
Loss of Flocculus Purkinje Cell Firing Precision Leads to Impaired Gaze Stabilization in a Mouse Model of Spinocerebellar Ataxia Type 6 (SCA6)




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
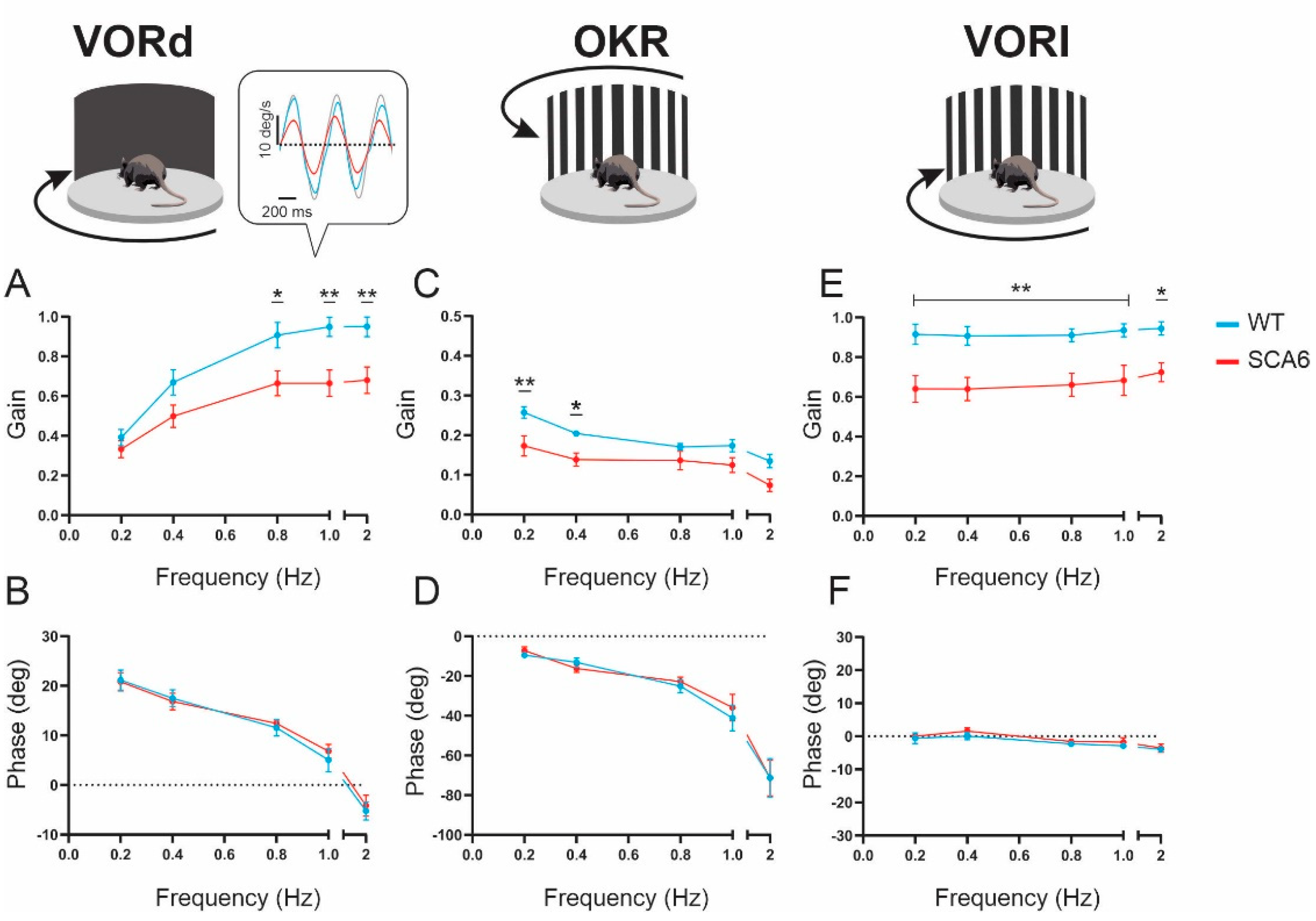
2.1. VOR and OKR Gains Are Reduced in SCA684Q Mice
2.2. Nystagmus Dynamics Are Comparable between Wild-Type and SCA684Q Mice
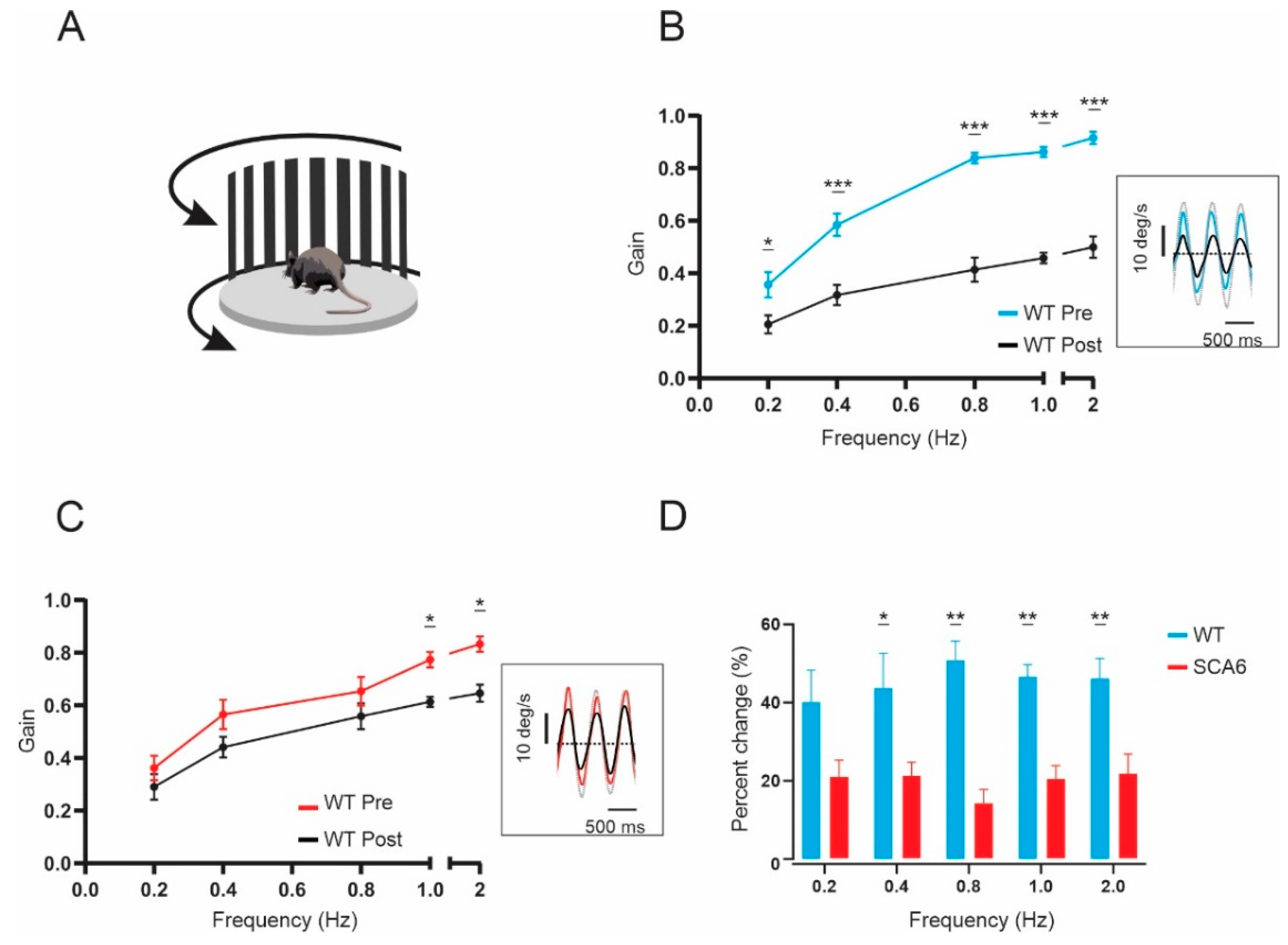
2.3. VOR Learning Is Impaired in SCA684Q Mice
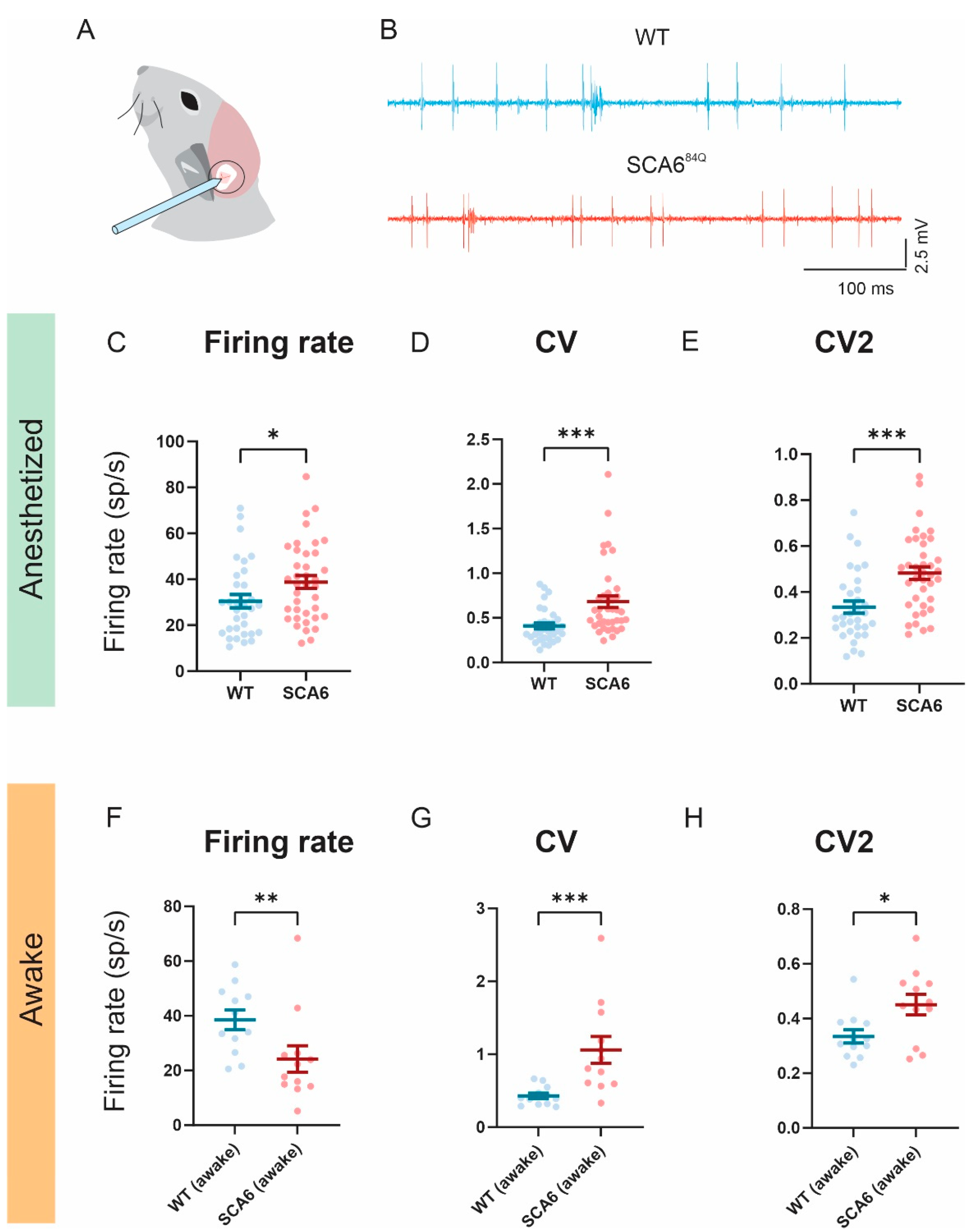
2.4. Firing Precision Is Altered in SCA684Q Mice
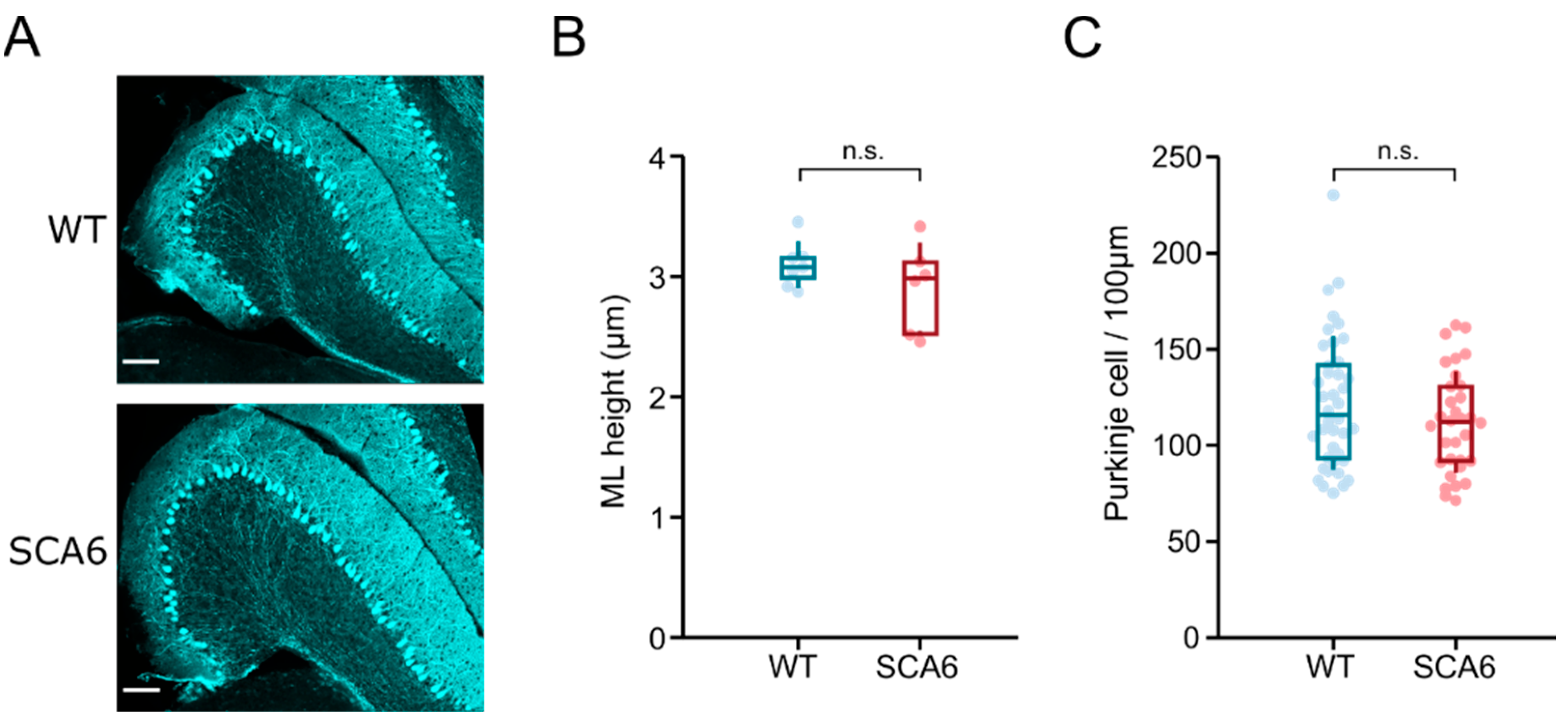
2.5. Flocculus Morphology Appears Normal in SCA684Q Mice
3. Discussion
3.1. Comparison of Oculomotor Deficits in SCA6 Patients and a Mouse Model of SCA6
3.2. Implication of the P/Q Channel Mutation on Cerebellar-Dependent Behaviors
3.3. Morphological Changes in the Purkinje Cells of Anterior Vermis and Flocculus in SCA684Q Mice
3.4. Clinical Implications
4. Methods
4.1. Animals
4.2. Quantification of VOR and OKR
4.3. In Vivo Single-Unit Recording Data Acquisition and Analysis
4.4. Immunohistochemistry
4.5. Image Acquisition and Analysis
4.6. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Klockgether, T.; Mariotti, C.; Paulson, H.L. Spinocerebellar ataxia. Nat. Rev. Dis. Primers 2019, 5, 24. [Google Scholar] [PubMed]
- Gomez, C.M.; Thompson, R.M.; Gammack, J.T.; Perlman, S.L.; Dobyns, W.B.; Truwit, C.L.; Zee, D.S.; Clark, H.B.; Anderson, J.H. Spinocerebellar ataxia type 6: Gaze-evoked and vertical nystagmus, Purkinje cell degeneration, and variable age of onset. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1997, 42, 933–950. [Google Scholar] [CrossRef] [PubMed]
