
Structure-Based Study to Overcome Cross-Reactivity of Novel Androgen Receptor Inhibitors
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Enhanced Green Fluorescent Protein (eGFP) Cellular AR Transcription Assay
2.2. Prostate-Specific Antigen (PSA) Assay
2.3. Luciferase Reporter Assay
2.4. Biolayer Interferometry (BLI) Assay
2.5. Protein Production for X-ray Crystallography
2.6. Crystallization, Data Collection and Structure Determination
2.7. DHT Displacement Assay
2.8. Computational Modelling
3. Results
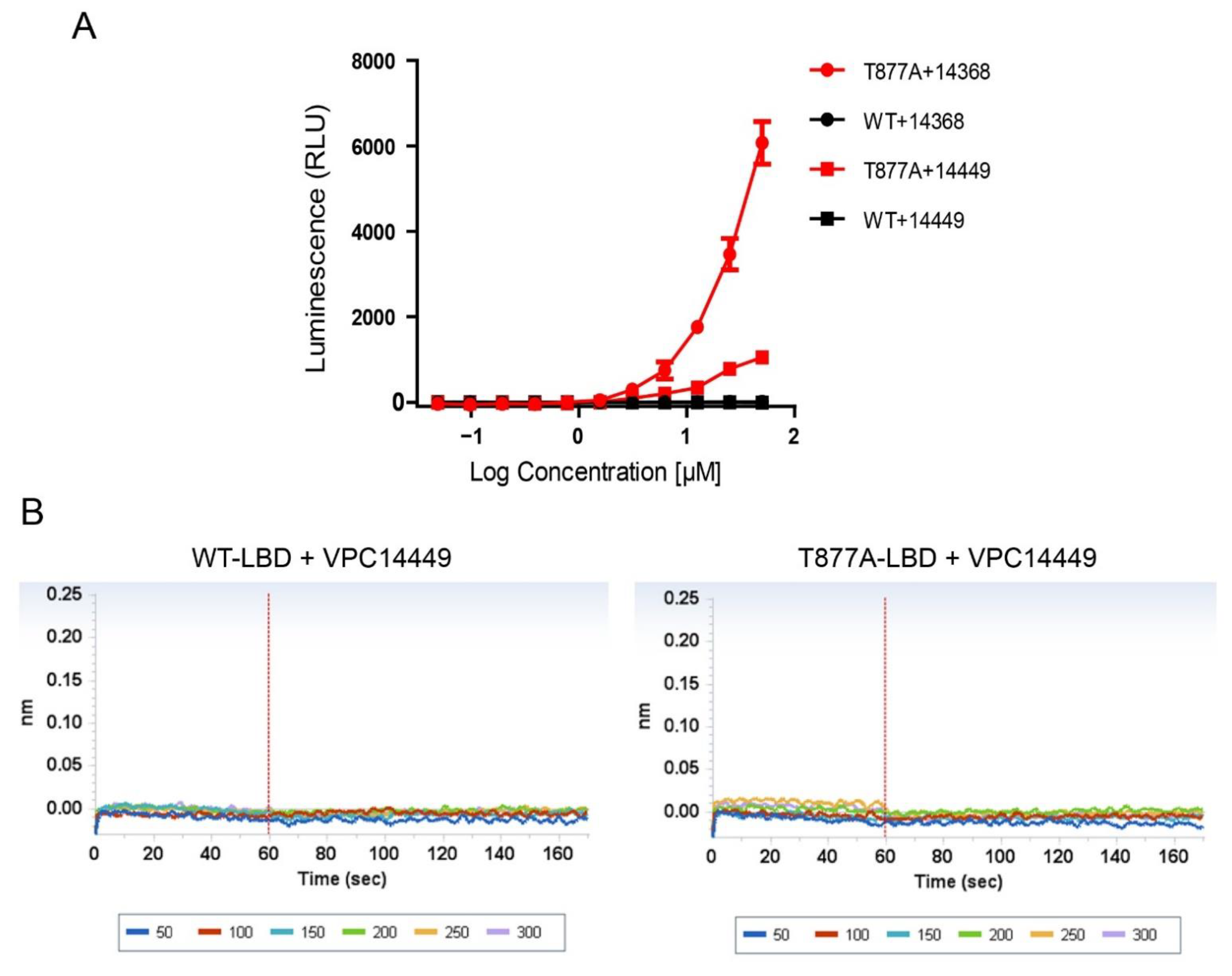
3.1. Identification of Partial AR Agonist Properties of the VPC14368 Compound
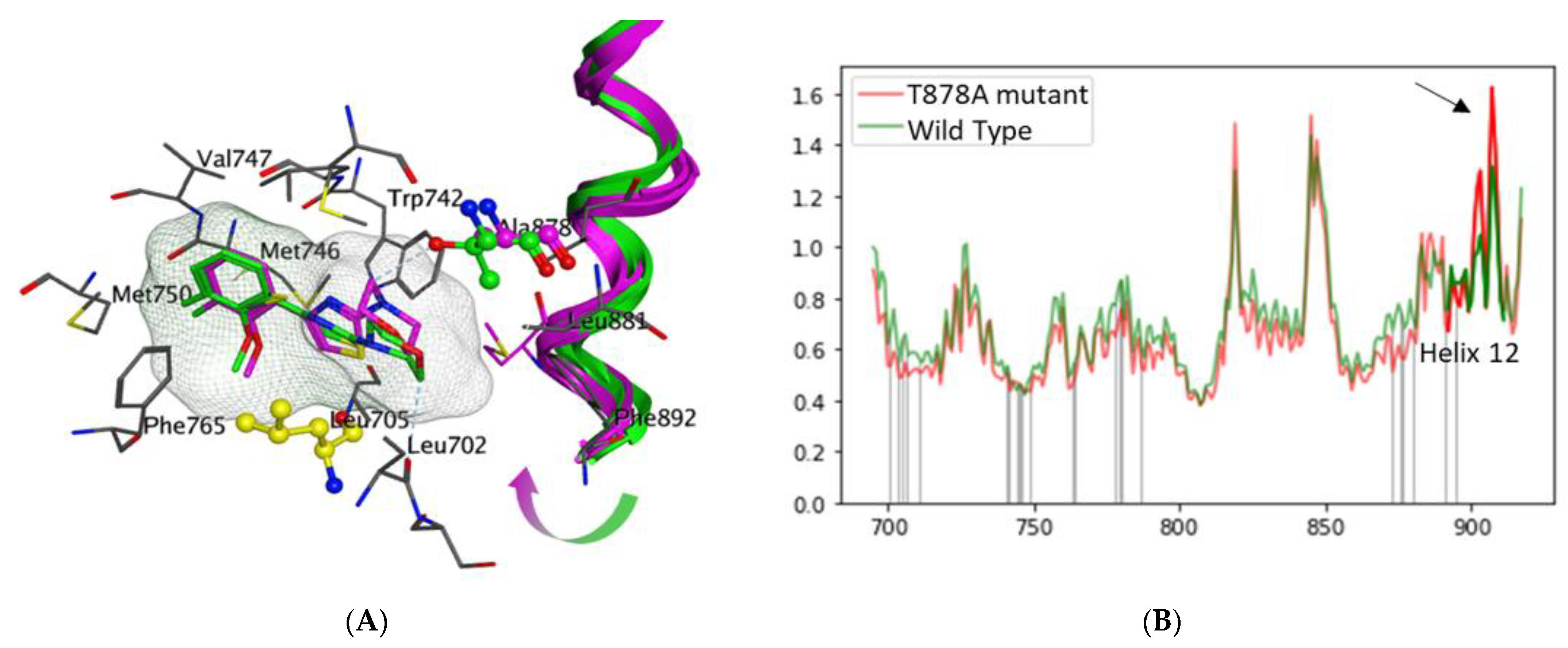
3.2. Structural Determination of the T878A-LBD-VPC14368 Complex
3.3. Computational Analysis of the Mechanism of Agonistic Switch in the VPC14368 Mutant T878A AR LBD
3.4. Elimination of Cross-Reactivity of 4-(4-Phenylthiazol-2-yl)morpholines
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jenster, G.; Van Der Korput, H.A.G.M.; Van Vroonhoven, C.; Van der Kwast, T.; Trapman, J.; Brinkmann, A.O. Domains of the human androgen receptor involved in steroid binding, transcriptional activation, and subcellular localization. Mol. Endocrinol. 1991, 5, 1396–1404. [Google Scholar] [CrossRef]
- Hiipakka, R.A.; Liao, S. Molecular mechanism of androgen action. Trends Endocrinol. Metab. 1998, 9, 317–324. [Google Scholar] [CrossRef]
- Lamont, K.R.; Tindall, D.J. Androgen regulation of gene expression. Adv. Cancer Res. 2010, 107, 137–162. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Nonomura, N. Role of androgen receptor in prostate cancer: A review. World J. Men's Health 2019, 37, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Attard, G.; Richards, J.; de Bono, J.S. New strategies in metastatic prostate cancer: Targeting the androgen receptor signaling pathway. Clin. Cancer Res. 2011, 17, 1649–1657. [Google Scholar] [CrossRef]
- Ferraldeschi, R.; de Bono, J. Agents that target androgen synthesis in castration-resistant prostate cancer. Cancer J. 2013, 19, 34–42. [Google Scholar] [CrossRef]
- Fizazi, K.; Scher, H.I.; Molina, A.; Logothetis, C.J.; Chi, K.N.; Jones, R.J.; Staffurth, J.N.; North, S.; Vogelzang, N.J.; Saad, F.; et al. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: Final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2012, 13, 983–992. [Google Scholar] [CrossRef]
