
Abnormal Cellular Phenotypes Induced by Three TMPO/LAP2 Variants Identified in Men with Cardiomyopathies
 , , , , and
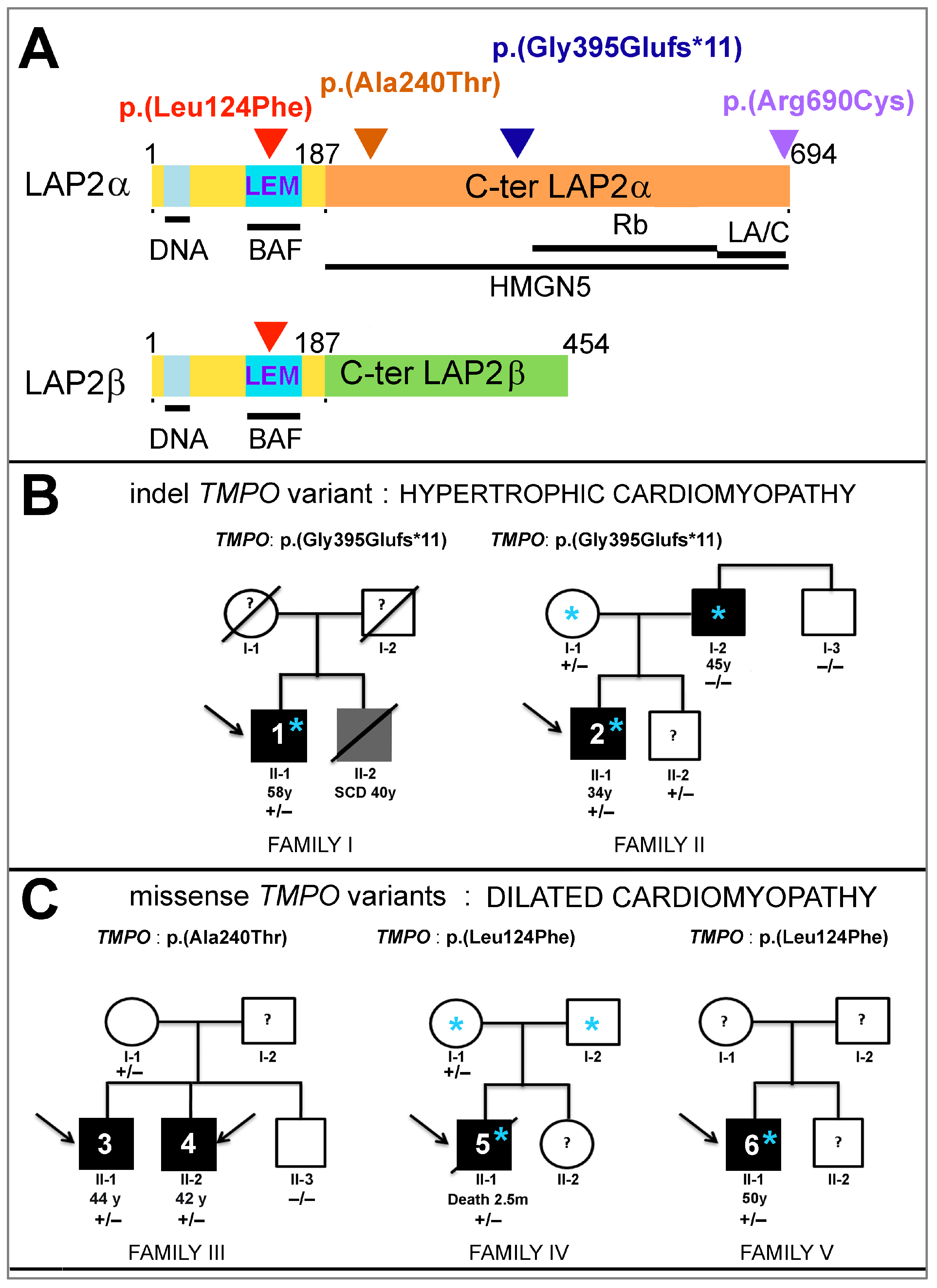
, , , , and 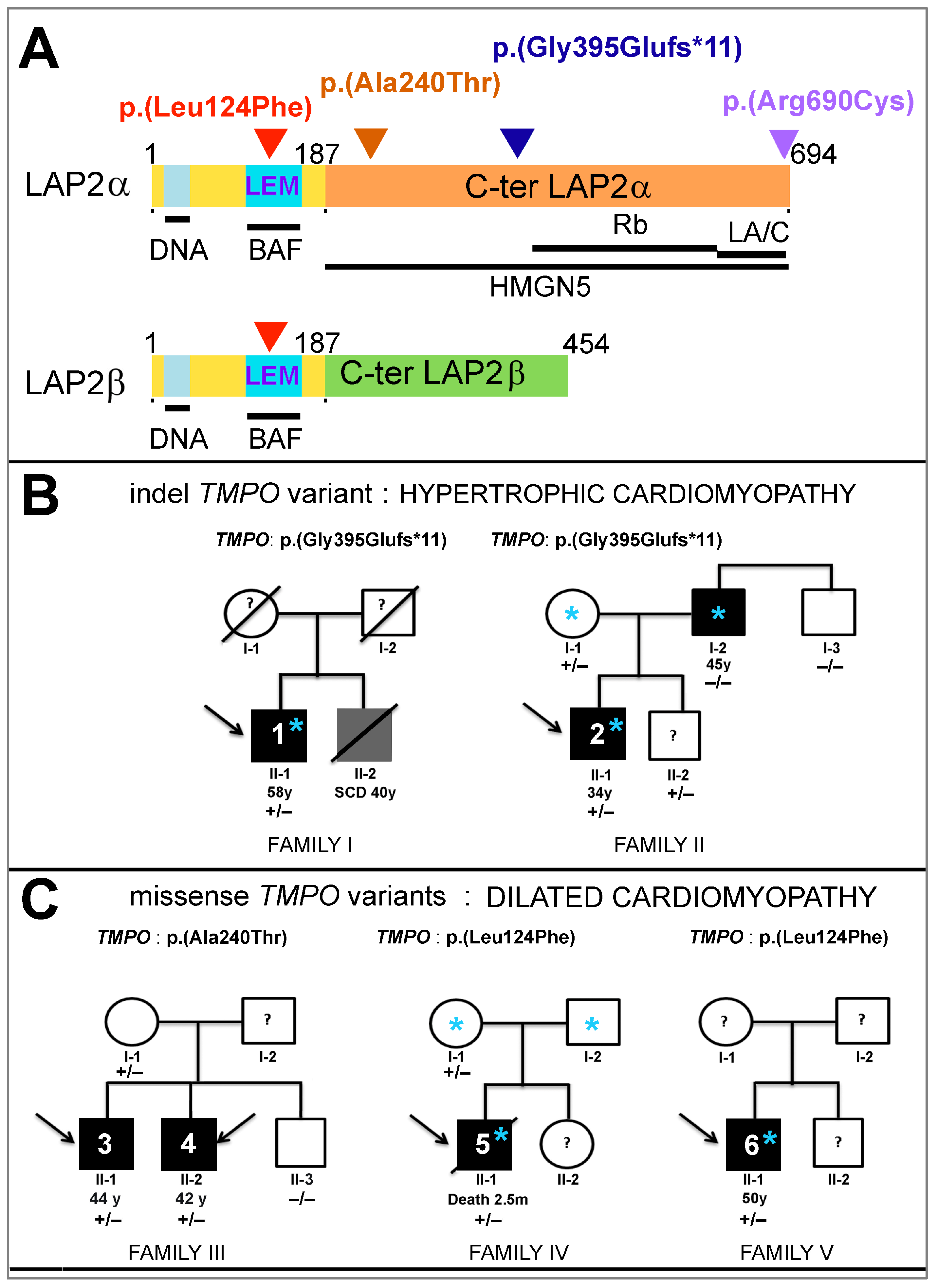{kind=link}
{kind=link}
{kind=link}
{kind=link}
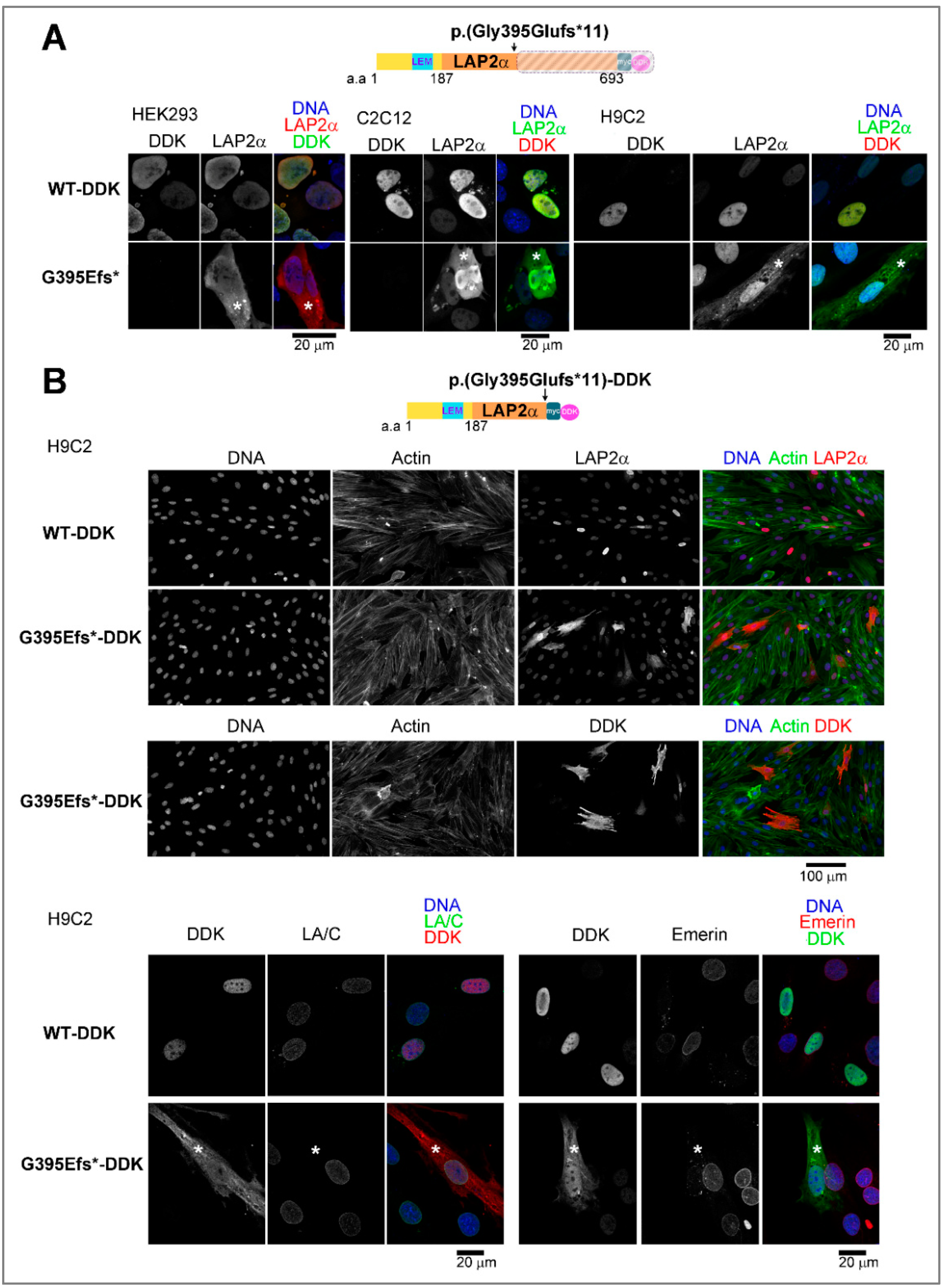{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cardiomyopathy Gene Panel Sequencing
2.2. Cell Culture and Transfection
2.3. Plasmids
2.4. qRT-PCR
2.5. Immunofluorescence Methods
2.6. Cell Protein Extract Preparation and Protein Analysis by Western Blot
2.7. Co-Immunoprecipitations
2.8. Antibodies
2.9. Reporter Assay
2.10. Proximity Ligation Assays (PLA)
2.11. Mouse Heart Sample Extract Preparation and Protein Analysis
2.12. Statistical Analyses
2.13. Collection of Biological Samples
3. Results
3.1. Three Novel TMPO Variants Identified in Patients with Cardiomyopathies
3.2. Clinical Phenotype of Patients Harboring New TMPO Variants
3.2.1. Patients Harboring the TMPO Frameshift Variant g.98927219del_c.1184del_p.(Gly395Glufs*11)
3.2.2. Clinical Phenotype of Patients Harboring the Missense Variant g.98926753G>A_c.718G>A_p.(Ala240Thr)
3.2.3. Clinical Phenotype of Patients Harboring the Missense Variant g.98921754C>T_c.370C>T_p.(Leu124Phe)
3.3. Sex and Cardiomyopathy Associated with TMPO Variants
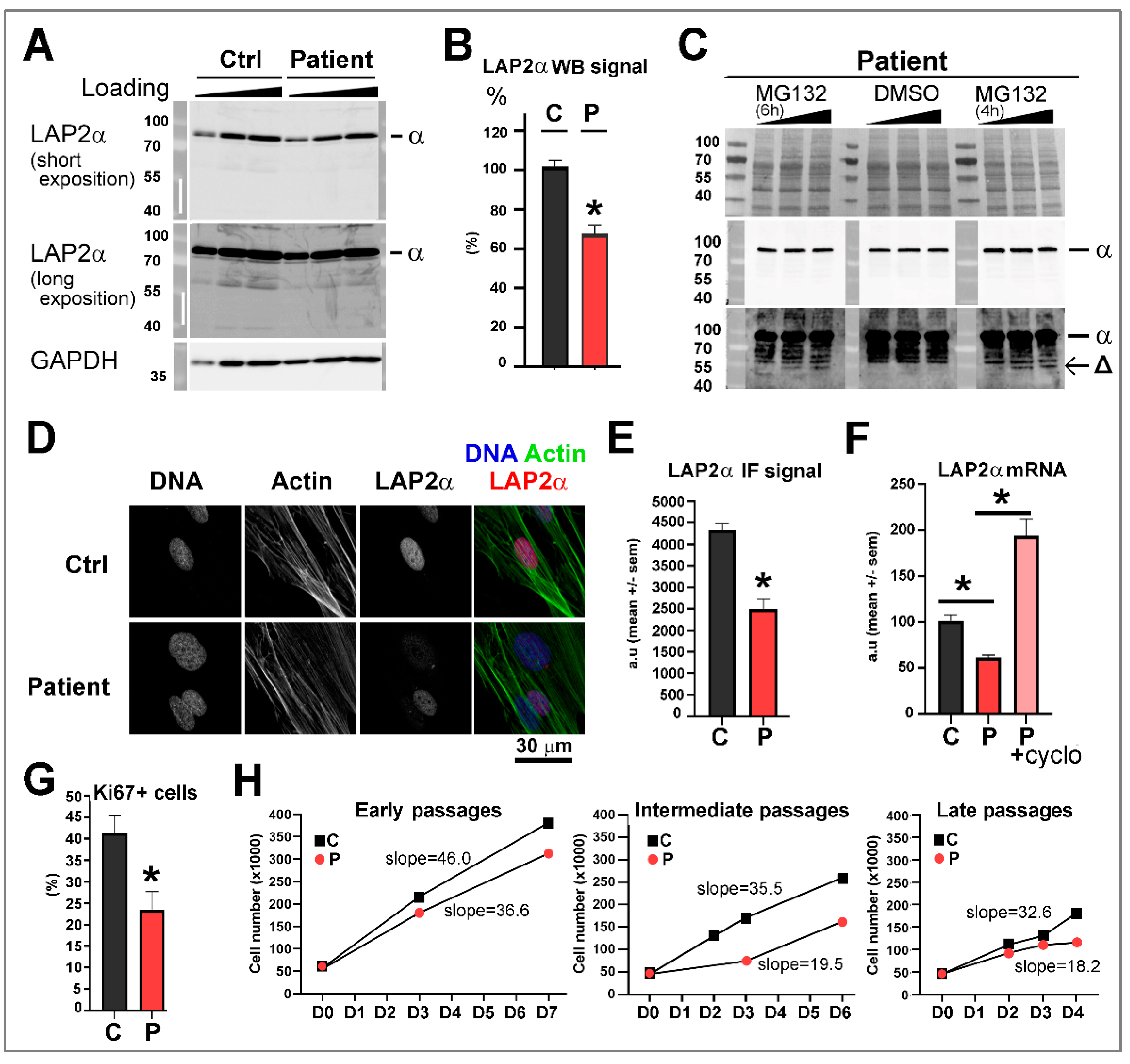
3.4. Dermal Fibroblasts from Patient 1 Harboring the TMPO Variant p.(Gly395Glufs*11), Present a Reduced LAP2α Expression Level Associated with a Decrease in Cell Proliferation
3.5. Overexpression of TMPO/LAP2 Variants Triggers Abnormal Distinct Cell Phenotypes
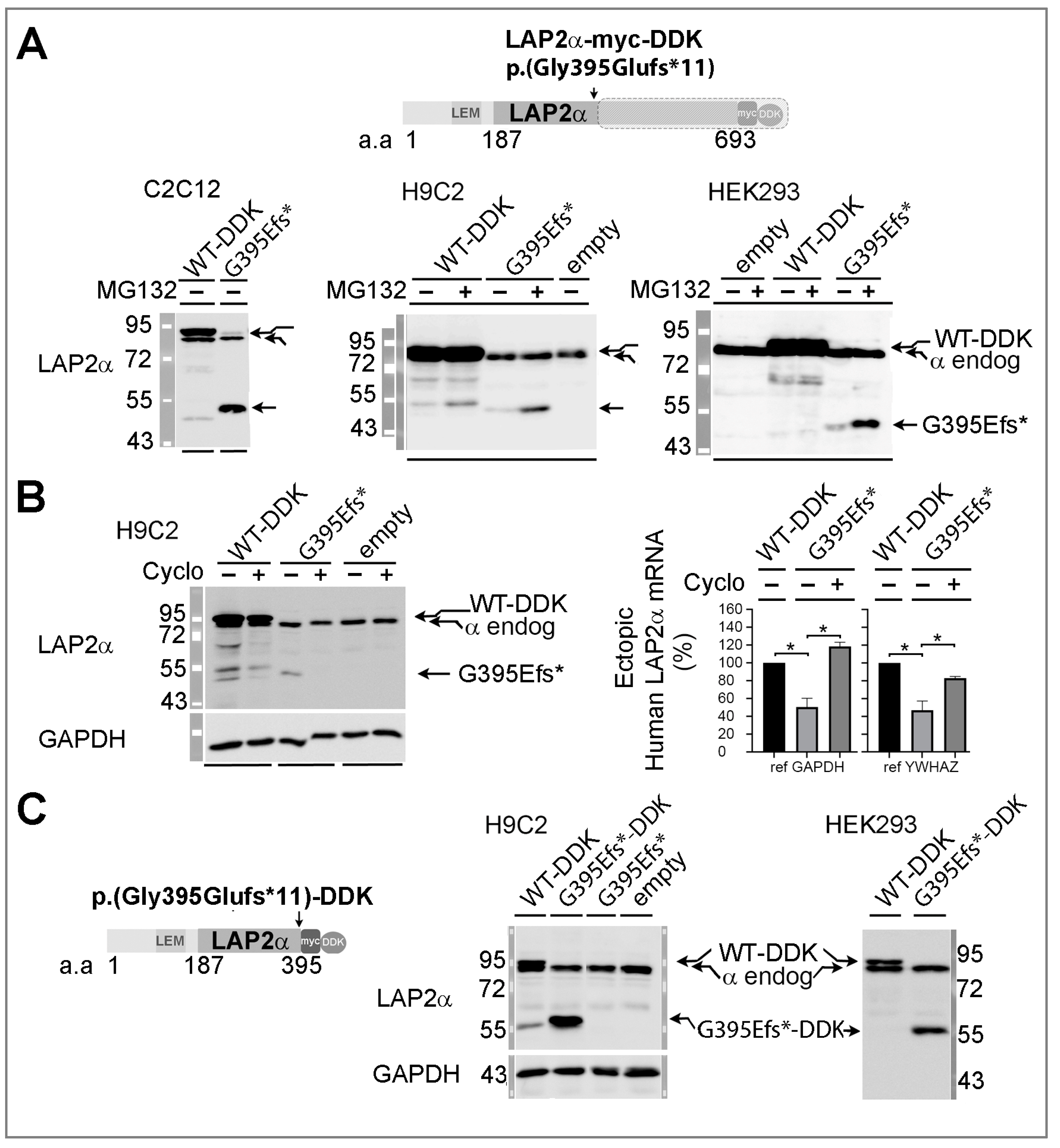
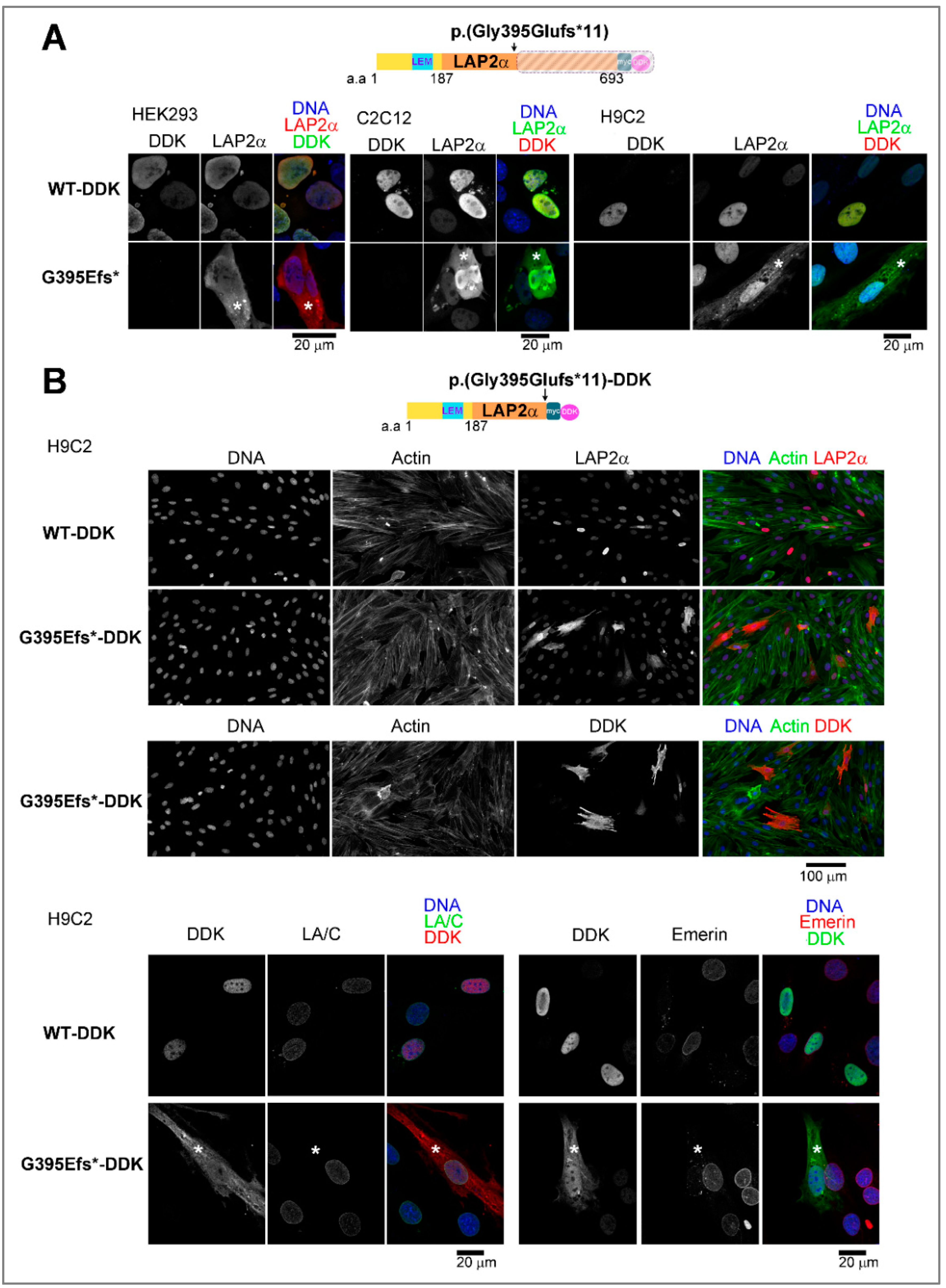
3.5.1. Ectopic Expression of the TMPO Indel Variant p.(Gly395Glufs*11) Leads to Full Length LAP2α Quantitative Defects and to Mislocalization of a Truncated LAP2α Protein in Cells
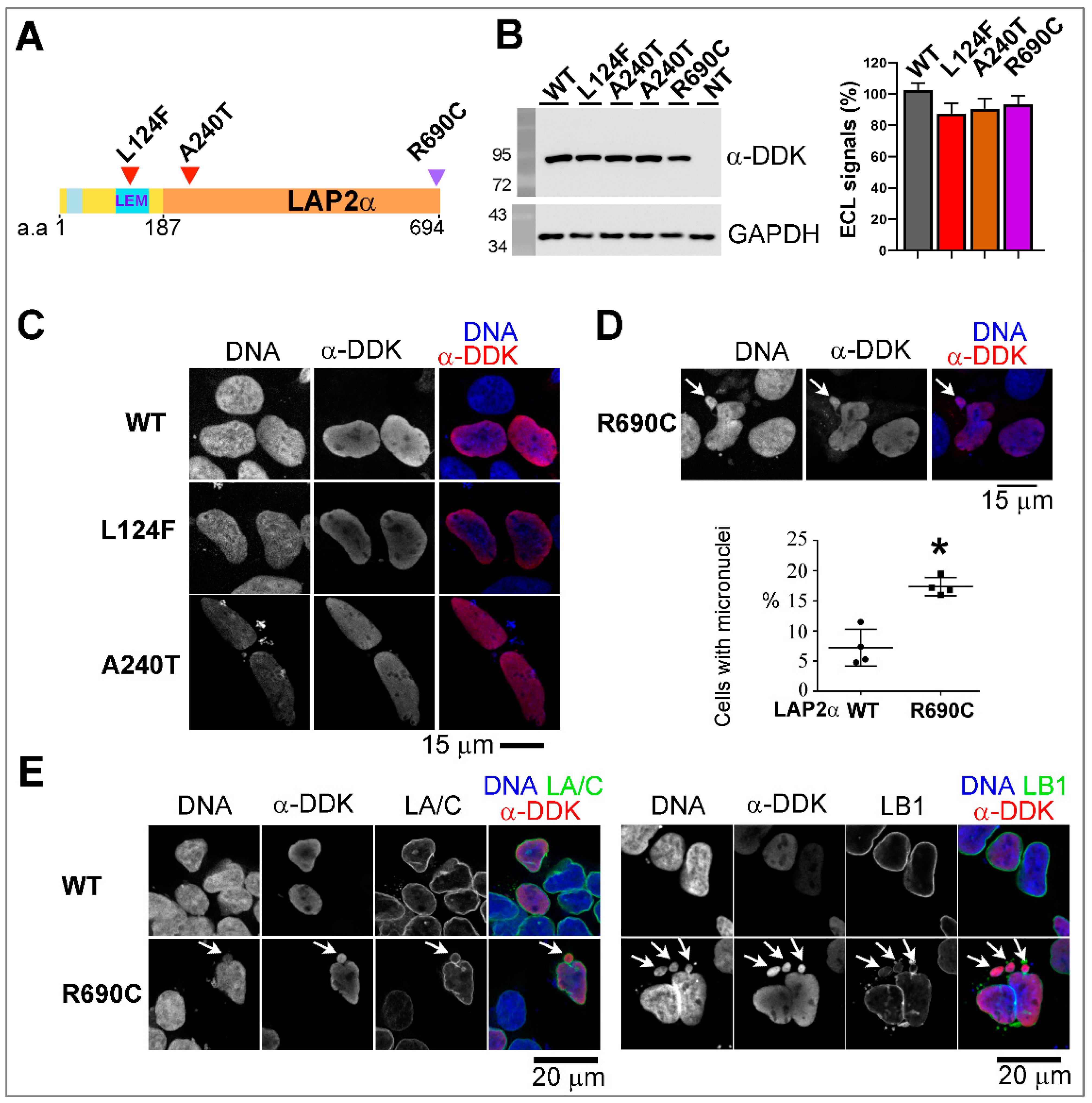
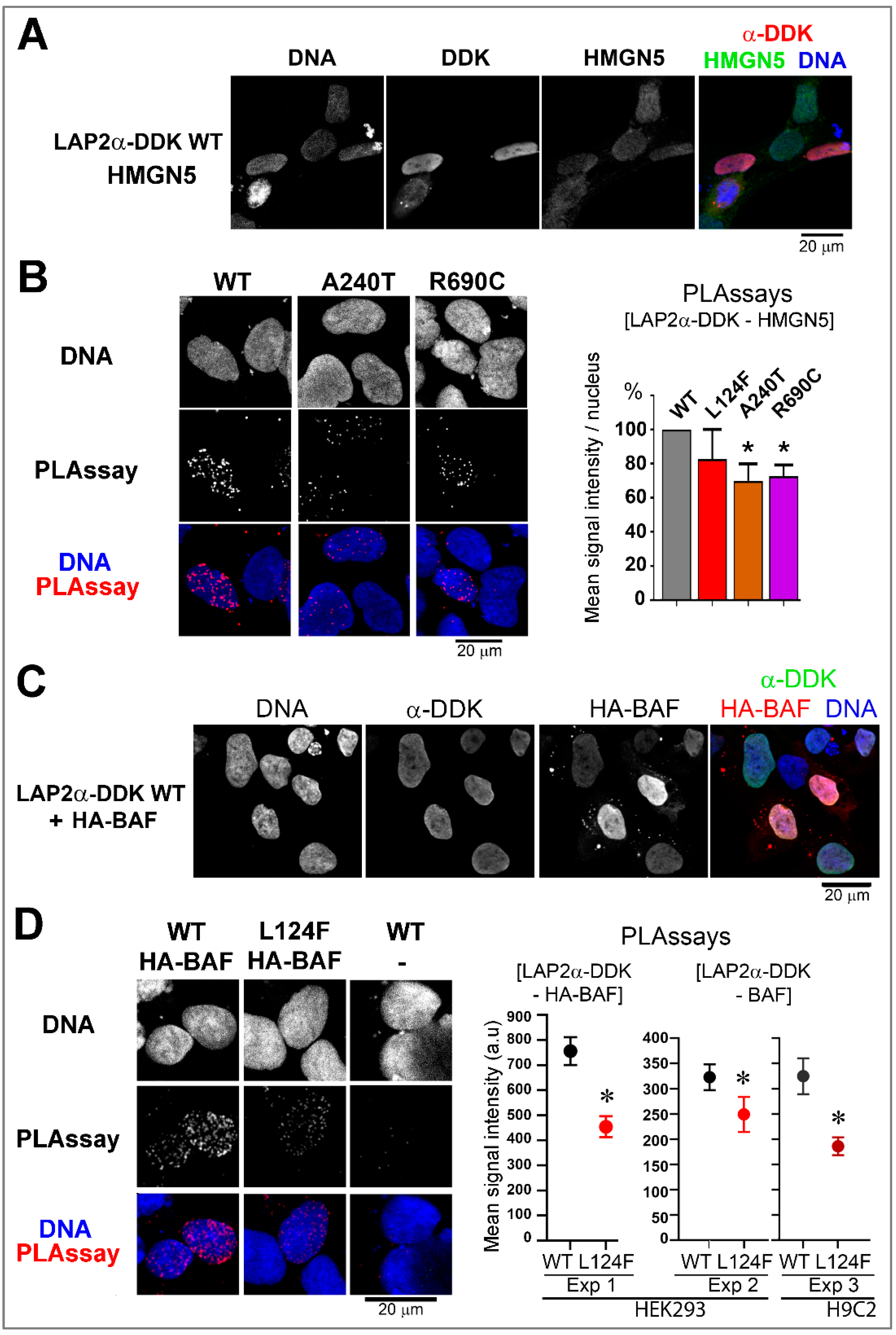
3.5.2. Ectopic Expression of the Two Rare Missense TMPO Variants Triggers Nuclear Defects Involving Chromatin-Associated Protein Partners
Nuclei Integrity and/or Association with HMGN5 or BAF Are Altered upon the Expression of LAP2 Variants
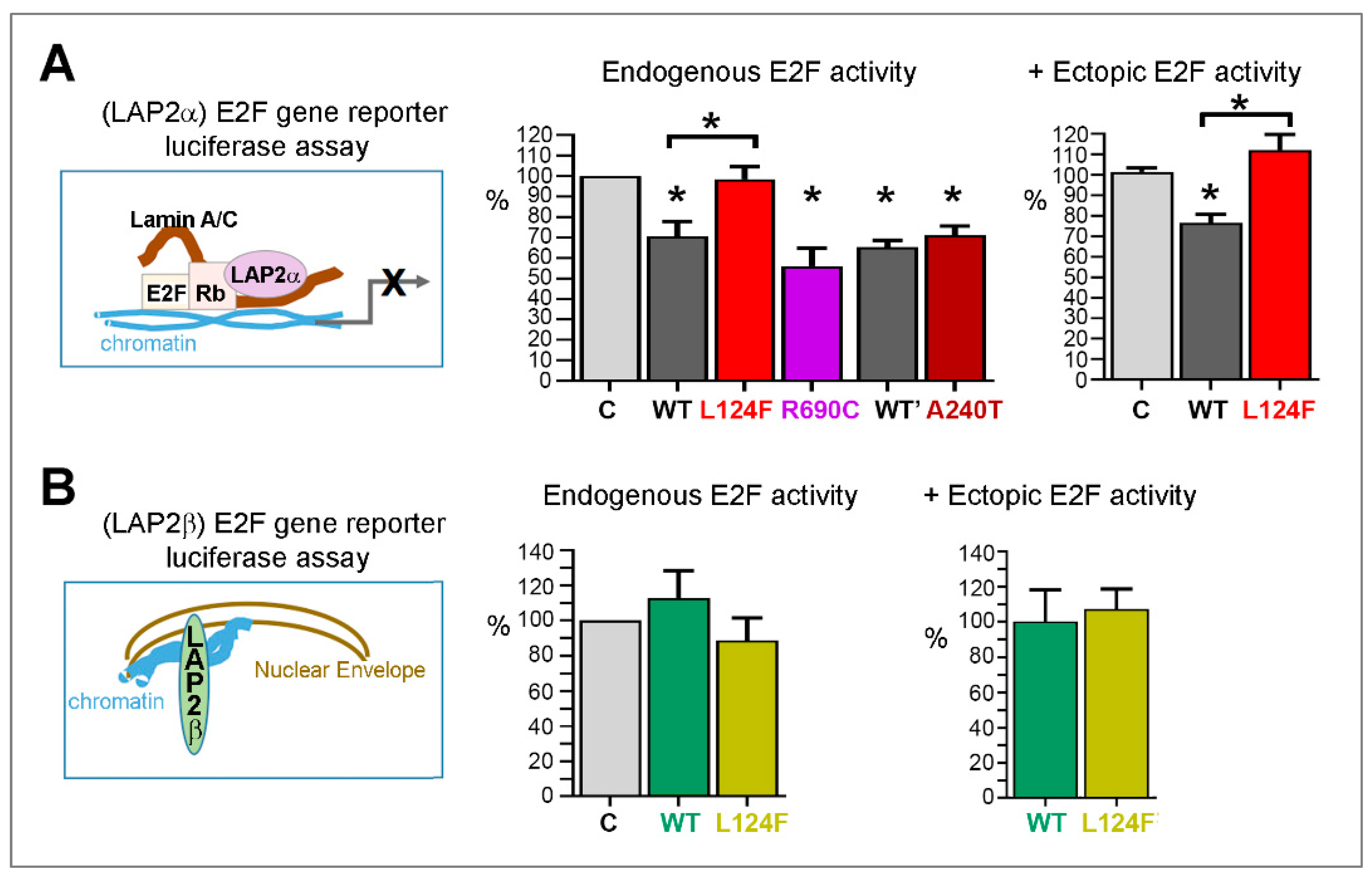
The Variant p.(Leu124Phe) Leads to Loss of LAP2α Capacity to Inhibit Gene Transcription Regulated by E2F1
4. Discussion
4.1. Combined Phenotyping and Gene Panel Sequencing Approaches Support an Association of TMPO with Cardiomyopathies
4.2. Sex influence on Disease Occurrence
4.3. Cellular Studies Reveal Pathogenic Mechanisms Induced by Three Novel Rare TMPO Variants