- Schöls, L.; Krüger, R.; Amoiridis, G.; Przuntek, H.; Epplen, J.T.; Riess, O. Spinocerebellar ataxia type 6: Genotype and phenotype in German kindreds. J. Neurol. Neurosurg. Psychiatry 1998, 64, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Watanabe, M.; Yoshizawa, K.; Fujita, T.; Iwamoto, H.; Yoshizawa, T.; Haradae, K.; Nakamagoea, K.; Komatsuzakid, Y.; Satoh, A. Clinical, neuropathological, and molecular study in two families with spinocerebellar ataxia type 6 (SCA6). J. Neurol. Neurosurg. Psychiatry 1999, 67, 86–89. [Google Scholar] [CrossRef]
- Sasaki, H.; Kojima, H.; Yabe, I.; Tashiro, K.; Hamada, T.; Sawa, H.; Hiraga, H.; Nagashima, K. Neuropathological and molecular studies of spinocerebellar ataxia type 6 (SCA6). Acta Neuropathol. 1998, 95, 199–204. [Google Scholar] [CrossRef]
- Yang, Q.; Hashizume, Y.; Yoshida, M.; Wang, Y.; Goto, Y.; Mitsuma, N.; Ishikawa, K.; Mizusawa, H. Morphological Purkinje cell changes in spinocerebellar ataxia type 6. Acta Neuropathol. 2000, 100, 371–376. [Google Scholar] [CrossRef]
- Murata, Y.; Kawakami, H.; Yamaguchi, S.; Nishimura, M.; Kohriyama, T.; Ishizaki, F.; Matsuyama, Z.; Mimori, Y.; Nakamura, S. Characteristic magnetic resonance imaging findings in spinocerebellar ataxia 6. Arch. Neurol. 1998, 55, 1348–1352. [Google Scholar] [CrossRef]
- Satoh, J.I.; Tokumoto, H.; Yukitake, M.; Matsui, M.; Matsuyama, Z.; Kawakami, H.; Nakamura, S.; Kuroda, Y. Spinocerebellar ataxia type 6: MRI of three Japanese patients. Neuroradiology 1998, 40, 222–227. [Google Scholar] [CrossRef]
- Butteriss, D.; Chinnery, P.; Birchall, D. Radiological characterization of spinocerebellar ataxia type 6. Br. J. Radiol. 2005, 78, 694–696. [Google Scholar] [CrossRef]
- Lukas, C.; Schöls, L.; Bellenberg, B.; Rüb, U.; Przuntek, H.; Schmid, G.; Köster, O.; Suchan, B. Dissociation of grey and white matter reduction in spinocerebellar ataxia type 3 and 6: A voxel-based morphometry study. Neurosci. Lett. 2006, 408, 230–235. [Google Scholar]