- Saad, F.; Miller, K. Treatment options in castration-resistant prostate cancer: Current therapies and emerging docetaxel-based regimens. Urol. Oncol. 2013, 32, 70–79. [Google Scholar] [CrossRef]
- Kim, T.J.; Lee, Y.H.; Koo, K.C. Current status and future perspectives of androgen receptor inhibition therapy for prostate cancer: A comprehensive review. Biomolecules 2021, 11, 492. [Google Scholar] [CrossRef]
- Nigro, M.C.; Mollica, V.; Marchetti, A.; Cheng, M.; Rosellini, M.; Montironi, R.; Cheng, L.; Massari, F. Current androgen receptor antagonists under investigation for resistant prostate cancer. Expert Rev. Anticancer. Ther. 2022, 22, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef]
- Raghow, S.; Kuliyev, E.; Steakley, M.; Greenberg, N.; Steiner, M.S. Efficacious chemoprevention of primary prostate cancer by flutamide in an autochthonous transgenic model. Cancer Res. 2000, 60, 4093–4097. [Google Scholar] [PubMed]
- Hoffman-Censits, J.; Kelly, W.K. Enzalutamide: A novel anti-androgen for patients with castrate resistant prostate cancer. Clin. Cancer Res. 2013, 19, 1335–1339. [Google Scholar] [CrossRef]
- Bohl, C.E.; Gao, W.; Miller, D.D.; Bell, C.E.; Dalton, J.T. Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 6201–6206. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, A.; Movahedpour, A.; Amiri, A.; Najaf, M.S.; Mostafavi-Pour, Z. Darolutamide as a second-generation androgen receptor inhibitor in the treatment of prostate cancer. Curr. Mol. Med. 2021, 21, 332–346. [Google Scholar] [CrossRef]
- Heinlein, C.A.; Chang, C. Androgen receptor (AR) coregulators: An overview. Endocr. Rev. 2002, 23, 175–200. [Google Scholar] [CrossRef]
- Heemers, H.V.; Tindall, D.J. Androgen receptor (AR) coregulators: A diversity of functions converging on and regulating the AR transcriptional complex. Endocr. Rev. 2007, 28, 778–808. [Google Scholar] [CrossRef] [PubMed]
- Bohl, C.E.; Wu, Z.; Chen, J.; Mohler, M.L.; Yang, J.; Hwang, D.J.; Mustafa, S.; Miller, D.D.; Bell, C.E.; Dalton, J.T. Effect of B-ring substitution pattern on binding mode of propionamide selective androgen receptor modulators. Bioorg. Med. Chem. Lett. 2008, 18, 5567–5570. [Google Scholar] [CrossRef]
- Nique, F.; Hebbe, S.; Peixoto, C.; Annoot, D.; Lefrançois, J.-M.; Duval, E.; Michoux, L.; Triballeau, N.; Lemoullec, J.-M.; Mollat, P.; et al. Discovery of diarylhydantoins as new selective androgen receptor modulators. J. Med. Chem. 2012, 55, 8225–8235. [Google Scholar] [CrossRef]