4.3.1. The p.(Gly395Glufs*11) LAP2α Variant Induces Haploinsufficiency
4.3.2. The LAP2alpha-Specific Variants p.(Ala240Thr) and p.(Arg690Cys) Modify LAP2α Association with the Genome Wide Organization Regulator, HMGN5
4.3.3. The LEM-Domain Variant p.(Leu124Phe) Modifies LAP2α-Mediated Regulation of BAF and E2F1
4.4. Relevance of the LEM Domain of Nuclear Proteins in Cardiac Pathophysiology
4.5. Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Worman:, H.J.; Östlund, C.; Wang, Y. Diseases of the Nuclear Envelope. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef]
- Janin, A.; Bauer, D.; Ratti, F.; Millat, G.; Méjat, A. Nuclear Envelopathies: A Complex LINC between Nuclear Envelope and Pathology. Orphanet J. Rare Dis. 2017, 12, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorboz, I.; Coutelier, M.; Bertrand, A.T.; Caberg, J.-H.; Elmaleh-Bergès, M.; Lainé, J.; Stevanin, G.; Bonne, G.; Boespflug-Tanguy, O.; Servais, L. Severe Dystonia, Cerebellar Atrophy, and Cardiomyopathy Likely Caused by a Missense Mutation in TOR1AIP1. Orphanet J. Rare Dis. 2014, 9. [Google Scholar] [CrossRef] [Green Version]
- McKenna, W.J.; Maron, B.J.; Thiene, G. Classification, Epidemiology, and Global Burden of Cardiomyopathies. Circ. Res. 2017, 121, 722–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondue, A.; Arbustini, E.; Bianco, A.; Ciccarelli, M.; Dawson, D.; De Rosa, M.; Hamdani, N.; Hilfiker-Kleiner, D.; Meder, B.; Leite-Moreira, A.F.; et al. Complex Roads from Genotype to Phenotype in Dilated Cardiomyopathy: Scientific Update from the Working Group of Myocardial Function of the European Society of Cardiology. Cardiovasc. Res. 2018, 114, 1287–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingles, J.; Burns, C.; Bagnall, R.D.; Lam, L.; Yeates, L.; Sarina, T.; Puranik, R.; Briffa, T.; Atherton, J.J.; Driscoll, T.; et al. Nonfamilial Hypertrophic Cardiomyopathy: Prevalence, Natural History, and Clinical Implications. Circ. Cardiovasc. Genet. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Walsh, R.; Buchan, R.; Wilk, A.; John, S.; Felkin, L.E.; Thomson, K.L.; Chiaw, T.H.; Loong, C.C.W.; Pua, C.J.; Raphael, C.; et al. Defining the Genetic Architecture of Hypertrophic Cardiomyopathy: Re-Evaluating the Role of Non-Sarcomeric Genes. Eur. Heart J. 2017, 38, 3461–3468. [Google Scholar] [CrossRef]
- Charron, P.; Arbustini, E.; Bonne, G. What Should the Cardiologist Know about Lamin Disease? Arrhythm. Electrophysiol. Rev. 2012, 1, 22–28. [Google Scholar] [CrossRef] [Green Version]