- Solodkin, A.; Gomez, C.M. Spinocerebellar ataxia type 6. In Handbook of Clinical Neurology, 1st ed.; Elsevier B.V.: Amsterdam, The Netherlands, 2012; Volume 103. [Google Scholar] [CrossRef]
- Zhuchenko, O.; Bailey, J.; Bonnen, P.; Ashizawa, T.; Stockton, D.W.; Amos, C.; Dobyns, W.B.; Subramony, S.H.; Zoghbi, H.Y.; Lee, C.C. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the α1A-voltage-dependent calcium channel. Nat. Genet. 1997, 15, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Hillman, D.; Chen, S.; Aung, T.T.; Cherksey, B.; Sugimori, M.; Llinas, R.R. Localization of P-type calcium channels in the central nervous system. Proc. Natl. Acad. Sci. USA 1991, 88, 7076–7080. [Google Scholar] [PubMed]
- Westenbroek, R.E.; Sakurai, T.; Elliott, E.M.; Hell, J.W.; Starr, T.V.; Snutch, T.P.; Catterall, W.A. Immunochemical identification and subcellular distribution of the alpha 1A subunits of brain calcium channels. J. Neurosci. 1995, 15, 6403–6418. [Google Scholar] [CrossRef] [PubMed]
- Womack, M.D.; Chevez, C.; Khodakhah, K. Calcium-activated potassium channels are selectively coupled to P/Q-type calcium channels in cerebellar Purkinje neurons. J. Neurosci. 2004, 24, 8818–8822. [Google Scholar] [CrossRef]
- Watase, K.; Barrett, C.F.; Miyazaki, T.; Ishiguro, T.; Ishikawa, K.; Hu, Y.; Unno, T.; Sun, Y.; Kasai, S.; Watanabe, M.; et al. Spinocerebellar ataxia type 6 knockin mice develop a progressive neuronal dysfunction with age-dependent accumulation of mutant CaV2. 1 channels. Proc. Natl. Acad. Sci. USA 2008, 105, 11987–11992. [Google Scholar] [CrossRef] [PubMed]