- Wang, F.; Liu, X.Q.; Li, H.; Liang, K.N.; Miner, J.N.; Hong, M.; Kallel, E.A.; Van Oeveren, A.; Zhi, L.; Jiang, T. Structure of the ligand-binding domain (LBD) of human androgen receptor in complex with a selective modulator LGD2226. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006, 62, 1067–1071. [Google Scholar] [CrossRef]
- Asano, M.; Hitaka, T.; Imada, T.; Yamada, M.; Morimoto, M.; Shinohara, H.; Hara, T.; Yamaoka, M.; Santou, T.; Nakayama, M.; et al. Synthesis and biological evaluation of novel selective androgen receptor modulators (SARMs). Part II: Optimization of 4-(pyrrolidin-1-yl) benzonitrile derivatives. Bioorg. Med. Chem. Lett. 2017, 27, 1897–1901. [Google Scholar] [CrossRef]
- Pereira de Jésus-Tran, K.; Côté, P.L.; Cantin, L.; Blanchet, J.; Labrie, F.; Breton, R. Comparison of crystal structures of human androgen receptor ligand-binding domain complexed with various agonists reveals molecular determinants responsible for binding affinity. Protein Sci. 2006, 15, 987–999. [Google Scholar] [CrossRef]
- Hu, X.; Chai, X.; Wang, X.; Duan, M.; Pang, J.; Fu, W.; Li, D.; Hou, T. Advances in the computational development of androgen receptor antagonists. Drug Discov. Today 2020, 25, 1453–1461. [Google Scholar] [CrossRef] [PubMed]
- Bohl, C.E.; Miller, D.D.; Chen, J.; Bell, C.E.; Dalton, J.T. Structural basis for accommodation of nonsteroidal ligands in the androgen receptor. J. Biol. Chem. 2005, 280, 37747–37754. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, A.W.; Gleave, M.E. Targeting the adaptive molecular landscape of castration-resistant prostate cancer. EMBO Mol. Med. 2015, 7, 878–894. [Google Scholar] [CrossRef]
- Snow, O.; Lallous, N.; Singh, K.; Lack, N.; Rennie, P.; Cherkasov, A. Androgen receptor plasticity and its implications for prostate cancer therapy. Cancer Treat. Rev. 2019, 81, 101871. [Google Scholar] [CrossRef] [PubMed]
- Ehsani, M.; David, F.O.; Baniahmad, A. Androgen receptor-dependent mechanisms mediating drug resistance in prostate cancer. Cancers 2021, 13, 1534. [Google Scholar] [CrossRef] [PubMed]
- Maitland, N.J. Resistance to antiandrogens in prostate cancer: Is it inevitable, intrinsic or induced? Cancers 2021, 13, 327. [Google Scholar] [CrossRef] [PubMed]