- Dorner, D.; Gotzmann, J.; Foisner, R. Nucleoplasmic Lamins and Their Interaction Partners, LAP2α, Rb, and BAF, in Transcriptional Regulation: Nucleoplasmic Lamins and Their Interactions. FEBS J. 2007, 274, 1362–1373. [Google Scholar] [CrossRef]
- Harris, C.A.; Andryuk, P.J.; Cline, S.W.; Mathew, S.; Siekierka, J.J.; Goldstein, G. Structure and Mapping of the Human Thymopoietin (TMPO) Gene and Relationship of Human TMPO Beta to Rat Lamin-Associated Polypeptide 2. Genomics 1995, 28, 198–205. [Google Scholar] [CrossRef]
- Dechat, T.; Korbei, B.; Vaughan, O.A.; Vlcek, S.; Hutchison, C.J.; Foisner, R. Lamina-Associated Polypeptide 2alpha Binds Intranuclear A-Type Lamins. J. Cell. Sci. 2000, 113 Pt. 19, 3473–3484. [Google Scholar] [CrossRef]
- Dechat, T.; Pfleghaar, K.; Sengupta, K.; Shimi, T.; Shumaker, D.K.; Solimando, L.; Goldman, R.D. Nuclear Lamins: Major Factors in the Structural Organization and Function of the Nucleus and Chromatin. Genes Dev. 2008, 22, 832–853. [Google Scholar] [CrossRef] [Green Version]
- Gruenbaum, Y.; Foisner, R. Lamins: Nuclear Intermediate Filament Proteins with Fundamental Functions in Nuclear Mechanics and Genome Regulation. Annu. Rev. Biochem. 2015, 84, 131–164. [Google Scholar] [CrossRef]
- Swift, J.; Ivanovska, I.L.; Buxboim, A.; Harada, T.; Dingal, P.C.D.P.; Pinter, J.; Pajerowski, J.D.; Spinler, K.R.; Shin, J.-W.; Tewari, M.; et al. Nuclear Lamin-A Scales with Tissue Stiffness and Enhances Matrix-Directed Differentiation. Science 2013, 341, 1240104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brachner, A. LEM2 Is a Novel MAN1-Related Inner Nuclear Membrane Protein Associated with A-Type Lamins. J. Cell Sci. 2005, 118, 5797–5810. [Google Scholar] [CrossRef] [Green Version]
- Herrada, I.; Bourgeois, B.; Samson, C.; Buendia, B.; Worman, H.J.; Zinn-Justin, S. Purification and Structural Analysis of LEM-Domain Proteins. Meth. Enzymol. 2016, 569, 43–61. [Google Scholar] [CrossRef]
- Cai, M.; Huang, Y.; Ghirlando, R.; Wilson, K.L.; Craigie, R.; Clore, G.M. Solution Structure of the Constant Region of Nuclear Envelope Protein LAP2 Reveals Two LEM-Domain Structures: One Binds BAF and the Other Binds DNA. EMBO J. 2001, 20, 4399–4407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dechat, T.; Vlcek, S.; Foisner, R. Review: Lamina-Associated Polypeptide 2 Isoforms and Related Proteins in Cell Cycle-Dependent Nuclear Structure Dynamics. J. Struct. Biol. 2000, 129, 335–345. [Google Scholar] [CrossRef]