- Unno, T.; Wakamori, M.; Koike, M.; Uchiyama, Y.; Ishikawa, K.; Kubota, H.; Yoshida, T.; Sasakawa, H.; Peters, C.; Mizusawa, H.; et al. Development of Purkinje cell degeneration in a knockin mouse model reveals lysosomal involvement in the pathogenesis of SCA6. Proc. Natl. Acad. Sci. USA 2012, 109, 17693–17698. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Wang, J.; Zhu, H.; Rinaldo, L.; Lamar, K.M.; Palmenberg, A.C.; Hansel, C.; Gomez, C.M. Second cistron in CACNA1A gene encodes a transcription factor mediating cerebellar development and SCA6. Cell 2013, 154, 118–133. [Google Scholar] [CrossRef]
- Mark, M.D.; Krause, M.; Boele, H.J.; Kruse, W.; Pollok, S.; Kuner, T.; Dalkara, D.; Koekkoek, S.; De Zeeuw, C.I.; Herlitze, S. Spinocerebellar ataxia type 6 protein aggregates cause deficits in motor learning and cerebellar plasticity. J. Neurosci. 2015, 35, 8882–8895. [Google Scholar] [CrossRef]
- Jayabal, S.; Ljungberg, L.; Erwes, T.; Cormier, A.; Quilez, S.; El Jaouhari, S.; Watt, A.J. Rapid onset of motor deficits in a mouse model of spinocerebellar ataxia type 6 precedes late cerebellar degeneration. Eneuro 2015, 2, 6. [Google Scholar] [CrossRef]
- Jayabal, S.; Chang HH, V.; Cullen, K.E.; Watt, A.J. 4-aminopyridine reverses ataxia and cerebellar firing deficiency in a mouse model of spinocerebellar ataxia type 6. Sci. Rep. 2016, 6, 29489. [Google Scholar]
- Cullen, K.E.; Van Horn, M.R. The neural control of fast vs. slow vergence eye movements. Eur. J. Neurosci. 2011, 33, 2147–2154. [Google Scholar] [PubMed]
- McMullen, C.A.; Andrade, F.H.; Stahl, J.S. Functional and genomic changes in the mouse ocular motor system in response to light deprivation from birth. J. Neurosci. 2004, 24, 161–169. [Google Scholar] [PubMed]
- Luebke, A.E.; Holt, J.C.; Jordan, P.M.; Wong, Y.S.; Caldwell, J.S.; Cullen, K.E. Loss of α-calcitonin gene-related peptide (αCGRP) reduces the efficacy of the vestibulo-ocular reflex (VOR). J. Neurosci. 2014, 34, 10453–10458. [Google Scholar] [CrossRef] [PubMed]
