
Clinical Spectrum of LMNA-Associated Type 2 Familial Partial Lipodystrophy: A Systematic Review
 , ,
, ,  ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Search Strategy
2.2. Study Selection
2.3. Identification and Inclusion of Studies
3. Results
3.1. Prevalence
3.2. Clinical Characteristics
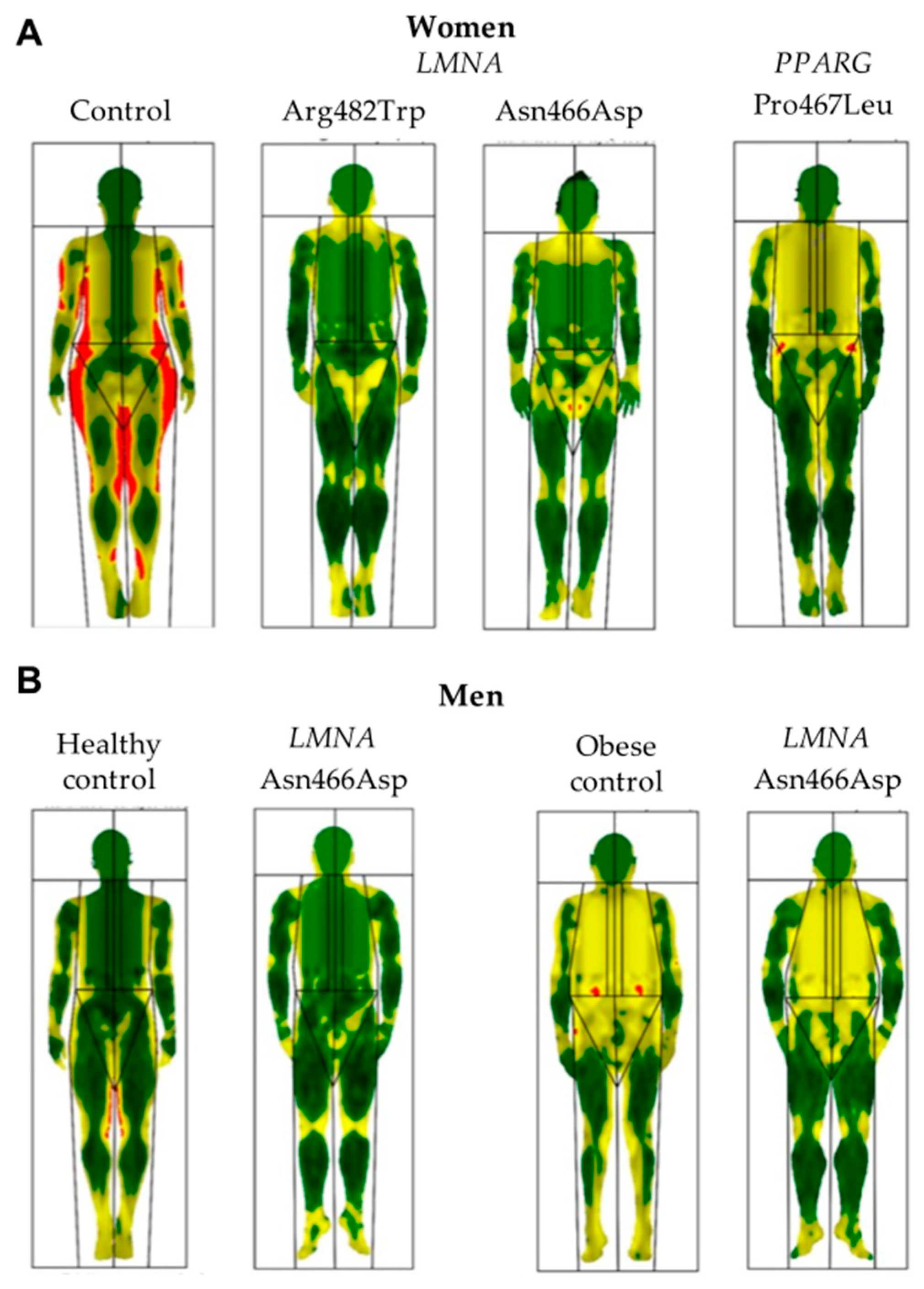
3.3. Body Composition
3.4. Comorbidities and Organ Abnormalities
3.4.1. Adipokine Disturbance
3.4.2. Insulin Resistance and Diabetes
3.4.3. Lipid Metabolism and Pancreatitis
3.4.4. Cardiovascular Disease
3.4.5. Liver Abnormalities
3.4.6. Gynaecological and Obstetric Disorders
3.4.7. Neuromuscular Disturbances
3.4.8. Renal Disease
3.4.9. Other Organ Abnormalities
3.5. Mortality
3.6. Treatment
3.7. Future Perspectives
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Broers, J.L.; Ramaekers, F.C.; Bonne, G.; Yaou, R.B.; Hutchison, C.J. Nuclear lamins: Laminopathies and their role in premature ageing. Physiol. Rev. 2006, 86, 967–1008. [Google Scholar] [CrossRef] [PubMed]
- Guillín-Amarelle, C.; Fernández-Pombo, A.; Sánchez-Iglesias, S.; Araújo-Vilar, D. Lipodystrophic laminopathies: Diagnostic clues. Nucleus 2018, 9, 249–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabanillas, R.; Cadiñanos, J.; Villameytide, J.A.; Pérez, M.; Longo, J.; Richard, J.M.; Álvarez, R.; Durán, N.S.; Illán, R.; González, D.J.; et al. Néstor-Guillermo progeria syndrome: A novel premature aging condition with early onset and chronic development caused by BANF1 mutations. Am. J. Med. Genet. A 2011, 155A, 2617–2625. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Peshock, R.M.; Fleckenstein, J.L. Adipose tissue distribution pattern in patients with familial partial lipodystrophy (Dunnigan variety). J. Clin. Endocrinol. Metab. 1999, 84, 170–174. [Google Scholar] [PubMed] [Green Version]
- Garg, A. Gender differences in the prevalence of metabolic complications in familial partial lipodystrophy (Dunnigan variety). J. Clin. Endocrinol. Metab. 2000, 85, 1776–1782. [Google Scholar]
- Garg, A.; Vinaitheerthan, M.; Weatherall, P.T.; Bowcock, A.M. Phenotypic heterogeneity in patients with familial partial lipodystrophy (dunnigan variety) related to the site of missense mutations in lamin a/c gene. J. Clin. Endocrinol. Metab. 2001, 86, 59–65. [Google Scholar]
- Fernández-Pombo, A.; Sánchez-Iglesias, S.; Cobelo-Gómez, S.; Hermida-Ameijeiras, Á.; Araújo-Vilar, D. Familial partial lipodystrophy syndromes. Presse Med. 2021, 50, 104071. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Dunnigan, M.G.; Cochrane, M.A.; Kelly, A.; Scott, J.W. Familial lipoatrophic diabetes with dominant transmission. A new syndrome. Q J. Med. 1974, 43, 33–48. [Google Scholar]
- Chiquette, E.; Oral, E.A.; Garg, A.; Araújo-Vilar, D.; Dhankhar, P. Estimating the prevalence of generalized and partial lipodystrophy: Findings and challenges. Diabetes Metab. Syndr. Obes. 2017, 10, 375–383. [Google Scholar] [CrossRef] [Green Version]
- Florwick, A.; Dharmaraj, T.; Jurgens, J.; Valle, D.; Wilson, K.L. LMNA Sequences of 60,706 Unrelated Individuals Reveal 132 Novel Missense Variants in A-Type Lamins and Suggest a Link between Variant p.G602S and Type 2 Diabetes. Front. Genet. 2017, 8, 79. [Google Scholar] [CrossRef] [Green Version]
- Gonzaga-Jauregui, C.; Ge, W.; Staples, J.; Van Hout, C.; Yadav, A.; Colonie, R.; Leader, J.B.; Kirchner, H.L.; Murray, M.F.; Reid, J.G.; et al. Geisinger-Regeneron DiscovEHR collaboration. Clinical and Molecular Prevalence of Lipodystrophy in an Unascertained Large Clinical Care Cohort. Diabetes 2020, 69, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Vigouroux, C.; Magré, J.; Vantyghem, M.C.; Bourut, C.; Lascols, O.; Shackleton, S.; Lloyd, D.J.; Guerci, B.; Padova, G.; Valensi, P.; et al. Lamin A/C gene: Sex-determined expression of mutations in Dunnigan-type familial partial lipodystrophy and absence of coding mutations in congenital and acquired generalized lipoatrophy. Diabetes 2000, 49, 1958–1962. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.N.; Howlett, T.A.; McNally, P.G.; O’Rahilly, S.; Trembath, R.C. Dunnigan-Kobberling syndrome: An autosomal dominant form of partial lipodystrophy. QJM 1997, 90, 27–36. [Google Scholar] [CrossRef]
- Köbberling, J.; Dunnigan, M.G. Familial partial lipodystrophy: Two types of an X linked dominant syndrome, lethal in the hemizygous state. J. Med. Genet. 1986, 23, 120–127. [Google Scholar] [CrossRef]
- Treiber, G.; Flaus Furmaniuk, A.; Guilleux, A.; Medjane, S.; Bonfanti, O.; Schneebeli, S.; Bernard, C.; Le-Moullec, N.; Bakiri, F.; Pholsena, M.; et al. A recurrent familial partial lipodystrophy due to a monoallelic or biallelic LMNA founder variant highlights the multifaceted cardiac manifestations of metabolic laminopathies. Eur. J. Endocrinol. 2021, 185, 453–462. [Google Scholar] [CrossRef]
- Drac, H.; Madej-Pilarczyk, A.; Gospodarczyk-Szot, K.; Gaweł, M.; Kwieciński, H.; Hausmanowa-Petrusewicz, I. Familial partial lipodystrophy associated with the heterozygous LMNA mutation 1445G>A (Arg482Gln) in a Polish family. Neurol. Neurochir. Pol. 2010, 44, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M.B.; Young, R.T. Metabolic studies in familial partial lipodystrophy of the lower trunk and extremities. Diabetologia 1975, 11, 561–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrini, C.; Columbaro, M.; Schena, E.; Prencipe, S.; Andrenacci, D.; Iozzo, P.; Angela Guzzardi, M.; Capanni, C.; Mattioli, E.; Loi, M.; et al. Altered adipocyte differentiation and unbalanced autophagy in type 2 Familial Partial Lipodystrophy: An in vitro and in vivo study of adipose tissue browning. Exp. Mol. Med. 2019, 51, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araújo-Vilar, D.; Victoria, B.; González-Méndez, B.; Barreiro, F.; Fernández-Rodríguez, B.; Cereijo, R.; Gallego-Escuredo, J.M.; Villarroya, F.; Pañeda-Menéndez, A. Histological and molecular features of lipomatous and nonlipomatous adipose tissue in familial partial lipodystrophy caused by LMNA mutations. Clin. Endocrinol. 2012, 76, 816–824. [Google Scholar] [CrossRef]