- Azhagiya Singam, E.R.; Tachachartvanich, P.; La Merrill, M.A.; Smith, M.T.; Durkin, K.A. Structural dynamics of agonist and antagonist binding to the androgen receptor. J. Phys. Chem. B 2019, 123, 7657–7666. [Google Scholar] [CrossRef]
- Duan, M.; Liu, N.; Zhou, W.; Li, D.; Yang, M.; Hou, T. Structural diversity of ligand-binding androgen receptors revealed by microsecond long molecular dynamics simulations and enhanced sampling. J. Chem. Theory Comput. 2016, 12, 4611–4619. [Google Scholar] [CrossRef]
- Bisson, W.H.; Abagyan, R.; Cavasotto, C.N. Molecular basis of agonicity and antagonicity in the androgen receptor studied by molecular dynamics simulations. J. Mol. Graph. Model. 2008, 27, 452–458. [Google Scholar] [CrossRef]
- Huang, P.; Chandra, V.; Rastinejad, F. Structural overview of the nuclear receptor superfamily: Insights into physiology and therapeutics. Annu. Rev. Physiol. 2010, 72, 247–272. [Google Scholar] [CrossRef] [PubMed]
- De Lera, A.R.; Bourguet, W.; Altucci, L.; Gronemeyer, H. Design of selective nuclear receptor modulators: RAR and RXR as a case study. Nat. Rev. Drug Discov. 2007, 6, 811–820. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, B.; Geng, G.; Wu, J.H. Study of the impact of the T877A mutation on ligand-induced helix-12 positioning of the androgen receptor resulted in design and synthesis of novel antiandrogens. Proteins Struct. Funct. Bioinform. 2010, 78, 623–637. [Google Scholar] [CrossRef]
- Liu, H.; Han, R.; Li, J.; Liu, H.; Zheng, L. Molecular mechanism of R-bicalutamide switching from androgen receptor antagonist to agonist induced by amino acid mutations using molecular dynamics simulations and free energy calculation. J. Comput. Aided Mol. Des. 2016, 30, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Lallous, N.; Dalal, K.; Cherkasov, A.; Rennie, P.S. Targeting alternative sites on the androgen receptor to treat castration-resistant prostate cancer. Int. J. Mol. Sci. 2013, 14, 12496–12519. [Google Scholar] [CrossRef]
- Caboni, L.; Lloyd, D.G. Beyond the Ligand-Binding Pocket: Targeting Alternate Sites in Nuclear Receptors. Med. Res. Rev. 2012, 33, 1081–1118. [Google Scholar] [CrossRef]
- Zhang, C.; Wu, J.; Chen, Q.; Tan, H.; Huang, F.; Guo, J.; Zhang, X.; Yu, H.; Shi, W. Allosteric binding on nuclear receptors: Insights on screening of non-competitive endocrine-disrupting chemicals. Environ. Int. 2022, 159, 107009. [Google Scholar] [CrossRef]