- Buttner, N.; Geschwind, D.; Cjen, J.; Pcrlman, S.; Pulst, S.M.; Baloh, R.W. Oculomotor phenotypes in autosomal dominant ataxias. Arch. Neurol. 1998, 55, 1353–1357. [Google Scholar]
- Holt, G.R.; Douglas, J. Comparison of Discharge Variability Visual Cortex Neurons In Vitro and In Vivo in Cat. J. Neurophysiol. 2022, 75, 1806–1814. [Google Scholar]
- Chopra, R.; Bushart, D.D.; Cooper, J.P.; Yellajoshyula, D.; Morrison, L.M.; Huang, H.; Handler, H.P.; Man, L.J.; Dansithong, W.; Scoles, D.R.; et al. Altered Capicua expression drives regional Purkinje neuron vulnerability through ion channel gene dysregulation in spinocerebellar ataxia type 1. Hum. Mol. Genet. 2020, 29, 3249–3265. [Google Scholar]
- Koeppen, A.H. The pathogenesis of spinocerebellar ataxia. Cerebellum 2005, 4, 62–73. [Google Scholar] [CrossRef]
- Rolfs, A.; Koeppen, A.H.; Bauer, I.; Bauer, P.; Buhlmann, S.; Topka, H.; Schöls, L.; Riess, O. Clinical features and neuropathology of autosomal dominant spinocerebellar ataxia (SCA17). Ann. Neurol. 2003, 54, 367–375. [Google Scholar]
- Blot, F.G.; Krijnen, W.H.; Den Hoedt, S.; Osório, C.; White, J.J.; Mulder, M.T.; Schonewille, M. Sphingolipid metabolism governs Purkinje cell patterned degeneration in Atxn1 [82Q]/+ mice. Proc. Natl. Acad. Sci. USA 2021, 118, e2016969118. [Google Scholar]
- Stoyas, C.A.; Bushart, D.D.; Switonski, P.M.; Ward, J.M.; Alaghatta, A.; Tang, M.; Niu, C.; Wadhwa, M.; Huang, H.; Savchenko, A.; et al. Nicotinamide Pathway-Dependent Sirt1 Activation Restores Calcium Homeostasis to Achieve Neuroprotection in Spinocerebellar Ataxia Type 7. Neuron 2020, 105, 630–644.e9. [Google Scholar]
