



Cells 2023, 12(10), 1404; https://doi.org/10.3390/cells12101404 - 17 May 2023
Cited by 5 | Viewed by 1799
Abstract
►
Show Figures
Machado-Joseph disease (MJD) is a dominant neurodegenerative disease caused by an expanded CAG repeat in the ATXN3 gene encoding the ataxin-3 protein. Several cellular processes, including transcription and apoptosis, are disrupted in MJD. To gain further insights into the extent of dysregulation of
[...] Read more.
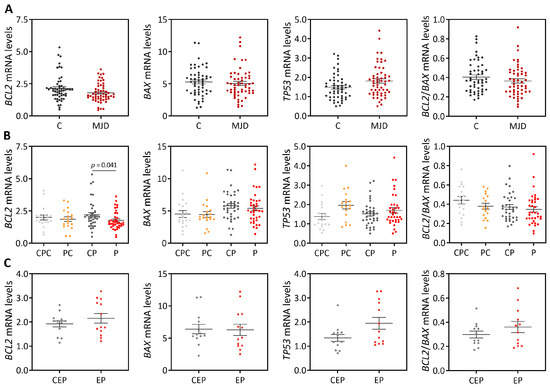
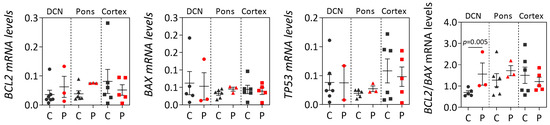
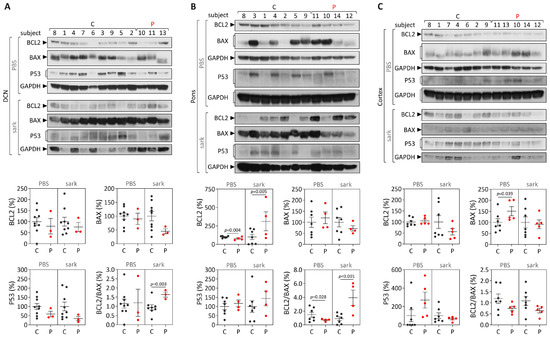
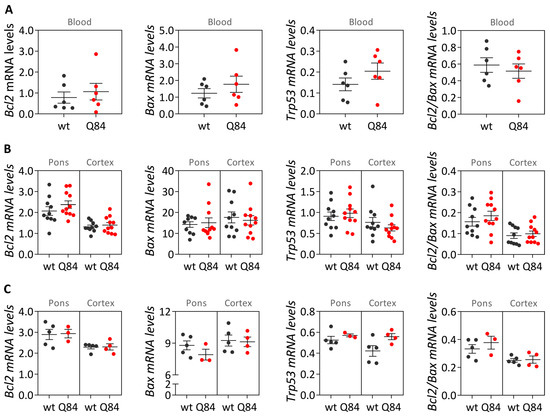
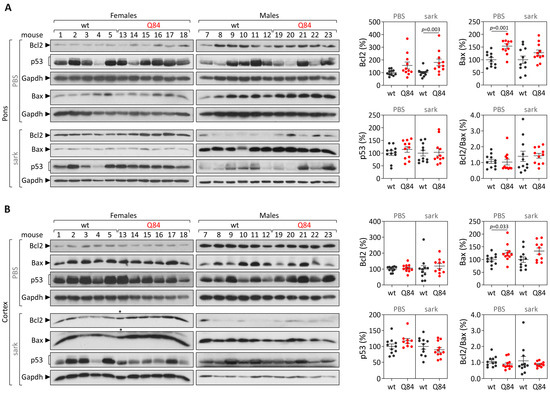
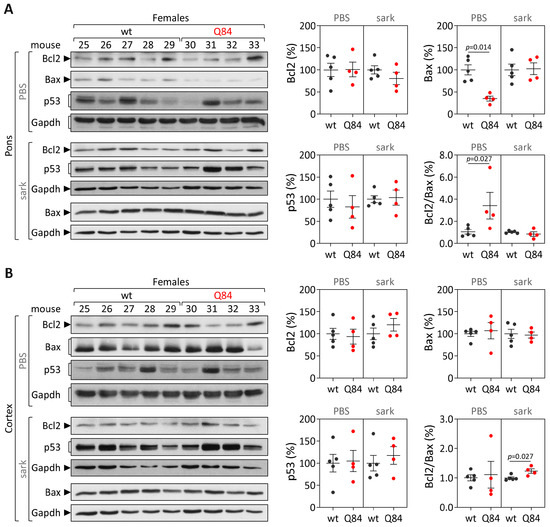
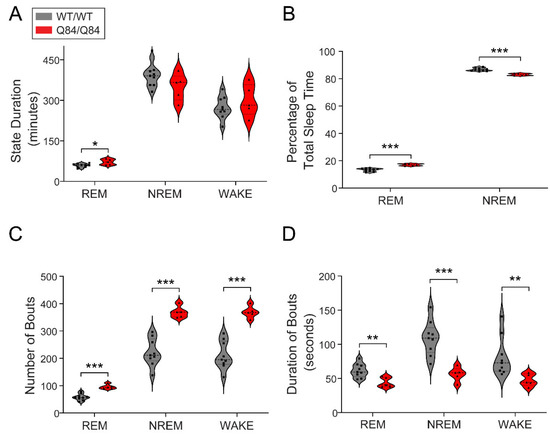
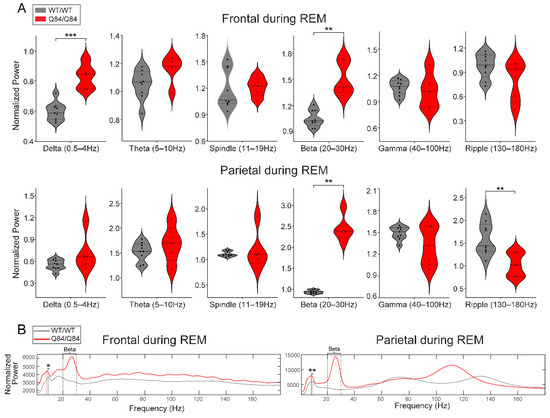
Machado-Joseph disease (MJD) is a dominant neurodegenerative disease caused by an expanded CAG repeat in the ATXN3 gene encoding the ataxin-3 protein. Several cellular processes, including transcription and apoptosis, are disrupted in MJD. To gain further insights into the extent of dysregulation of mitochondrial apoptosis in MJD and to evaluate if expression alterations of specific apoptosis genes/proteins can be used as transcriptional biomarkers of disease, the expression levels of BCL2, BAX and TP53 and the BCL2/BAX ratio (an indicator of susceptibility to apoptosis) were assessed in blood and post-mortem brain samples from MJD subjects and MJD transgenic mice and controls. While patients show reduced levels of blood BCL2 transcripts, this measurement displays low accuracy to discriminate patients from matched controls. However, increased levels of blood BAX transcripts and decreased BCL2/BAX ratio are associated with earlier onset of disease, indicating a possible association with MJD pathogenesis. Post-mortem MJD brains show increased BCL2/BAX transcript ratio in the dentate cerebellar nucleus (DCN) and increased BCL2/BAX insoluble protein ratio in the DCN and pons, suggesting that in these regions, severely affected by degeneration in MJD, cells show signs of apoptosis resistance. Interestingly, a follow-up study of 18 patients further shows that blood BCL2 and TP53 transcript levels increase over time in MJD patients. Furthermore, while the similar levels of blood BCL2, BAX, and TP53 transcripts observed in preclinical subjects and controls is mimicked by pre-symptomatic MJD mice, the expression profile of these genes in patient brains is partially replicated by symptomatic MJD mice. Globally, our findings indicate that there is tissue-specific vulnerability to apoptosis in MJD subjects and that this tissue-dependent behavior is partially replicated in a MJD mouse model.