- Zhang, S.; Schones, D.E.; Malicet, C.; Rochman, M.; Zhou, M.; Foisner, R.; Bustin, M. High Mobility Group Protein N5 (HMGN5) and Lamina-Associated Polypeptide 2α (LAP2α) Interact and Reciprocally Affect Their Genome-Wide Chromatin Organization. J. Biol. Chem. 2013, 288, 18104–18109. [Google Scholar] [CrossRef] [Green Version]
- Gotic, I.; Leschnik, M.; Kolm, U.; Markovic, M.; Haubner, B.J.; Biadasiewicz, K.; Metzler, B.; Stewart, C.L.; Foisner, R. Lamina-Associated Polypeptide 2alpha Loss Impairs Heart Function and Stress Response in Mice. Circ. Res. 2010, 106, 346–353. [Google Scholar] [CrossRef]
- Taylor, M.R.G.; Slavov, D.; Gajewski, A.; Vlcek, S.; Ku, L.; Fain, P.R.; Carniel, E.; Di Lenarda, A.; Sinagra, G.; Boucek, M.M.; et al. Thymopoietin (Lamina-Associated Polypeptide 2) Gene Mutation Associated with Dilated Cardiomyopathy. Hum. Mutat. 2005, 26, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.; Thomson, K.L.; Ware, J.S.; Funke, B.H.; Woodley, J.; McGuire, K.J.; Mazzarotto, F.; Blair, E.; Seller, A.; Taylor, J.C.; et al. Reassessment of Mendelian Gene Pathogenicity Using 7,855 Cardiomyopathy Cases and 60,706 Reference Samples. Genet. Med. 2017, 19, 192–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furusawa, T.; Rochman, M.; Taher, L.; Dimitriadis, E.K.; Nagashima, K.; Anderson, S.; Bustin, M. Chromatin De-Compaction By The Nucleosomal Binding Protein HMGN5 Impairs Nuclear Sturdiness. Nat. Commun. 2015, 6, 6138. [Google Scholar] [CrossRef] [Green Version]
- Dorner, D.; Vlcek, S.; Foeger, N.; Gajewski, A.; Makolm, C.; Gotzmann, J.; Hutchison, C.J.; Foisner, R. Lamina-Associated Polypeptide 2alpha Regulates Cell Cycle Progression and Differentiation via the Retinoblastoma-E2F Pathway. J. Cell Biol. 2006, 173, 83–93. [Google Scholar] [CrossRef] [Green Version]
- Denechaud, P.-D.; Fajas, L.; Giralt, A. E2F1, a Novel Regulator of Metabolism. Front. Endocrinol. (Lausanne) 2017, 8, 311. [Google Scholar] [CrossRef]
- Richard, P.; Ader, F.; Charron, P. Génétique des cardiomyopathies héréditaires. EMC-Cardiol. 2018, 13, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Samson, C.; Petitalot, A.; Celli, F.; Herrada, I.; Ropars, V.; Le Du, M.-H.; Nhiri, N.; Jacquet, E.; Arteni, A.-A.; Buendia, B.; et al. Structural Analysis of the Ternary Complex between Lamin A/C, BAF and Emerin Identifies an Interface Disrupted in Autosomal Recessive Progeroid Diseases. Nucleic Acids Res. 2018, 46, 10460–10473. [Google Scholar] [CrossRef] [Green Version]
- Favreau, C.; Higuet, D.; Courvalin, J.-C.; Buendia, B. Expression of a Mutant Lamin A That Causes Emery-Dreifuss Muscular Dystrophy Inhibits in Vitro Differentiation of C2C12 Myoblasts. Mol. Cell. Biol. 2004, 24, 1481–1492. [Google Scholar] [CrossRef] [Green Version]