- Larivière, R.; Gaudet, R.; Gentil, B.J.; Girard, M.; Conte, T.C.; Minotti, S.; Leclerc-Desaulniers, K.; Gehring, K.; Mckinney, R.A.; Shoubridge, E.A.; et al. Sacs knockout mice present pathophysiological defects underlying autosomal recessive spastic ataxia of charlevoix-saguenay. Hum. Mol. Genet. 2015, 24, 727–739. [Google Scholar]
- Toscano Márquez, B.; Cook, A.A.; Rice, M.; Smileski, A.; Vieira-Lomasney, K.; Charron, F.; McKinney, R.A.; Watt, A.J. Molecular Identity and Location Influence Purkinje Cell Vulnerability in Autosomal-Recessive Spastic Ataxia of Charlevoix-Saguenay Mice. Front. Cell. Neurosci. 2021, 15, 1–15. [Google Scholar]
- Takeichi, N.; Fukushima, K.; Sasaki, H.; Yabe, I.; Tashiro, K.; Inuyama, Y. Dissociation of smooth pursuit and vestibulo-ocular reflex cancellation in SCA-6. Neurology 2000, 54, 860–866. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Gorman, G.; Bateman, D.E.; Leigh, R.J.; Chinnery, P.F. Vertigo and vestibular abnormalities in spinocerebellar ataxia type 6. J. Neurol. 2009, 256, 78–82. [Google Scholar]
- Wiest, G.; Tian, J.R.; Baloh, R.W.; Crane, B.T.; Demer, J.L. Otolith function in cerebellar ataxia due to mutations in the calcium channel gene CACNA1A. Brain 2001, 124, 2407–2416. [Google Scholar] [CrossRef]
- Huh, Y.E.; Kim, J.S.; Kim, H.J.; Park, S.H.; Jeon, B.S.; Kim, J.M.; Cho, J.W.; Zee, D.S. Vestibular performance during high-acceleration stimuli correlates with clinical decline in SCA6. Cerebellum 2015, 14, 284–291. [Google Scholar]
- Lemos, J.; Manto, M. Pharmacotherapy of cerebellar and vestibular disorders. Curr. Opin. Neurol. 2022, 35, 118–125. [Google Scholar]
- Raman, I.M.; Bean, B.P. Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J. Neurosci. 1999, 19, 1663–1674. [Google Scholar]
- Womack, M.D.; Khodakhah, K. Characterization of large conductance Ca2+-activated K+ channels in cerebellar Purkinje neurons. Eur. J. Neurosci. 2002, 16, 1214–1222. [Google Scholar]