Full article
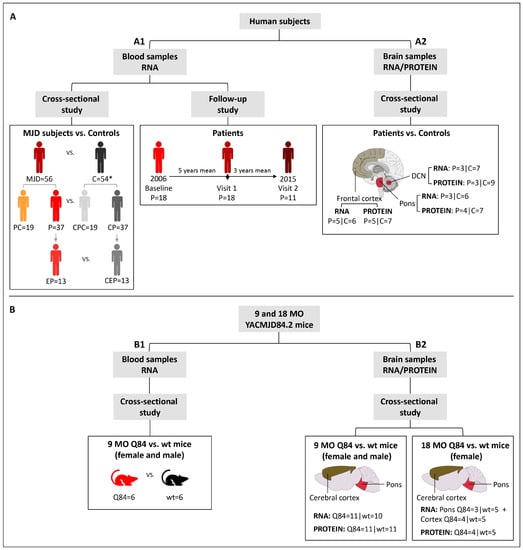
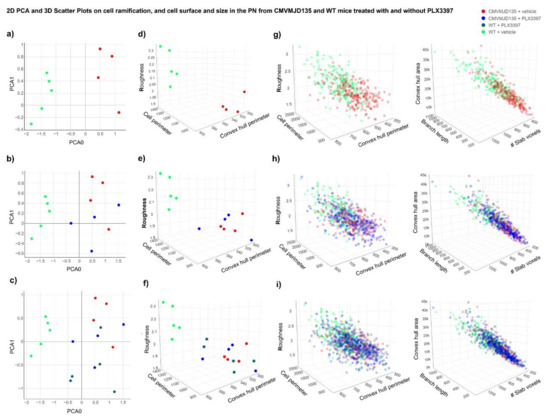
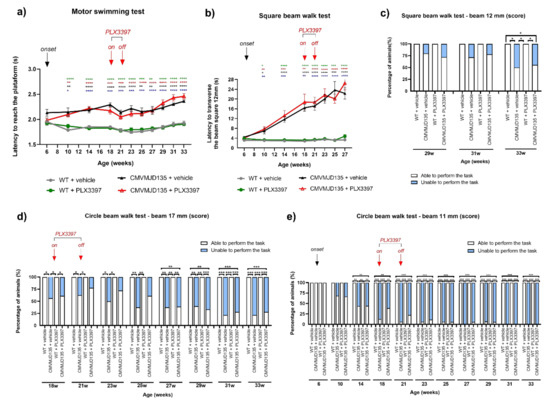
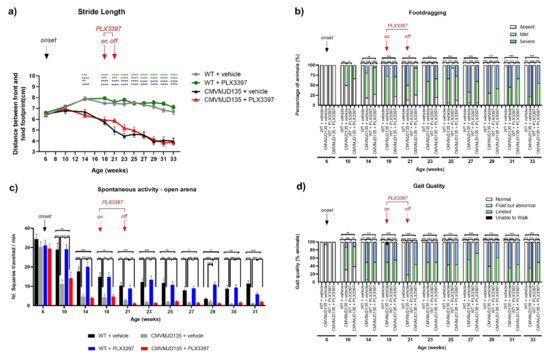
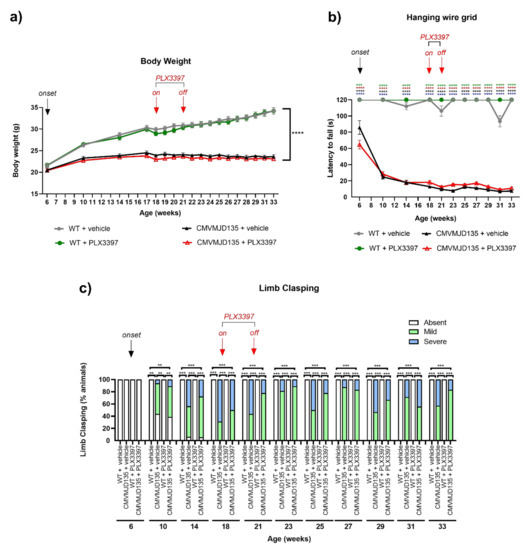
Figure 1


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}