- Spuler, S.; Kalbhenn, T.; Zabojszcza, J.; van Landeghem, F.; Ludtke, A.; Wenzel, K.; Koehnlein, M.; Schuelke, M.; Ludemann, L.; Schmidt, H.H. Muscle and nerve pathology in Dunnigan familial partial lipodystrophy. Neurology 2007, 68, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Akinci, G.; Topaloglu, H.; Demir, T.; Danyeli, A.E.; Talim, B.; Keskin, F.E.; Kadioglu, P.; Talip, E.; Altay, C.; Yaylali, G.F.; et al. Clinical spectra of neuromuscular manifestations in patients with lipodystrophy: A multicenter study. Neuromuscul. Disord. 2017, 27, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Vantyghem, M.C.; Pigny, P.; Maurage, C.A.; Rouaix-Emery, N.; Stojkovic, T.; Cuisset, J.M.; Millaire, A.; Lascols, O.; Vermersch, P.; Wemeau, J.L.; et al. Patients with familial partial lipodystrophy of the Dunnigan type due to a LMNA R482W mutation show muscular and cardiac abnormalities. J. Clin. Endocrinol. Metab. 2004, 89, 5337–5346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burn, J.; Baraitser, M. Partial lipoatrophy with insulin resistant diabetes and hyperlipidaemia (Dunnigan syndrome). J. Med. Genet. 1986, 23, 128–130. [Google Scholar] [CrossRef] [Green Version]
- Morel, C.F.; Thomas, M.A.; Cao, H.; O’Neil, C.H.; Pickering, J.G.; Foulkes, W.D.; Hegele, R.A. A LMNA splicing mutation in two sisters with severe Dunnigan-type familial partial lipodystrophy type 2. J. Clin. Endocrinol. Metab. 2006, 91, 2689–2695. [Google Scholar] [CrossRef] [Green Version]
- Vaidya, R.A.; Vaidya, A.D.B.; Talwalkar, S.C.; Mehtalia, S.D.; Shringi, M.S.; Pandey, S.N.; Shah, S.J.; Godse, C.; Joshi, J.V.; Sheth, J.; et al. Clinical, endocrine and metabolic studies in the kindred of familial partial lipodystrophy--a syndrome of insulin resistance. J. Assoc. Physicians India 2002, 50, 773–776. [Google Scholar]
- Schmidt, H.H.; Genschel, J.; Baier, P.; Schmidt, M.; Ockenga, J.; Tietge, U.J.; Propsting, M.; Buttner, C.; Manns, M.P.; Lochs, H.; et al. Dyslipemia in familial partial lipodystrophy caused by an R482W mutation in the LMNA gene. J. Clin. Endocrinol. Metab. 2001, 86, 2289–2295. [Google Scholar] [CrossRef]
- Patni, N.; Li, X.; Adams-Huet, B.; Vasandani, C.; Gomez-Diaz, R.A.; Garg, A. Regional Body Fat Changes and Metabolic Complications in Children With Dunnigan Lipodystrophy-Causing LMNA Variants. J. Clin. Endocrinol. Metab. 2019, 104, 1099–1108. [Google Scholar] [CrossRef] [Green Version]
- Hegele, R.A.; Cao, H.; Anderson, C.M.; Hramiak, I.M. Heterogeneity of nuclear lamin A mutations in Dunnigan-type familial partial lipodystrophy. J. Clin. Endocrinol. Metab. 2000, 85, 3431–3435. [Google Scholar]
- Resende, A.T.P.; Martins, C.S.; Bueno, A.C.; Moreira, A.C.; Foss-Freitas, M.C.; de Castro, M. Phenotypic diversity and glucocorticoid sensitivity in patients with familial partial lipodystrophy type 2. Clin. Endocrinol. 2019, 91, 94–103. [Google Scholar] [CrossRef]
- Sorkina, E.L.; Kalashnikova, M.F.; Melnichenko, G.A.; Tyulpakov, A.N. [Familial partial lipodystrophy (Dunnigan syndrome) due to LMNA gene mutation: The first description of its clinical case in Russia]. Ter Arkh 2015, 87, 83–87. [Google Scholar] [CrossRef]
- Araújo-Vilar, D.; Fernández-Pombo, A.; Victoria, B.; Mosquera-Orgueira, A.; Cobelo-Gómez, S.; Castro-Pais, A.; Hermida-Ameijeiras, A.; Loidi, L.; Sanchez-Iglesias, S. Variable Expressivity and Allelic Heterogeneity in Type 2 Familial Partial Lipodystrophy: The p.(Thr528Met) LMNA Variant. J. Clin. Med. 2021, 10, 1497. [Google Scholar] [CrossRef]
- Mory, P.B.; Crispim, F.; Freire, M.B.; Salles, J.E.; Valério, C.M.; Godoy-Matos, A.F.; Dib, S.A. Phenotypic diversity in patients with lipodystrophy associated with LMNA mutations. Eur. J. Endocrinol. 2012, 167, 423–431. [Google Scholar] [CrossRef] [Green Version]
- Montenegro, R.M., Jr.; Costa-Riquetto, A.D.; Fernandes, V.O.; Montenegro, A.P.D.R.; de Santana, L.S.; Jorge, A.A.L.; Karbage, L.B.A.S.; Aguiar, L.B.; Carvalho, F.H.C.; Teles, M.G.; et al. Homozygous and Heterozygous Nuclear Lamin A p.R582C Mutation: Different Lipodystrophic Phenotypes in the Same Kindred. Front. Endocrinol. 2018, 9, 458. [Google Scholar] [CrossRef] [Green Version]
- Muschke, P.; Kölsch, U.; Jakubiczka, S.; Wieland, I.; Brune, T.; Wieacker, P. The heterozygous LMNA mutation p.R471G causes a variable phenotype with features of two types of familial partial lipodystrophy. Am. J. Med. Genet. A. 2007, 143A, 2810–2814. [Google Scholar] [PubMed]
- Akinci, B.; Onay, H.; Demir, T.; Savas-Erdeve, Ş.; Gen, R.; Simsir, I.Y.; Keskin, F.E.; Ertürk, M.S.; Uzum, A.K.; Yaylali, G.F.; et al. Clinical presentations, metabolic abnormalities and end-organ complications in patients with familial partial lipodystrophy. Metabolism 2017, 72, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Decaudain, A.; Vantyghem, M.C.; Guerci, B.; Hécart, A.C.; Auclair, M.; Reznik, Y.; Narbonne, H.; Ducluzeau, P.H.; Donadille, B.; Lebbe, C.; et al. New metabolic phenotypes in laminopathies: LMNA mutations in patients with severe metabolic syndrome. J. Clin. Endocrinol. Metab. 2007, 92, 4835–4844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Andrade, N.X.S.; Adiyaman, S.C.; Yuksel, B.D.; Ferrari, C.T.; Eldin, A.J.; Saydam, B.O.; Altay, C.; Sharma, P.; Bhave, N.; Little, A.; et al. Unusual presentations of LMNA-associated lipodystrophy with complex phenotypes and generalized fat loss: When the genetic diagnosis uncovers novel features. AACE Clin. Case Rep. 2020, 6, e79–e85. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, P.D.; Ryan, J.; Hodnett, P.A.; O’Halloran, D.; Maher, M.M. Quantitative whole-body MRI in familial partial lipodystrophy type 2: Changes in adipose tissue distribution coincide with biochemical improvement. AJR Am. J. Roentgenol. 2012, 199, W602–W606. [Google Scholar] [CrossRef]