- Swensen, A.M.; Bean, B.P. Robustness of burst firing in dissociated purkinje neurons with acute or long-term reductions in sodium conductance. J. Neurosci. 2005, 25, 3509–3520. [Google Scholar]
- Womack, M.D.; Khodakhah, K. Somatic and dendritic small-conductance calcium-activated potassium channels regulate the output of cerebellar Purkinje neurons. J. Neurosci. 2003, 23, 2600–2607. [Google Scholar] [CrossRef]
- Pietrobon, D. Calcium channels and channelopathies of the central nervous system. Mol. Neurobiol. 2002, 25, 31–50. [Google Scholar] [CrossRef]
- Stahl, J.S. Eye movements of the murine P/Q calcium channel mutant rocker, and the impact of aging. J. Neurophysiol. 2004, 91, 2066–2078. [Google Scholar] [CrossRef] [PubMed]
- Stahl, J.S.; James, R.A.; Oommen, B.S.; Hoebeek, F.E.; De Zeeuw, C.I. Eye movements of the murine P/Q calcium channel mutant tottering, and the impact of aging. J. Neurophysiol. 2006, 95, 1588–1607. [Google Scholar] [CrossRef] [PubMed]
- Katoh, A.; Jindal, J.A.; Raymond, J.L. Motor deficits in homozygous and heterozygous p/q-type calcium channel mutants. J. Neurophysiol. 2007, 97, 1280–1287. [Google Scholar] [CrossRef] [Green Version]
- Stahl, J.S.; Thumser, Z.C. Flocculus Purkinje cell signals in mouse Cacna1a calcium channel mutants of escalating severity: An investigation of the role of firing irregularity in ataxia. J. Neurophysiol. 2014, 112, 2647–2663. [Google Scholar] [CrossRef]
- Walter, J.T.; Alviña, K.; Womack, M.D.; Chevez, C.; Khodakhah, K. Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat. Neurosci. 2006, 9, 389–397. [Google Scholar] [CrossRef]
- Alviña, K.; Khodakhah, K. The therapeutic mode of action of 4-aminopyridine in cerebellar ataxia. J. Neurosci. 2010, 30, 7258–7268. [Google Scholar] [CrossRef]