- Axerio-Cilies, P.; Lack, N.A.; Nayana, M.R.S.; Chan, K.H.; Yeung, A.; Leblanc, E.; Guns, E.S.T.; Rennie, P.S.; Cherkasov, A. Inhibitors of androgen receptor activation function-2 (AF2) site identified through virtual screening. J. Med. Chem. 2011, 54, 6197–6205. [Google Scholar] [CrossRef] [PubMed]
- Estébanez-Perpiñá, E.; Arnold, L.A.; Nguyen, P.; Rodrigues, E.D.; Mar, E.; Bateman, R.; Pallai, P.; Shokat, K.M.; Baxter, J.D.; Guy, R.K.; et al. A surface on the androgen receptor that allosterically regulates coactivator binding. Proc. Natl. Acad. Sci. USA 2007, 104, 16074–16079. [Google Scholar] [CrossRef] [Green Version]
- Lack, N.A.; Axerio-Cilies, P.; Tavassoli, P.; Han, F.Q.; Chan, K.H.; Feau, C.; LeBlanc, E.; Guns, E.T.; Guy, R.K.; Rennie, P.S.; et al. Targeting the binding function 3 (BF3) site of the human androgen receptor through virtual screening. J. Med. Chem. 2011, 54, 8563–8573. [Google Scholar] [CrossRef] [PubMed]
- Munuganti, R.S.N.; Leblanc, E.; Axerio-Cilies, P.; Labriere, C.; Frewin, K.; Singh, K.; Hassona, M.D.H.; Lack, N.A.; Li, H.; Ban, F.; et al. Targeting the Binding Function 3 (BF3) Site of the Androgen Receptor Through Virtual Screening. 2. Development of 2-((2-phenoxyethyl) thio)-1H-benzimidazole derivatives. J. Med. Chem. 2013, 56, 1136–1148. [Google Scholar] [CrossRef] [PubMed]
- Munuganti, R.S.; Hassona, M.D.; Leblanc, E.; Frewin, K.; Singh, K.; Ma, D.; Ban, F.; Hsing, M.; Adomat, H.; Lallous, N.; et al. Identification of a Potent Antiandrogen that Targets the BF3 Site of the Androgen Receptor and Inhibits Enzalutamide-Resistant Prostate Cancer. Chem. Biol. 2014, 21, 1476–1485. [Google Scholar] [CrossRef]
- Jehle, K.; Cato, L.; Neeb, A.; Muhle-Goll, C.; Jung, N.; Smith, E.; Buzon, V.; Carbó, L.R.; Estébanez-Perpiñá, E.; Schmitz, K.; et al. Coregulator Control of Androgen Receptor Action by a Novel Nuclear Receptor-Binding Motif. J. Biol. Chem. 2014, 289, 8839–8851. [Google Scholar] [CrossRef]
- Ban, F.; Leblanc, E.; Li, H.; Munuganti, R.S.N.; Frewin, K.; Rennie, P.S.; Cherkasov, A. Discovery of 1H-indole-2-carboxamides as novel inhibitors of the androgen receptor binding function 3 (BF3). J. Med. Chem. 2014, 57, 6867–6872. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Qiu, Y. A new trick of an old molecule: Androgen receptor splice variants taking the stage?! Int. J. Biol. Sci. 2011, 7, 815–822. [Google Scholar] [CrossRef]