- Monteiro, L.Z.; Foss-Freitas, M.C.; Júnior Montenegro, R.M.; Foss, M.C. Body fat distribution in women with familial partial lipodystrophy caused by mutation in the lamin A/C gene. Indian J. Endocrinol. Metab. 2012, 16, 136–138. [Google Scholar] [CrossRef]
- Godoy-Matos, A.F.; Valerio, C.M.; Moreira, R.O.; Momesso, D.P.; Bittencourt, L.K. Pancreatic fat deposition is increased and related to beta-cell function in women with familial partial lipodystrophy. Diabetol. Metab. Syndr. 2018, 10, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bensmaïne, F.; Benomar, K.; Espiard, S.; Vahe, C.; Le Mapihan, K.; Lion, G.; Lemdani, M.; Chazard, E.; Ernst, O.; Vigouroux, C.; et al. Irisin levels in LMNA-associated partial lipodystrophies. Diabetes Metab. 2019, 45, 67–75. [Google Scholar] [CrossRef]
- Ajluni, N.; Meral, R.; Neidert, A.H.; Brady, G.F.; Buras, E.; McKenna, B.; DiPaola, F.; Chenevert, T.L.; Horowitz, J.F.; Buggs-Saxton, C.; et al. Spectrum of disease associated with partial lipodystrophy: Lessons from a trial cohort. Clin. Endocrinol. 2017, 86, 698–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Attar, S.A.; Pollex, R.L.; Robinson, J.F.; A Miskie, B.; Walcarius, R.; Little, C.H.; Rutt, B.K.; Hegele, R.A. Quantitative and qualitative differences in subcutaneous adipose tissue stores across lipodystrophy types shown by magnetic resonance imaging. BMC. Med. Imaging 2007, 7, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joy, T.; Kennedy, B.A.; Al-Attar, S.; Rutt, B.K.; Hegele, R.A. Predicting abdominal adipose tissue among women with familial partial lipodystrophy. Metabolism 2009, 58, 828–834. [Google Scholar] [CrossRef] [PubMed]
- Altay, C.; Secil, M.; Demir, T.; Atik, T.; Akinci, G.; Kutbay, N.O.; Temeloglu, E.K.; Simsir, I.Y.; Ozisik, S.; Demir, L.; et al. Determining residual adipose tissue characteristics with MRI in patients with various subtypes of lipodystrophy. Diagn. Interv. Radiol. 2017, 23, 428–434. [Google Scholar] [CrossRef] [Green Version]
- Vasandani, C.; Li, X.; Sekizkardes, H.; Brown, R.J.; Garg, A. Phenotypic Differences Among Familial Partial Lipodystrophy Due to LMNA or PPARG Variants. J. Endocr. Soc. 2022, 6, bvac155. [Google Scholar] [CrossRef]
- Al-Attar, S.A.; Pollex, R.L.; Robinson, J.F.; A Miskie, B.; Walcarius, R.; Rutt, B.K.; Hegele, R.A. Semi-automated segmentation and quantification of adipose tissue in calf and thigh by MRI: A preliminary study in patients with monogenic metabolic syndrome. BMC. Med. Imaging 2006, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Demir, T.; Onay, H.; Savage, D.B.; Temeloglu, E.; Uzum, A.K.; Kadioglu, P.; Altay, C.; Ozen, S.; Demir, L.; Cavdar, U.; et al. Familial partial lipodystrophy linked to a novel peroxisome proliferator activator receptor -γ (PPARG) mutation, H449L: A comparison of people with this mutation and those with classic codon 482 Lamin A/C (LMNA) mutations. Diabet. Med. 2016, 33, 1445–1450. [Google Scholar] [CrossRef] [Green Version]
- Vasandani, C.; Li, X.; Sekizkardes, H.; Adams-Huet, B.; Brown, R.J.; Garg, A. Diagnostic Value of Anthropometric Measurements for Familial Partial Lipodystrophy, Dunnigan Variety. J. Clin. Endocrinol. Metab. 2020, 105, 2132–2141. [Google Scholar] [CrossRef]
- Valerio, C.M.; Zajdenverg, L.; de Oliveira, J.E.; Mory, P.B.; Moyses, R.S.; Godoy-Matos, A.F. Body composition study by dual-energy x-ray absorptiometry in familial partial lipodystrophy: Finding new tools for an objective evaluation. Diabetol. Metab. Syndr. 2012, 4, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godoy-Matos, A.F.; Moreira, R.O.; Valerio, C.M.; Mory, P.B.; Moises, R.S. A new method for body fat evaluation, body adiposity index, is useful in women with familial partial lipodystrophy. Obesity 2012, 20, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Meral, R.; Ryan, B.J.; Malandrino, N.; Jalal, A.; Neidert, A.H.; Muniyappa, R.; Akıncı, B.; Horowitz, J.F.; Brown, R.J.; Oral, E.A. “Fat Shadows” From DXA for the Qualitative Assessment of Lipodystrophy: When a Picture Is Worth a Thousand Numbers. Diabetes Care 2018, 41, 2255–2258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, H.; Weatherall, P.; Adams-Huet, B.; Garg, A. Increased skeletal muscle volume in women with familial partial lipodystrophy, Dunnigan variety. J. Clin. Endocrinol. Metab. 2013, 98, E1410–E1413. [Google Scholar] [CrossRef] [Green Version]
- Simha, V.; Lanza, I.R.; Dasari, S.; Klaus, K.A.; Le Brasseur, N.; Vuckovic, I.; Laurenti, M.C.; Cobelli, C.; Port, J.D.; Nair, K.S. Impaired Muscle Mitochondrial Function in Familial Partial Lipodystrophy. J. Clin. Endocrinol. Metab. 2022, 107, 346–362. [Google Scholar] [CrossRef]
- Hennekam, R.C. Hutchinson-Gilford progeria syndrome: Review of the phenotype. Am. J. Med. Genet. A. 2006, 140, 2603–2624. [Google Scholar] [CrossRef] [Green Version]
- Kosho, T.; Takahashi, J.; Momose, T.; Nakamura, A.; Sakurai, A.; Wada, T.; Yoshida, K.; Wakui, K.; Suzuki, T.; Kasuga, K.; et al. Mandibuloacral dysplasia and a novel LMNA mutation in a woman with severe progressive skeletal changes. Am. J. Med. Genet. A. 2007, 143A, 2598–2603. [Google Scholar] [CrossRef]
- Cunningham, V.J.; D’Apice, M.R.; Licata, N.; Novelli, G.; Cundy, T. Skeletal phenotype of mandibuloacral dysplasia associated with mutations in ZMPSTE24. Bone 2010, 47, 591–597. [Google Scholar] [CrossRef]
- Motegi, S.; Yokoyama, Y.; Uchiyama, A.; Ogino, S.; Takeuchi, Y.; Yamada, K.; Hattori, T.; Hashizume, H.; Ishikawa, Y.; Goto, M.; et al. First Japanese case of atypical progeroid syndrome/atypical Werner syndrome with heterozygous LMNA mutation. J. Dermatol. 2014, 41, 1047–1052. [Google Scholar] [CrossRef]