- Vadrot, N.; Duband-Goulet, I.; Cabet, E.; Attanda, W.; Barateau, A.; Vicart, P.; Gerbal, F.; Briand, N.; Vigouroux, C.; Oldenburg, A.R.; et al. The p.R482W Substitution in A-Type Lamins Deregulates SREBP1 Activity in Dunnigan-Type Familial Partial Lipodystrophy. Hum. Mol. Genet. 2015, 24, 2096–2109. [Google Scholar] [CrossRef] [Green Version]
- Mittnacht, S.; Weinberg, R.A. G1/S Phosphorylation of the Retinoblastoma Protein Is Associated with an Altered Affinity for the Nuclear Compartment. Cell 1991, 65, 381–393. [Google Scholar] [CrossRef]
- Barateau, A.; Buendia, B. In Situ Detection of Interactions Between Nuclear Envelope Proteins and Partners. Methods Mol. Biol. 2016, 1411, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Barateau, A.; Vadrot, N.; Vicart, P.; Ferreiro, A.; Mayer, M.; Héron, D.; Vigouroux, C.; Buendia, B. A Novel Lamin A Mutant Responsible for Congenital Muscular Dystrophy Causes Distinct Abnormalities of the Cell Nucleus. PLoS ONE 2017, 12, e0169189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, L.; Lv, Q.; Zhang, Y. STRUM: Structure-Based Prediction of Protein Stability Changes upon Single-Point Mutation. Bioinformatics 2016, 32, 2936–2946. [Google Scholar] [CrossRef] [Green Version]
- Bradley, C.M.; Jones, S.; Huang, Y.; Suzuki, Y.; Kvaratskhelia, M.; Hickman, A.B.; Craigie, R.; Dyda, F. Structural Basis for Dimerization of LAP2α, a Component of the Nuclear Lamina. Structure 2007, 15, 643–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markiewicz, E.; Dechat, T.; Foisner, R.; Quinlan, R.A.; Hutchison, C.J. Lamin A/C Binding Protein LAP2α Is Required for Nuclear Anchorage of Retinoblastoma Protein. MBoC 2002, 13, 4401–4413. [Google Scholar] [CrossRef] [Green Version]
- Roncarati, R.; Viviani Anselmi, C.; Krawitz, P.; Lattanzi, G.; von Kodolitsch, Y.; Perrot, A.; di Pasquale, E.; Papa, L.; Portararo, P.; Columbaro, M.; et al. Doubly Heterozygous LMNA and TTN Mutations Revealed by Exome Sequencing in a Severe Form of Dilated Cardiomyopathy. Eur. J. Hum. Genet. 2013, 21, 1105–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdallah, A.M.; Carlus, S.J.; Al-Mazroea, A.H.; Alluqmani, M.; Almohammadi, Y.; Bhuiyan, Z.A.; Al-Harbi, K.M. Digenic Inheritance of LAMA4 and MYH7 Mutations in Patient with Infantile Dilated Cardiomyopathy. Medicina 2019, 55, 17. [Google Scholar] [CrossRef] [Green Version]
- Ren, M.; Chai, X.; Li, L.; Wang, X.; Yin, C. Potential Digenic Inheritance of Familial Hypertrophic Cardiomyopathy Identified by Whole-exome Sequencing. Mol. Genet. Genom. Med. 2020, 8. [Google Scholar] [CrossRef]