- Dehm, S.M.; Schmidt, L.J.; Heemers, H.V.; Vessella, R.L.; Tindall, D.J. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008, 68, 5469–5477. [Google Scholar] [CrossRef]
- Hu, R.; Dunn, T.A.; Wei, S.; Isharwal, S.; Veltri, R.W.; Humphreys, E.; Han, M.; Partin, A.W.; Vessella, R.L.; Isaacs, W.B.; et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009, 69, 16–22. [Google Scholar] [CrossRef]
- Dalal, K.M.; Roshan-Moniri, M.; Li, H.; Sharma, A.; Ban, F.; Leblanc, E.; Dehm, S.; Cherkasov, A.; Rennie, P.S. Selectively Targeting the DNA Binding Domain of the Androgen Receptor as a Prospective Therapy for Prostate Cancer. J. Biol. Chem. 2014, 289, 26417–26429. [Google Scholar] [CrossRef]
- Dalal, K.; Che, M.; Que, N.S.; Sharma, A.; Yang, R.; Lallous, N.; Borgmann, H.; Ozistanbullu, D.; Tse, R.; Ban, F.; et al. Bypassing Drug Resistance Mechanisms of Prostate Cancer with Small Molecules that Target Androgen Receptor–Chromatin Interactions. Mol. Cancer Ther. 2017, 16, 2281–2291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ban, F.; Leblanc, E.; Cavga, A.; Huang, C.-C.; Flory, M.; Zhang, F.; Chang, M.; Morin, H.; Lallous, N.; Singh, K.; et al. Development of an androgen receptor inhibitor targeting the N-Terminal domain of androgen receptor for treatment of castration resistant prostate cancer. Cancers 2021, 13, 3488. [Google Scholar] [CrossRef]
- Radaeva, M.; Ban, F.; Zhang, F.; LeBlanc, E.; Lallous, N.; Rennie, P.; Gleave, M.; Cherkasov, A. Development of novel inhibitors targeting the d-box of the dna binding domain of androgen receptor. Int. J. Mol. Sci. 2021, 22, 2493. [Google Scholar] [CrossRef]
- Dalal, K.; Ban, F.; Li, H.; Morin, H.; Roshan-Moniri, M.; Tam, K.J.; Shepherd, A.; Sharma, A.; Peacock, J.; Carlson, M.L.; et al. Selectively targeting the dimerization interface of human androgen receptor with small-molecules to treat castration-resistant prostate cancer. Cancer Lett. 2018, 437, 35–43. [Google Scholar] [CrossRef]
- Li, H.; Ban, F.; Dalal, K.; Leblanc, E.; Frewin, K.; Ma, D.; Adomat, H.; Rennie, P.S.; Cherkasov, A. Discovery of small-molecule inhibitors selectively targeting the DNA-binding domain of the human androgen receptor. J. Med. Chem. 2014, 57, 6458–6467. [Google Scholar] [CrossRef] [PubMed]
- Tavassoli, P.; Snoek, R.; Ray, M.; Rao, L.G.; Rennie, P.S. Rapid, non-destructive, cell-based screening assays for agents that modulate growth, death, and androgen receptor activation in prostate cancer cells. Prostate 2007, 67, 416–426. [Google Scholar] [CrossRef]