- Fernandez-Pombo, A.; Ossandon-Otero, J.A.; Guillín-Amarelle, C.; Sánchez-Iglesias, S.; Castro, A.I.; González-Méndez, B.; Rodríguez-García, S.; Rodriguez-Cañete, L.; Casanueva, F.F.; Araújo-Vilar, D. Bone mineral density in familial partial lipodystrophy. Clin. Endocrinol. 2018, 88, 44–50. [Google Scholar] [CrossRef]
- Teboul-Coré, S.; Rey-Jouvin, C.; Miquel, A.; Vatier, C.; Capeau, J.; Robert, J.-J.; Pham, T.; Lascols, O.; Berenbaum, F.; Laredo, J.-D.; et al. Bone imaging findings in genetic and acquired lipodystrophic syndromes: An imaging study of 24 cases. Skeletal. Radiol. 2016, 45, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Koo, E.; Foss-Freitas, M.C.; Meral, R.; Ozer, M.; Eldin, A.J.; Akinci, B.; Miller, N.; Rothberg, A.E.; Oral, E.A. The Metabolic Equivalent BMI in Patients with Familial Partial Lipodystrophy (FPLD) Compared with Those with Severe Obesity. Obesity 2021, 29, 274–278. [Google Scholar] [CrossRef]
- Guillín-Amarelle, C.; Sánchez-Iglesias, S.; Castro-Pais, A.; Rodriguez-Cañete, L.; Ordóñez-Mayán, L.; Pazos, M.; González-Méndez, B.; Rodríguez-García, S.; Casanueva, F.F.; Fernández-Marmiesse, A.; et al. Type 1 familial partial lipodystrophy: Understanding the Köbberling syndrome. Endocrine 2016, 54, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Herbst, K.L.; Tannock, L.R.; Deeb, S.S.; Purnell, J.Q.; Brunzell, J.D.; Chait, A. Köbberling type of familial partial lipodystrophy: An underrecognized syndrome. Diabetes Care 2003, 26, 1819–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Shali, K.; Cao, H.; Knoers, N.; Hermus, A.R.; Tack, C.J.; Hegele, R.A. A single-base mutation in the peroxisome proliferator-activated receptor gamma4 promoter associated with altered in vitro expression and partial lipodystrophy. J. Clin. Endocrinol. Metab. 2004, 89, 5655–5660. [Google Scholar] [CrossRef] [Green Version]
- Hegele, R.A.; Ur, E.; Ransom, T.P.; Cao, H. A frameshift mutation in peroxisome-proliferator-activated receptor-gamma in familial partial lipodystrophy subtype 3 (FPLD3; MIM 604367). Clin. Genet. 2006, 70, 360–362. [Google Scholar] [CrossRef]
- Francis, G.A.; Li, G.; Casey, R.; Wang, J.; Cao, H.; Leff, T.; Hegele, R.A. Peroxisomal proliferator activated receptor-gamma deficiency in a Canadian kindred with familial partial lipodystrophy type 3 (FPLD3). BMC Med. Genet. 2006, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Auclair, M.; Vigouroux, C.; Boccara, F.; Capel, E.; Vigeral, C.; Guerci, B.; Lascols, O.; Capeau, J.; Caron-Debarle, M. Peroxisome proliferator-activated receptor-γ mutations responsible for lipodystrophy with severe hypertension activate the cellular renin-angiotensin system. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 829–838. [Google Scholar] [CrossRef]
- Semple, R.K.; Chatterjee, V.K.; O’Rahilly, S. PPAR gamma and human metabolic disease. J. Clin. Investig. 2006, 116, 581–589. [Google Scholar] [CrossRef] [Green Version]
- Gandotra, S.; Le Dour, C.; Bottomley, W.; Cervera, P.; Giral, P.; Reznik, Y.; Charpentier, G.; Auclair, M.; Delépine, M.; Barroso, I.; et al. Perilipin deficiency and autosomal dominant partial lipodystrophy. N. Engl. J. Med. 2011, 364, 740–748. [Google Scholar] [CrossRef] [Green Version]
- Kozusko, K.; Tsang, V.; Bottomley, W.; Cho, Y.H.; Gandotra, S.; Mimmack, M.L.; Lim, K.; Isaac, I.; Patel, S.; Saudek, V.; et al. Clinical and molecular characterization of a novel PLIN1 frameshift mutation identified in patients with familial partial lipodystrophy. Diabetes 2015, 64, 299–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laver, T.W.; Patel, K.A.; Colclough, K.; Curran, J.; Dale, J.; Davis, N.; Savage, D.B.; Flanagan, S.E.; Ellard, S.; Hattersley, A.T.; et al. PLIN1 Haploinsufficiency Is Not Associated With Lipodystrophy. J. Clin. Endocrinol. Metab. 2018, 103, 3225–3230. [Google Scholar] [CrossRef] [PubMed]
- Jéru, I.; Vantyghem, M.C.; Bismuth, E.; Cervera, P.; Barraud, S.; PLIN1-Study Group; Auclair, M.; Vatier, C.; Lascols, O.; Savage, D.B.; et al. Diagnostic Challenge in PLIN1-Associated Familial Partial Lipodystrophy. J. Clin. Endocrinol. Metab. 2019, 104, 6025–6032. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Cabezas, O.; Puri, V.; Murano, I.; Saudek, V.; Semple, R.K.; Dash, S.; Hyden, C.S.S.; Bottomley, W.; Vigouroux, C.; Magré, J.; et al. Partial lipodystrophy and insulin resistant diabetes in a patient with a homozygous nonsense mutation in CIDEC. EMBO Mol. Med. 2009, 1, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Farhan, S.M.; Robinson, J.F.; McIntyre, A.D.; Marrosu, M.G.; Ticca, A.F.; Loddo, S.; Carboni, N.; Brancati, F.; Hegele, R.A. A novel LIPE nonsense mutation found using exome sequencing in siblings with late-onset familial partial lipodystrophy. Can. J. Cardiol. 2014, 30, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Carboni, N.; Brancati, F.; Cocco, E.; Solla, E.; D’Apice, M.R.; Mateddu, A.; McIntyre, A.; Fadda, E.; Mura, M.; Lattanzi, G.; et al. A partial lipodystrophy associated with muscular dystrophy of unknown genetic origin. Muscle Nerve 2014, 49, 928–930. [Google Scholar] [CrossRef]
- Zolotov, S.; Xing, C.; Mahamid, R.; Shalata, A.; Sheikh-Ahmad, M.; Garg, A. Homozygous LIPE mutation in siblings with multiple symmetric lipomatosis, partial lipodystrophy, and myopathy. Am. J. Med. Genet. A. 2017, 173, 190–194. [Google Scholar] [CrossRef] [Green Version]
- Sollier, C.; Capel, E.; Aguilhon, C.; Smirnov, V.; Auclair, M.; Douillard, C.; Ladsous, M.; Defoort-Dhellemmes, S.; Gorwood, J.; Braud, L.; et al. LIPE-related lipodystrophic syndrome: Clinical features and disease modeling using adipose stem cells. Eur. J. Endocrinol. 2021, 184, 155–168. [Google Scholar] [CrossRef]