- Slemmer, J.E.; Haasdijk, E.D.; Engel, D.C.; Plesnila, N.; Weber, J.T. Aldolase C-positive cerebellar Purkinje cells are resistant to delayed death after cerebral trauma and AMPA-mediated excitotoxicity. Eur. J. Neurosci. 2007, 26, 649–656. [Google Scholar] [CrossRef]
- Ying, S.H.; Choi, S.I.; Lee, M.; Perlman, S.L.; Baloh, R.W.; Toga, A.W.; Zee, D.S. Relative atrophy of the flocculus and ocular motor dysfunction in SCA2 and SCA6. Ann. N. Y. Acad. Sci. 2005, 1039, 430–435. [Google Scholar] [CrossRef]
- Cook, A.A.; Fields, E.; Watt, A.J. Losing the beat: Contribution of Purkinje cell firing dysfunction to disease, and its reversal. Neuroscience 2021, 462, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Beraneck, M.; Cullen, K.E. Activity of vestibular nuclei neurons during vestibular and optokinetic stimulation in the alert mouse. J. Neurophysiol. 2007, 98, 1549–1565. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, H.H.V.; Cook, A.A.; Watt, A.J.; Cullen, K.E. Loss of Flocculus Purkinje Cell Firing Precision Leads to Impaired Gaze Stabilization in a Mouse Model of Spinocerebellar Ataxia Type 6 (SCA6). Cells 2022, 11, 2739. https://doi.org/10.3390/cells11172739
Chang HHV, Cook AA, Watt AJ, Cullen KE. Loss of Flocculus Purkinje Cell Firing Precision Leads to Impaired Gaze Stabilization in a Mouse Model of Spinocerebellar Ataxia Type 6 (SCA6). Cells. 2022; 11(17):2739. https://doi.org/10.3390/cells11172739
Chicago/Turabian StyleChang, Hui Ho Vanessa, Anna A. Cook, Alanna J. Watt, and Kathleen E. Cullen. 2022. "Loss of Flocculus Purkinje Cell Firing Precision Leads to Impaired Gaze Stabilization in a Mouse Model of Spinocerebellar Ataxia Type 6 (SCA6)" Cells 11, no. 17: 2739. https://doi.org/10.3390/cells11172739