- Snoek, R.; Bruchovsky, N.; Kasper, S.; Matusik, R.J.; Gleave, M.; Sato, N.; Mawji, N.R.; Rennie, P.S. Differential transactivation by the androgen receptor in prostate cancer cells. Prostate 1998, 36, 256–263. [Google Scholar] [CrossRef]
- Kabsch, W. Xds. Acta Crystallographica. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallographica. Sect. D Biol. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE); Chemical Computing Group (ULC): Montreal, QC, Canada, 2019.
- Schrödinger Release 2020-4: Glide; Schrödinger, LLC: New York, NY, USA, 2020.
- Berendsen, H.J.; Postma, J.P.; van Gunsteren, W.F.; Hermans, J. Intermolecular Forces; Springer: Berlin/Heidelberg, Germany, 1981; pp. 331–342. [Google Scholar]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending force field coverage for drug-like small molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hou, T.; Xu, X. Recent advances in free energy calculations with a combination of molecular mechanics and continuum models. Curr. Comput. Aided Drug Des. 2006, 2, 287–306. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins Struct. Funct. Bioinform. 2011, 79, 2794–2812. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Nyquist, M.D.; Li, Y.; Hwang, T.H.; Manlove, L.S.; Vessella, R.L.; Silverstein, K.A.T.; Voytas, D.F.; Dehm, S.M. TALEN-engineered AR gene rearrangements reveal endocrine uncoupling of androgen receptor in prostate cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 17492–17497. [Google Scholar] [CrossRef]
- Hara, T.; Miyazaki, J.-I.; Araki, H.; Yamaoka, M.; Kanzaki, N.; Kusaka, M.; Miyamoto, M. Novel mutations of androgen receptor: A possible mechanism of bicalutamide withdrawal syndrome. Cancer Res. 2003, 63, 149–153. [Google Scholar] [PubMed]
- Thevis, M.; Kohler, M.; Schlörer, N.; Kamber, M.; Kühn, A.; Linscheid, M.W.; Schänzer, W. Mass spectrometry of hydantoin-derived selective androgen receptor modulators. J. Mass Spectrom. 2008, 43, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Salvati, M.E.; Balog, A.; Shan, W.; Wei, D.D.; Pickering, D.; Attar, R.M.; Geng, J.; Rizzo, C.A.; Gottardis, M.M.; Weinmann, R.; et al. Structure based approach to the design of bicyclic-1H-isoindole-1,3(2H)-dione based androgen receptor antagonists. Bioorg. Med. Chem. Lett. 2005, 15, 271–276. [Google Scholar] [CrossRef]
