ABE44 |
| H3K9me3 | Rabbit | 1:500 | Abcam | ab8898 |
| γ-H2AX | Mouse IgG1 | 1:500 | Millipore | 05-636 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schuster, K.H.; Putka, A.F.; McLoughlin, H.S. Pathogenetic Mechanisms Underlying Spinocerebellar Ataxia Type 3 Are Altered in Primary Oligodendrocyte Culture. Cells 2022, 11, 2615. https://doi.org/10.3390/cells11162615
Schuster KH, Putka AF, McLoughlin HS. Pathogenetic Mechanisms Underlying Spinocerebellar Ataxia Type 3 Are Altered in Primary Oligodendrocyte Culture. Cells. 2022; 11(16):2615. https://doi.org/10.3390/cells11162615
Chicago/Turabian StyleSchuster, Kristen H., Alexandra F. Putka, and Hayley S. McLoughlin. 2022. "Pathogenetic Mechanisms Underlying Spinocerebellar Ataxia Type 3 Are Altered in Primary Oligodendrocyte Culture" Cells 11, no. 16: 2615. https://doi.org/10.3390/cells11162615