- Berger, J.R.; Oral, E.A.; Taylor, S.I. Familial lipodystrophy associated with neurodegeneration and congenital cataracts. Neurology 2002, 58, 43–47. [Google Scholar] [CrossRef]
- Garg, A.; Kircher, M.; Del Campo, M.; Amato, R.S.; Agarwal, A.K.; University of Washington Center for Mendelian Genomics. Whole exome sequencing identifies de novo heterozygous CAV1 mutations associated with a novel neonatal onset lipodystrophy syndrome. Am. J. Med. Genet. A. 2015, 167A, 1796–1806. [Google Scholar] [PubMed] [Green Version]
- Shearin, A.L.; Monks, B.R.; Seale, P.; Birnbaum, M.J. Lack of AKT in adipocytes causes severe lipodystrophy. Mol. Metab. 2016, 5, 472–479. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Rochford, J.J.; Wolfrum, C.; Gray, S.L.; Schinner, S.; Wilson, J.C.; Soos, M.A.; Murgatroyd, P.R.; Williams, R.M.; Acerini, C.L.; et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science 2004, 304, 1325–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, F.; Lim, K.; Girousse, A.; Brown, R.J.; Kory, N.; Robbins, A.; Xue, Y.; Sleigh, A.; Cochran, E.; Adams, C.; et al. Mutations disrupting the Kennedy phosphatidylcholine pathway in humans with congenital lipodystrophy and fatty liver disease. Proc. Natl. Acad. Sci. USA 2014, 111, 8901–8906. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.; Sankella, S.; Xing, C.; Agarwal, A.K. Whole-exome sequencing identifies ADRA2A mutation in atypical familial partial lipodystrophy. JCI Insight 2016, 1, e86870. [Google Scholar] [CrossRef] [Green Version]
- Rocha, N.; Bulger, D.A.; Frontini, A.; Titheradge, H.; Gribsholt, S.B.; Knox, R.; Page, M.; Harris, J.; Payne, F.; Adams, C.; et al. Human biallelic MFN2 mutations induce mitochondrial dysfunction, upper body adipose hyperplasia, and suppression of leptin expression. Elife 2017, 6, e23813. [Google Scholar] [CrossRef]
- Sawyer, S.L.; Cheuk-Him Ng, A.; Innes, A.M.; Wagner, J.D.; Dyment, D.A.; Tetreault, M.; Consortium, C.R.C.; Majewski, J.; Boycott, K.M.; Screaton, R.A.; et al. Homozygous mutations in MFN2 cause multiple symmetric lipomatosis associated with neuropathy. Hum. Mol. Genet. 2015, 24, 5109–5114. [Google Scholar] [CrossRef] [Green Version]
- Capel, E.; Vatier, C.; Cervera, P.; Stojkovic, T.; Disse, E.; Cottereau, A.-S.; Auclair, M.; Verpont, M.-C.; Mosbah, H.; Gourdy, P.; et al. MFN2-associated lipomatosis: Clinical spectrum and impact on adipose tissue. J. Clin. Lipidol. 2018, 12, 1420–1435. [Google Scholar] [CrossRef]
- Zhong, Z.X.; Harris, J.; Wilber, E.; Gorman, S.; Savage, D.B.; O’Rahilly, S.; Stears, A.; Williams, R.M. Describing the natural history of clinical, biochemical and radiological outcomes of children with familial partial lipodystrophy type 2 (FPLD2) from the United Kingdom: A retrospective case series. Clin. Endocrinol. 2022, 97, 755–762. [Google Scholar] [CrossRef]
- Jeru, I.; Vatier, C.; Vantyghem, M.C.; Lascols, O.; Vigouroux, C. LMNA-associated partial lipodystrophy: Anticipation of metabolic complications. J. Med. Genet. 2017, 54, 413–416. [Google Scholar] [CrossRef]
- Wong, S.P.; Huda, M.; English, P.; Bargiota, A.; Wilding, J.P.; Corrall, R.; Pinkney, J.H. Adipokines and the insulin resistance syndrome in familial partial lipodystrophy caused by a mutation in lamin A/C. Diabetologia 2005, 48, 2641–2649. [Google Scholar] [CrossRef]
- Hegele, R.A.; Cao, H.; Huff, M.W.; Anderson, C.M. LMNA R482Q mutation in partial lipodystrophy associated with reduced plasma leptin concentration. J. Clin. Endocrinol. Metab. 2000, 85, 3089–3093. [Google Scholar]
- Hegele, R.A.; Kraw, M.E.; Ban, M.R.; Miskie, B.A.; Huff, M.W.; Cao, H. Elevated serum C-reactive protein and free fatty acids among nondiabetic carriers of missense mutations in the gene encoding lamin A/C (LMNA) with partial lipodystrophy. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Haque, W.A.; Shimomura, I.; Matsuzawa, Y.; Garg, A. Serum adiponectin and leptin levels in patients with lipodystrophies. J. Clin. Endocrinol. Metab. 2002, 87, 2395. [Google Scholar] [CrossRef]
- Treiber, G.; Guilleux, A.; Huynh, K.; Bonfanti, O.; Flaus–Furmaniuk, A.; Couret, D.; Mellet, N.; Bernard, C.; Le-Moullec, N.; Doray, B.; et al. Lipoatrophic diabetes in familial partial lipodystrophy type 2: From insulin resistance to diabetes. Diabetes Metab. 2022, 49, 101409. [Google Scholar] [CrossRef]
- Foss-Freitas, M.C.; Ferraz, R.C.; Monteiro, L.Z.; Gomes, P.M.; Iwakura, R.; de Freitas, L.C.C.; Foss, M.C. Endoplasmic reticulum stress activation in adipose tissue induces metabolic syndrome in individuals with familial partial lipodystrophy of the Dunnigan type. Diabetol. Metab. Syndr. 2018, 10, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godoy-Matos, A.F.; Moreira, R.O.; MacDowell, R.; Bendet, I.; Mory, P.B.; Moises, R.S. Serum retinol binding protein 4 in patients with familial partial lipodystrophy. Clin. Biochem. 2009, 42, 1183–1186. [Google Scholar] [CrossRef] [PubMed]
- Boschmann, M.; Engeli, S.; Moro, C.; Luedtke, A.; Adams, F.; Gorzelniak, K.; Rahn, G.; Mähler, A.; Dobberstein, K.; Kruger, A.; et al. LMNA mutations, skeletal muscle lipid metabolism, and insulin resistance. J. Clin. Endocrinol. Metab. 2010, 95, 1634–1643. [Google Scholar] [CrossRef] [PubMed]
- Araujo-Vilar, D.; Lattanzi, G.; Gonzalez-Mendez, B.; Costa-Freitas, A.T.; Prieto, D.; Columbaro, M.; Mattioli, E.; Victoria, B.; Martinez-Sanchez, N.; Ramazanova, A.; et al. Site-dependent differences in both prelamin A and adipogenic genes in subcutaneous adipose tissue of patients with type 2 familial partial lipodystrophy. J. Med. Genet. 2009, 46, 40–48. [Google Scholar] [CrossRef]
- UUrsich, M.J.; Fukui, R.T.; Galvão, M.S.; Marcondes, J.A.; Santomauro, A.T.; Silva, M.E.; Rocha, D.M.; Wajchenberg, B.L. Insulin resistance in limb and trunk partial lipodystrophy (type 2 Köbberling-Dunnigan syndrome). Metabolism 1997, 46, 159–163. [Google Scholar] [CrossRef]