- Liu, H.; An, X.; Li, S.; Wang, Y.; Li, J.; Liu, H. Interaction mechanism exploration of R-bicalutamide/S-1 with WT/W741L AR using molecular dynamics simulations. Mol. Biosyst. 2015, 11, 3347–3354. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Structure | Reporter eGFP IC50 (µM) | Androgen Displacement |
|---|---|---|---|
| 14368 |  | 0.035 ± 0.02 | Yes |
| 14449 |  | 0.15 ± 0.04 | <10% |
| 14404 |  | 0.27 ± 0.02 | <10% |
| 14439 |  | 0.16 ± 0.03 | Yes |
| 14435 |  | 3.87 ± 1.26 | No |
| 14436 |  | 4.53 ± 0.26 | No |
| 14403 |  | inactive | - |
| 14406 |  | inactive | - |
| 14291 |  | inactive | - |
| T878A AR-LBD + VPC14368 | |
|---|---|
| PDB code | 8E1A |
| Expression system | E. coli |
| pH of crystallization | 7.5 |
| Data collection | |
| Wavelength (Å) | 0.972452 |
| Space group | P212121 |
| Unit cell | a = 55.01 Å b = 65.68 Å c = 69.07 Å α = β = γ = 90° |
| Resolution (Å) * | 40–1.2 (1.27–1.20) |
| Rsym (I) (%) * | 4.0 (39.7) |
| I/σ(I) * | 22.84 (3.83) |
| Completeness (%) * | 97 (86.4) |
| Redundancy * | 6.2 (4.7) |
| Refinement | |
| Resolution (Å) | 40–1.2 |
| No. reflections | 72,860 |
| Rcrys/Rfree (%) | 12.4/15.1 |
| AR molecule/AU | 1 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.023 |
| Bond angles (°) | 2.15 |
| Average B factor (Å2) | 17.60 |
| Protein (Chain A) | 15.60 |
| VPC14368 (Chain B) | 13.30 |
| Water (Chain D) | 33.30 |
| Ramachandran plot | |
| Ramachandran plot | |
| Favoured (%) | 97.7 |
| Allowed (%) | 2.3 |
| Outliers (%) | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radaeva, M.; Li, H.; LeBlanc, E.; Dalal, K.; Ban, F.; Ciesielski, F.; Chow, B.; Morin, H.; Awrey, S.; Singh, K.; et al. Structure-Based Study to Overcome Cross-Reactivity of Novel Androgen Receptor Inhibitors. Cells 2022, 11, 2785. https://doi.org/10.3390/cells11182785
Radaeva M, Li H, LeBlanc E, Dalal K, Ban F, Ciesielski F, Chow B, Morin H, Awrey S, Singh K, et al. Structure-Based Study to Overcome Cross-Reactivity of Novel Androgen Receptor Inhibitors. Cells. 2022; 11(18):2785. https://doi.org/10.3390/cells11182785
Chicago/Turabian StyleRadaeva, Mariia, Huifang Li, Eric LeBlanc, Kush Dalal, Fuqiang Ban, Fabrice Ciesielski, Bonny Chow, Helene Morin, Shannon Awrey, Kriti Singh, and et al. 2022. "Structure-Based Study to Overcome Cross-Reactivity of Novel Androgen Receptor Inhibitors" Cells 11, no. 18: 2785. https://doi.org/10.3390/cells11182785