- Johnson, R.; Peters, S.; Ingles, J.; Correnti, G.; Ingrey, A.; Mountain, H.; Zentner, D.; Thompson, T.; Oates, E.; Ronan, A.; et al. Penetrance of Dilated Cardiomyopathy in Families with Truncating TTN Variants: A National Perspective. Heart Lung Circ. 2019, 28, S140. [Google Scholar] [CrossRef] [Green Version]
- Nijenkamp, L.L.A.M.; Bollen, I.A.E.; van Velzen, H.G.; Regan, J.A.; van Slegtenhorst, M.; Niessen, H.W.M.; Schinkel, A.F.L.; Krüger, M.; Poggesi, C.; Ho, C.Y.; et al. Sex Differences at the Time of Myectomy in Hypertrophic Cardiomyopathy. Circ. Heart Fail. 2018, 11, e004133. [Google Scholar] [CrossRef]
- Bione, S.; Maestrini, E.; Rivella, S.; Mancini, M.; Regis, S.; Romeo, G.; Toniolo, D. Identification of a Novel X-Linked Gene Responsible for Emery-Dreifuss Muscular Dystrophy. Nat. Genet. 1994, 8, 323–327. [Google Scholar] [CrossRef]
- Pelliccia, F.; Limongelli, G.; Autore, C.; Gimeno-Blanes, J.R.; Basso, C.; Elliott, P. Sex-Related Differences in Cardiomyopathies. Int. J. Cardiol. 2019, 286, 239–243. [Google Scholar] [CrossRef] [PubMed]
- InanlooRahatloo, K.; Liang, G.; Vo, D.; Ebert, A.; Nguyen, I.; Nguyen, P.K. Sex-Based Differences in Myocardial Gene Expression in Recently Deceased Organ Donors with No Prior Cardiovascular Disease. PLoS ONE 2017, 12, e0183874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naetar, N.; Georgiou, K.; Knapp, C.; Bronshtein, I.; Zier, E.; Fichtinger, P.; Dechat, T.; Garini, Y.; Foisner, R. LAP2alpha Maintains a Mobile and Low Assembly State of A-Type Lamins in the Nuclear Interior. eLife 2021, 10, e63476. [Google Scholar] [CrossRef]
- González-Garrido, A.; Rosas-Madrigal, S.; Rojo-Domínguez, A.; Arellanes-Robledo, J.; López-Mora, E.; Carnevale, A.; Arregui, L.; Rosendo-Gutiérrez, R.; Romero-Hidalgo, S.; Villarreal-Molina, M.T. Leukocyte Nuclear Morphology Alterations in Dilated Cardiomyopathy Caused by a Lamin AC Truncating Mutation (LMNA/Ser431*) Are Modified by the Presence of a LAP2 Missense Polymorphism (TMPO/Arg690Cys). IJMS 2022, 23, 13626. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Madrigal, S.; Villarreal-Molina, M.T.; Flores-Rivera, J.; Rivas-Alonso, V.; Macias-Kauffer, L.R.; Ordoñez, G.; Acuña-Alonzo, V.; Chima-Galán, M.D.C.; Macín-Pérez, G.; Barquera, R. Interaction of HLA Class II Rs9272219 and TMPO Rs17028450 (Arg690Cys) Variants Affects Neuromyelitis Optica Spectrum Disorder Susceptibility in an Admixed Mexican Population. Front. Genet. 2021, 12, 647343. [Google Scholar] [CrossRef] [PubMed]