- Araújo-Vilar, D.; Loidi, L.; Domínguez, F.; Cabezas-Cerrato, J. Phenotypic gender differences in subjects with familial partial lipodystrophy (Dunnigan variety) due to a nuclear lamin A/C R482W mutation. Horm. Metab. Res. 2003, 35, 29–35. [Google Scholar] [CrossRef]
- Haque, W.A.; Oral, E.A.; Dietz, K.; Bowcock, A.M.; Agarwal, A.K.; Garg, A. Risk factors for diabetes in familial partial lipodystrophy, Dunnigan variety. Diabetes Care 2003, 26, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Hegele, R.A. Nuclear lamin A/C R482Q mutation in canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum. Mol. Genet. 2000, 9, 109–112. [Google Scholar] [CrossRef] [Green Version]
- Caldas, D.; Silva Júnior, W.S.; Simonetti, J.P.; Costa, E.V.; Farias, M.L. Biochemical, hormonal and genetic evaluation of the families of two Brazilian patients with type 2 familial partial lipodystrophy. Arq. Bras. Endocrinol. Metabol. 2013, 57, 583–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarte, J.; Wang, J.; McIntyre, A.D.; Hegele, R.A. Prevalence of severe hypertriglyceridemia and pancreatitis in familial partial lipodystrophy type 2. J. Clin. Lipidol. 2021, 15, 653–657. [Google Scholar] [CrossRef]
- Ludtke, A.; Genschel, J.; Brabant, G.; Bauditz, J.; Taupitz, M.; Koch, M.; Wermke, W.; Worman, H.J.; Schmidt, H.H.-J. Hepatic steatosis in Dunnigan-type familial partial lipodystrophy. Am. J. Gastroenterol. 2005, 100, 2218–2224. [Google Scholar] [CrossRef]
- Speckman, R.A.; Garg, A.; Du, F.; Bennett, L.; Veile, R.; Arioglu, E.; Taylor, S.I.; Lovett, M.; Bowcock, A.M. Mutational and haplotype analyses of families with familial partial lipodystrophy (Dunnigan variety) reveal recurrent missense mutations in the globular C-terminal domain of lamin A/C. Am. J. Hum. Genet. 2000, 66, 1192–1198. [Google Scholar] [CrossRef] [Green Version]
- Kutbay, N.O.; Yurekli, B.S.; Onay, H.; Altay, C.T.; Atik, T.; Hekimsoy, Z.; Saygili, F.; Akinci, B. A case of familial partial lipodystrophy caused by a novel lamin A/C (LMNA) mutation in exon 1 (D47N). Eur. J. Intern. Med. 2016, 29, 37–39. [Google Scholar] [CrossRef]
- Haque, W.A.; Vuitch, F.; Garg, A. Post-mortem findings in familial partial lipodystrophy, Dunnigan variety. Diabetes Med. 2002, 19, 1022–1025. [Google Scholar] [CrossRef] [PubMed]
- Nabrdalik, K.; Strózik, A.; Minkina-Pędras, M.; Jarosz-Chobot, P.; Młynarski, W.; Grzeszczak, W.; Gumprecht, J. Dunnigan-type familial partial lipodystrophy associated with the heterozygous R482W mutation in LMNA gene—Case study of three women from one family. Endokrynol. Pol. 2013, 64, 306–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araújo-Vilar, D.; Sánchez-Iglesias, S.; Castro, A.; Cobelo-Gómez, S.; Hermida-Ameijeiras, A.; Rodríguez-Carnero, G.; Casanueva, F.; Fernández-Pombo, A. Variable Expressivity in Type 2 Familial Partial Lipodystrophy Related to R482 and N466 Variants in the LMNA Gene. J. Clin. Med. 2021, 10, 1259. [Google Scholar] [CrossRef]
- Araújo-Vilar, D.; Lado-Abeal, J.; Palos-Paz, F.; Lattanzi, G.; Bandín, M.A.; Bellido, D.; Domínguez-Gerpe, L.; Calvo, C.; Pérez, O.; Ramazanova, A.; et al. A novel phenotypic expression associated with a new mutation in LMNA gene, characterized by partial lipodystrophy, insulin resistance, aortic stenosis and hypertrophic cardiomyopathy. Clin. Endocrinol. 2008, 69, 61–68. [Google Scholar] [CrossRef]
- Garg, A.; Speckman, R.A.; Bowcock, A.M. Multisystem dystrophy syndrome due to novel missense mutations in the amino-terminal head and alpha-helical rod domains of the lamin A/C gene. Am. J. Med. 2002, 112, 549–555. [Google Scholar] [CrossRef]
- Subramanyam, L.; Simha, V.; Garg, A. Overlapping syndrome with familial partial lipodystrophy, Dunnigan variety and cardiomyopathy due to amino-terminal heterozygous missense lamin A/C mutations. Clin. Genet. 2010, 78, 66–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bidault, G.; Garcia, M.; Vantyghem, M.-C.; Ducluzeau, P.-H.; Morichon, R.; Thiyagarajah, K.; Moritz, S.; Capeau, J.; Vigouroux, C.; Béréziat, V. Lipodystrophy-linked LMNA p.R482W mutation induces clinical early atherosclerosis and in vitro endothelial dysfunction. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2162–2171. [Google Scholar] [CrossRef] [Green Version]
- Hegele, R.A. Premature atherosclerosis associated with monogenic insulin resistance. Circulation 2001, 103, 2225–2229. [Google Scholar] [CrossRef] [Green Version]
- Weterings, A.A.; van Rijsingen, I.A.; Plomp, A.S.; Zwinderman, A.H.; Lekanne Deprez, R.H.; Mannens, M.M.; van den Bergh Weerman, M.A.; van der Wal, A.C.; Pinto-Sietsma, S.J. A novel lamin A/C mutation in a Dutch family with premature atherosclerosis. Atherosclerosis 2013, 229, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Eldin, A.J.; Akinci, B.; da Rocha, A.M.; Meral, R.; Simsir, I.Y.; Adiyaman, S.C.; Ozpelit, E.; Bhave, N.; Gen, R.; Yurekli, B.; et al. Cardiac phenotype in familial partial lipodystrophy. Clin. Endocrinol. 2021, 94, 1043–1053. [Google Scholar] [CrossRef]
- Godoy-Matos, A.F.; Valério, C.M.; Bragança, J.B.; Oliveira Rde, A.; Zagury, R.L.; Lustosa Rde, P.; Camargo, G.C.; Nascimento, C.A.; Moreira, R.O. Evaluation of epicardial adipose tissue in familial partial lipodystrophy. Diabetol. Metab. Syndr. 2015, 7, 29. [Google Scholar] [CrossRef] [Green Version]
- Kwapich, M.; Lacroix, D.; Espiard, S.; Ninni, S.; Brigadeau, F.; Kouakam, C.; Degroote, P.; Laurent, J.M.; Tiffreau, V.; Jannin, A.; et al. Diamenord–AEDNL Working Group. Cardiometabolic assessment of lamin A/C gene mutation carriers: A phenotype-genotype correlation. Diabetes Metab. 2019, 45, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Andre, P.; Schneebeli, S.; Vigouroux, C.; Lascols, O.; Schaaf, M.; Chevalier, P. Metabolic and cardiac phenotype characterization in 37 atypical Dunnigan patients with nonfarnesylated mutated prelamin A. Am. Heart J. 2015, 169, 587–593. [Google Scholar] [CrossRef]