- Pekovic, V.; Harborth, J.; Broers, J.L.V.; Ramaekers, F.C.S.; van Engelen, B.; Lammens, M.; von Zglinicki, T.; Foisner, R.; Hutchison, C.; Markiewicz, E. Nucleoplasmic LAP2α–Lamin A Complexes Are Required to Maintain a Proliferative State in Human Fibroblasts. J. Cell Biol. 2007, 176, 163–172. [Google Scholar] [CrossRef]
- Vignier, N.; Schlossarek, S.; Fraysse, B.; Mearini, G.; Krämer, E.; Pointu, H.; Mougenot, N.; Guiard, J.; Reimer, R.; Hohenberg, H.; et al. Nonsense-Mediated MRNA Decay and Ubiquitin–Proteasome System Regulate Cardiac Myosin-Binding Protein C Mutant Levels in Cardiomyopathic Mice. Circ. Res. 2009, 105, 239–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gesson, K.; Rescheneder, P.; Skoruppa, M.P.; von Haeseler, A.; Dechat, T.; Foisner, R. A-Type Lamins Bind Both Hetero- and Euchromatin, the Latter Being Regulated by Lamina-Associated Polypeptide 2 Alpha. Genome Res. 2016, 26, 462–473. [Google Scholar] [CrossRef] [Green Version]
- Bulteau, A.-L.; Szweda, L.I.; Friguet, B. Age-Dependent Declines in Proteasome Activity in the Heart. Arch. Biochem. Biophys. 2002, 397, 298–304. [Google Scholar] [CrossRef]
- Marian, A.J. Modifier Genes for Hypertrophic Cardiomyopathy. Curr. Opin. Cardiol. 2002, 17, 242–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brady, G.F.; Kwan, R.; Ulintz, P.J.; Nguyen, P.; Bassirian, S.; Basrur, V.; Nesvizhskii, A.I.; Loomba, R.; Omary, M.B. Nuclear Lamina Genetic Variants, Including a Truncated LAP2, in Twins and Siblings with Nonalcoholic Fatty Liver Disease. Hepatology 2018, 67, 1710–1725. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Yoon, M.-H.; Park, B.-J. Laminopathies; Mutations on Single Gene and Various Human Genetic Diseases. BMB Rep. 2018, 51, 327–337. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Kwon, M.; Mannino, M.; Yang, N.; Renda, F.; Khodjakov, A.; Pellman, D. Nuclear Envelope Assembly Defects Link Mitotic Errors to Chromothripsis. Nature 2018, 561, 551–555. [Google Scholar] [CrossRef]
- Abdelfatah, N.; Chen, R.; Duff, H.J.; Seifer, C.M.; Buffo, I.; Huculak, C.; Clarke, S.; Clegg, R.; Jassal, D.S.; Gordon, P.M.K.; et al. Characterization of a Unique Form of Arrhythmic Cardiomyopathy Caused by Recessive Mutation in LEMD2. JACC: Basic Transl. Sci. 2019, 4, 204–221. [Google Scholar] [CrossRef] [PubMed]
- Poleshko, A.; Shah, P.P.; Gupta, M.; Babu, A.; Morley, M.P.; Manderfield, L.J.; Ifkovits, J.L.; Calderon, D.; Aghajanian, H.; Sierra-Pagán, J.E.; et al. Genome-Nuclear Lamina Interactions Regulate Cardiac Stem Cell Lineage Restriction. Cell 2017, 171, 573–587. [Google Scholar] [CrossRef] [Green Version]