- Vantyghem, M.C.; Vincent-Desplanques, D.; Defrance-Faivre, F.; Capeau, J.; Fermon, C.; Valat, A.S.; Lascols, O.; Hecart, A.C.; Pigny, P.; Delemer, B.; et al. Fertility and obstetrical complications in women with LMNA-related familial partial lipodystrophy. J. Clin. Endocrinol. Metab. 2008, 93, 2223–2229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambineri, A.; Semple, R.K.; Forlani, G.; Genghini, S.; Grassi, I.; Hyden, C.S.S.; Pagotto, U.; O’Rahilly, S.; Pasquali, R. Monogenic polycystic ovary syndrome due to a mutation in the lamin A/C gene is sensitive to thiazolidinediones but not to metformin. Eur. J. Endocrinol. 2008, 159, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Joy, T.R.; Hegele, R.A. Prevalence of reproductive abnormalities among women with familial partial lipodystrophy. Endocr. Pract. 2008, 14, 1126–1132. [Google Scholar] [CrossRef] [Green Version]
- Hegele, R.A. Lessons from human mutations in PPARgamma. Int. J. Obes. 2005, 29 (Suppl. S1), S31–S35. [Google Scholar] [CrossRef] [Green Version]
- Akinci, B.; Unlu, S.M.; Celik, A.; Simsir, I.Y.; Sen, S.; Nur, B.; Keskin, F.E.; Saydam, B.O.; Ozdemir, N.K.; Yurekli, B.S.; et al. Renal complications of lipodystrophy: A closer look at the natural history of kidney disease. Clin. Endocrinol. 2018, 89, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Thong, K.M.; Xu, Y.; Cook, J.; Takou, A.; Wagner, B.; Kawar, B.; Ong, A.C. Cosegregation of focal segmental glomerulosclerosis in a family with familial partial lipodystrophy due to a mutation in LMNA. Nephron. Clin. Pract. 2013, 124, 31–37. [Google Scholar] [CrossRef]
- Rankin, J.; Auer-Grumbach, M.; Bagg, W.; Colclough, K.; Duong, N.T.; Fenton-May, J.; Hattersley, A.; Hudson, J.; Jardine, P.; Josifova, D.; et al. Extreme phenotypic diversity and nonpenetrance in families with the LMNA gene mutation R644C. Am. J. Med. Genet. A. 2008, 146A, 1530–1542. [Google Scholar] [CrossRef] [PubMed]
- Lado-Abeal, J.; Calvo, R.M.; Victoria, B.; Castro, I.; Obregon, M.J.; Araujo-Vilar, D. Regional decrease of subcutaneous adipose tissue in patients with type 2 familial partial lipodystrophy is associated with changes in thyroid hormone metabolism. Thyroid 2010, 20, 419–424. [Google Scholar] [CrossRef] [Green Version]
- Youssef, S.J.; Macielak, R.J.; Schimmenti, L.A.; Chatzopoulos, K.; Price, D.L. Hypopharyngeal Squamous Cell Carcinoma in Sisters with LMNA Associated Familial Partial Lipodystrophy: A Case Report and Review of the Literature. Ann. Otol. Rhinol. Laryngol. 2020, 129, 1243–1246. [Google Scholar] [CrossRef]
- Akinci, B.; A Oral, E.; Neidert, A.; Rus, D.; Cheng, W.Y.; Thompson-Leduc, P.; Cheung, H.C.; Bradt, P.; De Freitas, M.C.F.; Montenegro, R.M.; et al. Comorbidities and Survival in Patients With Lipodystrophy: An International Chart Review Study. J. Clin. Endocrinol. Metab. 2019, 104, 5120–5135. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.; Ali, O.; Akinci, B.; de Freitas, M.C.F.; Montenegro, R.M.; Fernandes, V.O.; Gupta, D.; Lou, K.-J.; Tuttle, E.; A Oral, E.; et al. Effect of Leptin Therapy on Survival in Generalized and Partial Lipodystrophy: A Matched Cohort Analysis. J. Clin. Endocrinol. Metab. 2021, 106, e2953–e2967. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, L.; Foss-Freitas, M.C.; Navarro, A.; Pereira, F.; Coeli, F.; Carneseca, E.; Junior, R.M.; Foss, M. Evaluation of Dietary Intake, Leisure-Time Physical Activity, and Metabolic Profile in Women with Mutation in the LMNA Gene. J. Am. Coll. Nutr. 2017, 36, 248–252. [Google Scholar] [CrossRef]
- Brown, R.J.; Araujo-Vilar, D.; Cheung, P.T.; Dunger, D.; Garg, A.; Jack, M.; Mungai, L.; Oral, E.A.; Patni, N.; Rother, K.I.; et al. The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline. J. Clin. Endocrinol. Metab. 2016, 101, 4500–4511. [Google Scholar] [CrossRef]
- Luedtke, A.; Boschmann, M.; Colpe, C.; Engeli, S.; Adams, F.; Birkenfeld, A.L.; Haufe, S.; Rahn, G.; Luft, F.C.; Schmidt, H.H.; et al. Thiazolidinedione response in familial lipodystrophy patients with LMNA mutations: A case series. Horm. Metab. Res. 2012, 44, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Valerio, C.M.; de Almeida, J.S.; Moreira, R.O.; Aguiar, L.B.S.; Siciliano, P.O.; Carvalho, D.P.; Godoy-Matos, A.F. Dipeptidyl peptidase-4 levels are increased and partially related to body fat distribution in patients with familial partial lipodystrophy type 2. Diabetol. Metab. Syndr. 2017, 9, 26. [Google Scholar] [CrossRef] [Green Version]
- Oral, E.A.; Garg, A.; Tami, J.; Huang, E.A.; O’Dea, L.S.L.; Schmidt, H.; Tiulpakov, A.; Mertens, A.; Alexander, V.J.; Watts, L.; et al. Assessment of efficacy and safety of volanesorsen for treatment of metabolic complications in patients with familial partial lipodystrophy: Results of the BROADEN study: Volanesorsen in FPLD; The BROADEN Study. J. Clin. Lipidol. 2022, 16, 833–849. [Google Scholar] [CrossRef] [PubMed]
- Foss-Freitas, M.C.; Akinci, B.; Neidert, A.; Bartlett, V.J.; Hurh, E.; Karwatowska-Prokopczuk, E.; Oral, E.A. Selective targeting of angiopoietin-like 3 (ANGPTL3) with vupanorsen for the treatment of patients with familial partial lipodystrophy (FPLD): Results of a proof-of-concept study. Lipids Health Dis. 2021, 20, 174. [Google Scholar] [CrossRef]
- Park, J.Y.; Javor, E.D.; Cochran, E.K.; DePaoli, A.M.; Gorden, P. Long-term efficacy of leptin replacement in patients with Dunnigan-type familial partial lipodystrophy. Metabolism 2007, 56, 508–516. [Google Scholar] [CrossRef] [Green Version]
- Mosbah, H.; Vantyghem, M.C.; Nobécourt, E.; Andreelli, F.; Archambeaud, F.; Bismuth, E.; Briet, C.; Cartigny, M.; Chevalier, B.; Donadille, B.; et al. Therapeutic indications and metabolic effects of metreleptin in patients with lipodystrophy syndromes: Real-life experience from a national reference network. Diabetes Obes. Metab. 2022, 24, 1565–1577. [Google Scholar] [CrossRef]
- Simha, V.; Subramanyam, L.; Szczepaniak, L.; Quittner, C.; Adams-Huet, B.; Snell, P.; Garg, A. Comparison of efficacy and safety of leptin replacement therapy in moderately and severely hypoleptinemic patients with familial partial lipodystrophy of the Dunnigan variety. J. Clin. Endocrinol. Metab. 2012, 97, 785–792. [Google Scholar] [CrossRef] [Green Version]
- Oral, E.A.; Gorden, P.; Cochran, E.; Araújo-Vilar, D.; Savage, D.B.; Long, A.; Fine, G.; Salinardi, T.; Brown, R.J. Long-term effectiveness and safety of metreleptin in the treatment of patients with partial lipodystrophy. Endocrine 2019, 64, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Vatier, C.; Fetita, S.; Boudou, P.; Tchankou, C.; Deville, L.; Riveline, J.; Young, J.; Mathivon, L.; Travert, F.; Morin, D.; et al. One-year metreleptin improves insulin secretion in patients with diabetes linked to genetic lipodystrophic syndromes. Diabetes Obes. Metab. 2016, 18, 693–697. [Google Scholar] [CrossRef] [Green Version]
- Javor, E.D.; Ghany, M.G.; Cochran, E.K.; Oral, E.A.; DePaoli, A.M.; Premkumar, A.; Kleiner, D.E.; Gorden, P. Leptin reverses nonalcoholic steatohepatitis in patients with severe lipodystrophy. Hepatology 2005, 41, 753–760. [Google Scholar] [CrossRef]
- Miehle, K.; Stumvoll, M.; Fasshauer, M.; Hierl, T. Facial soft tissue volume decreases during metreleptin treatment in patients with partial and generalized lipodystrophy. Endocrine 2017. 58, 262–266. [CrossRef]
- Meral, R.; Malandrino, N.; Walter, M.; Neidert, A.H.; Muniyappa, R.; Oral, E.A.; Brown, R.J. Endogenous Leptin Concentrations Poorly Predict Metreleptin Response in Patients With Partial Lipodystrophy. J. Clin. Endocrinol. Metab. 2022, 107, e1739–e1751. [Google Scholar] [CrossRef]
- Sekizkardes, H.; Cochran, E.; Malandrino, N.; Garg, A.; Brown, R.J. Efficacy of Metreleptin Treatment in Familial Partial Lipodystrophy Due to PPARG vs LMNA Pathogenic Variants. J. Clin. Endocrinol. Metab. 2019, 104, 3068–3076. [Google Scholar] [CrossRef] [PubMed]
- Oldenburg, A.; Briand, N.; Sørensen, A.L.; Cahyani, I.; Shah, A.; Moskaug, J.Ø.; Collas, P. A lipodystrophy-causing lamin A mutant alters conformation and epigenetic regulation of the anti-adipogenic MIR335 locus. J. Cell Biol. 2017, 216, 2731–2743. [Google Scholar] [CrossRef] [Green Version]
- Vadrot, N.; Duband-Goulet, I.; Cabet, E.; Attanda, W.; Barateau, A.; Vicart, P.; Gerbal, F.; Briand, N.; Vigouroux, C.; Oldenburg, A.R.; et al. The p.R482W substitution in A-type lamins deregulates SREBP1 activity in Dunnigan-type familial partial lipodystrophy. Hum. Mol. Genet. 2015, 24, 2096–2109. [Google Scholar] [CrossRef] [Green Version]
- Kuo, F.-C.; Neville, M.J.; Sabaratnam, R.; Wesolowska-Andersen, A.; Phillips, D.; Wittemans, L.B.; van Dam, A.D.; Loh, N.Y.; Todorčević, M.; Denton, N.; et al. HOTAIR interacts with PRC2 complex regulating the regional preadipocyte transcriptome and human fat distribution. Cell Rep. 2022, 40, 111136. [Google Scholar] [CrossRef]
- Araújo-Vilar, D.; Fernández-Pombo, A.; Rodríguez-Carnero, G.; Martínez-Olmos, M.; Cantón, A.; Villar-Taibo, R.; Hermida-Ameijeiras, Á.; Santamaría-Nieto, A.; Díaz-Ortega, C.; Martínez-Rey, C.; et al. LipoDDx: A mobile application for identification of rare lipodystrophy syndromes. Orphanet J. Rare Dis. 2020, 15, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Cunha Olegario, N.B.; da Cunha Neto, J.S.; Barbosa, P.C.S.; Pinheiro, P.R.; Landim, P.L.A.; Montenegro, A.P.D.R.; Fernandes, V.O.; de Albuquerque, V.H.C.; Duarte, J.B.F.; da Cruz Paiva Lima, G.E.; et al. Identifying congenital generalized lipodystrophy using deep learning-DEEPLIPO. Sci. Rep. 2023, 13, 2176. [Google Scholar] [CrossRef] [PubMed]
- von Schnurbein, J.; Adams, C.; Akinci, B.; Ceccarini, G.; D’Apice, M.R.; Gambineri, A.; Hennekam, R.C.M.; Jeru, I.; Lattanzi, G.; Miehle, K.; et al. European lipodystrophy registry: Background and structure. Orphanet J. Rare Dis. 2020, 15, 17. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FPLD2 Main Characteristics [4,5,6,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128] | Differential Characteristics of other FPLD Syndromes [63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87] | |
| Gene | LMNA | Unknown (FPLD1), PPARG (FPLD3), PLIN1 (FPLD4), CIDEC (FPLD5), LIPE (FPLD6), CAV1 (FPLD7), AKT2, PCYT1A, ADRA2A, MFN2. |
| Inheritance | AD (homozygous cases have also been described). | AR in FPLD5, FPLD6, PCYT1A- and MFN2-related FPLD |
| Onset of phenotype | Puberty in women, later in men. |
|
| Abnormal fat distribution |
|
|
| Clinical features |
|
|
| Analytical parameters |
|
|
| Main comorbidities |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandez-Pombo, A.; Diaz-Lopez, E.J.; Castro, A.I.; Sanchez-Iglesias, S.; Cobelo-Gomez, S.; Prado-Moraña, T.; Araujo-Vilar, D. Clinical Spectrum of LMNA-Associated Type 2 Familial Partial Lipodystrophy: A Systematic Review. Cells 2023, 12, 725. https://doi.org/10.3390/cells12050725
Fernandez-Pombo A, Diaz-Lopez EJ, Castro AI, Sanchez-Iglesias S, Cobelo-Gomez S, Prado-Moraña T, Araujo-Vilar D. Clinical Spectrum of LMNA-Associated Type 2 Familial Partial Lipodystrophy: A Systematic Review. Cells. 2023; 12(5):725. https://doi.org/10.3390/cells12050725
Chicago/Turabian StyleFernandez-Pombo, Antia, Everardo Josue Diaz-Lopez, Ana I. Castro, Sofia Sanchez-Iglesias, Silvia Cobelo-Gomez, Teresa Prado-Moraña, and David Araujo-Vilar. 2023. "Clinical Spectrum of LMNA-Associated Type 2 Familial Partial Lipodystrophy: A Systematic Review" Cells 12, no. 5: 725. https://doi.org/10.3390/cells12050725