- Rebelo, S.; da Cruz e Silva, E.F.; da Cruz e Silva, O.A.B. Genetic Mutations Strengthen Functional Association of LAP1 with DYT1 Dystonia and Muscular Dystrophy. Mutat. Res. /Rev. Mutat. Res. 2015, 766, 42–47. [Google Scholar] [CrossRef]
- Berk, J.M.; Tifft, K.E.; Wilson, K.L. The Nuclear Envelope LEM-Domain Protein Emerin. Nucleus 2013, 4, 298–314. [Google Scholar] [CrossRef] [Green Version]
- Ben Yaou, R.; Toutain, A.; Arimura, T.; Demay, L.; Massart, C.; Peccate, C.; Muchir, A.; Llense, S.; Deburgrave, N.; Leturcq, F.; et al. Multitissular Involvement in a Family with LMNA and EMD Mutations: Role of Digenic Mechanism? Neurology 2007, 68, 1883–1894. [Google Scholar] [CrossRef] [PubMed]
- Karst, M.L.; Herron, K.J.; Olson, T.M. X-Linked Nonsyndromic Sinus Node Dysfunction and Atrial Fibrillation Caused by Emerin Mutation. J. Cardiovasc. Electrophysiol. 2008, 19, 510–515. [Google Scholar] [CrossRef]
- Essawy, N.; Samson, C.; Petitalot, A.; Moog, S.; Bigot, A.; Herrada, I.; Marcelot, A.; Arteni, A.-A.; Coirault, C.; Zinn-Justin, S. An Emerin LEM-Domain Mutation Impairs Cell Response to Mechanical Stress. Cells 2019, 8, 570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naetar, N.; Korbei, B.; Kozlov, S.; Kerenyi, M.A.; Dorner, D.; Kral, R.; Gotic, I.; Fuchs, P.; Cohen, T.V.; Bittner, R.; et al. Loss of Nucleoplasmic LAP2α–Lamin A Complexes Causes Erythroid and Epidermal Progenitor Hyperproliferation. Nat. Cell Biol. 2008, 10, 1341–1348. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vadrot, N.; Ader, F.; Moulin, M.; Merlant, M.; Chapon, F.; Gandjbakhch, E.; Labombarda, F.; Maragnes, P.; Réant, P.; Rooryck, C.; et al. Abnormal Cellular Phenotypes Induced by Three TMPO/LAP2 Variants Identified in Men with Cardiomyopathies. Cells 2023, 12, 337. https://doi.org/10.3390/cells12020337
Vadrot N, Ader F, Moulin M, Merlant M, Chapon F, Gandjbakhch E, Labombarda F, Maragnes P, Réant P, Rooryck C, et al. Abnormal Cellular Phenotypes Induced by Three TMPO/LAP2 Variants Identified in Men with Cardiomyopathies. Cells. 2023; 12(2):337. https://doi.org/10.3390/cells12020337
Chicago/Turabian StyleVadrot, Nathalie, Flavie Ader, Maryline Moulin, Marie Merlant, Françoise Chapon, Estelle Gandjbakhch, Fabien Labombarda, Pascale Maragnes, Patricia Réant, Caroline Rooryck, and et al. 2023. "Abnormal Cellular Phenotypes Induced by Three TMPO/LAP2 Variants Identified in Men with Cardiomyopathies" Cells 12, no. 2: 337. https://doi.org/10.3390/cells12020337