
DRAM1 Promotes Lysosomal Delivery of Mycobacterium marinum in Macrophages
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Macrophage Cell Culture
2.2. Mm Culture and Infection Experiments
2.3. Immunofluorescence
2.4. LysoTracker Labelling
2.5. DRAM1 shRNA Knockdown
2.6. Quantitative PCR
2.7. Western Blot
2.8. Confocal Laser Scanning Microscopy and Analysis of Colocalisation Patterns
2.9. CFU Assay
2.10. SYTOX Green Staining
2.11. Statistical Analyses
3. Results
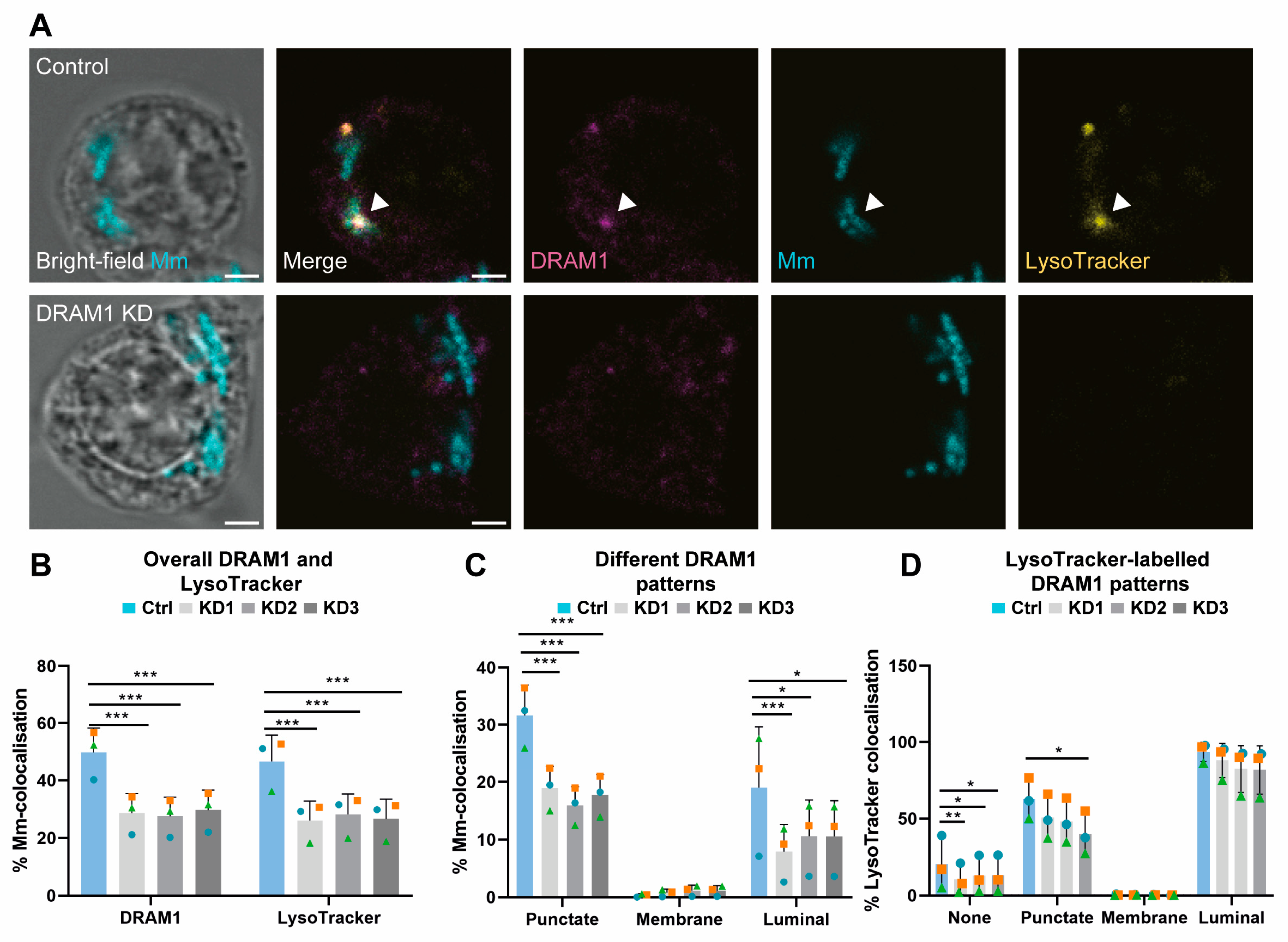
3.1. DRAM1 Colocalises with Acidified Mm-Containing Vesicles
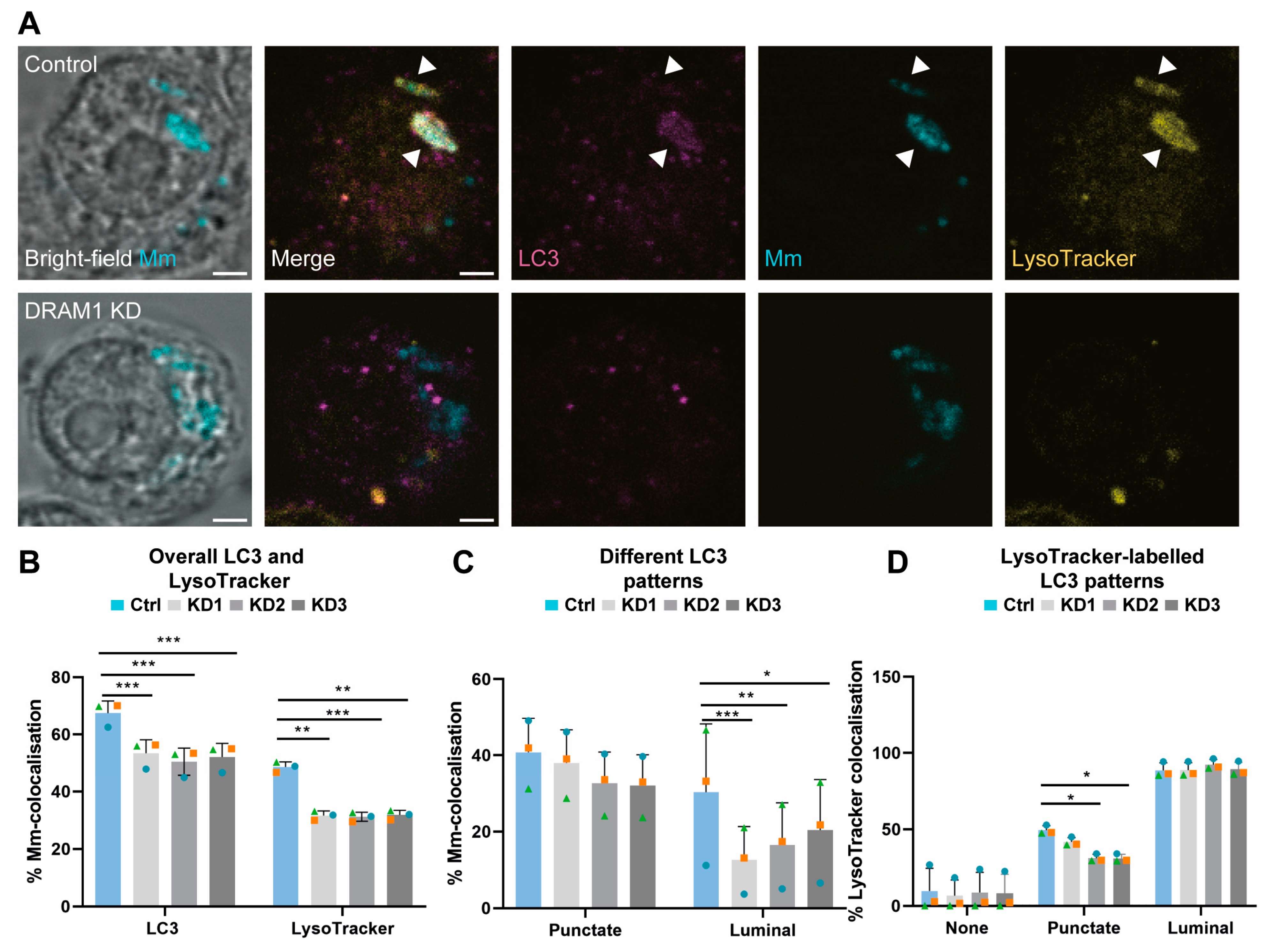
3.2. Acidified Mm-Containing Vesicles Are Positive for the Autophagy Marker LC3
3.3. DRAM1 Promotes the Acidification of Mm-Containing Vesicles
3.4. DRAM1 Mediates LC3 Trafficking to Mm
3.5. DRAM1 Promotes the Fusion of LAMP1-Positive Lysosomes with Mm-Containing Vesicles
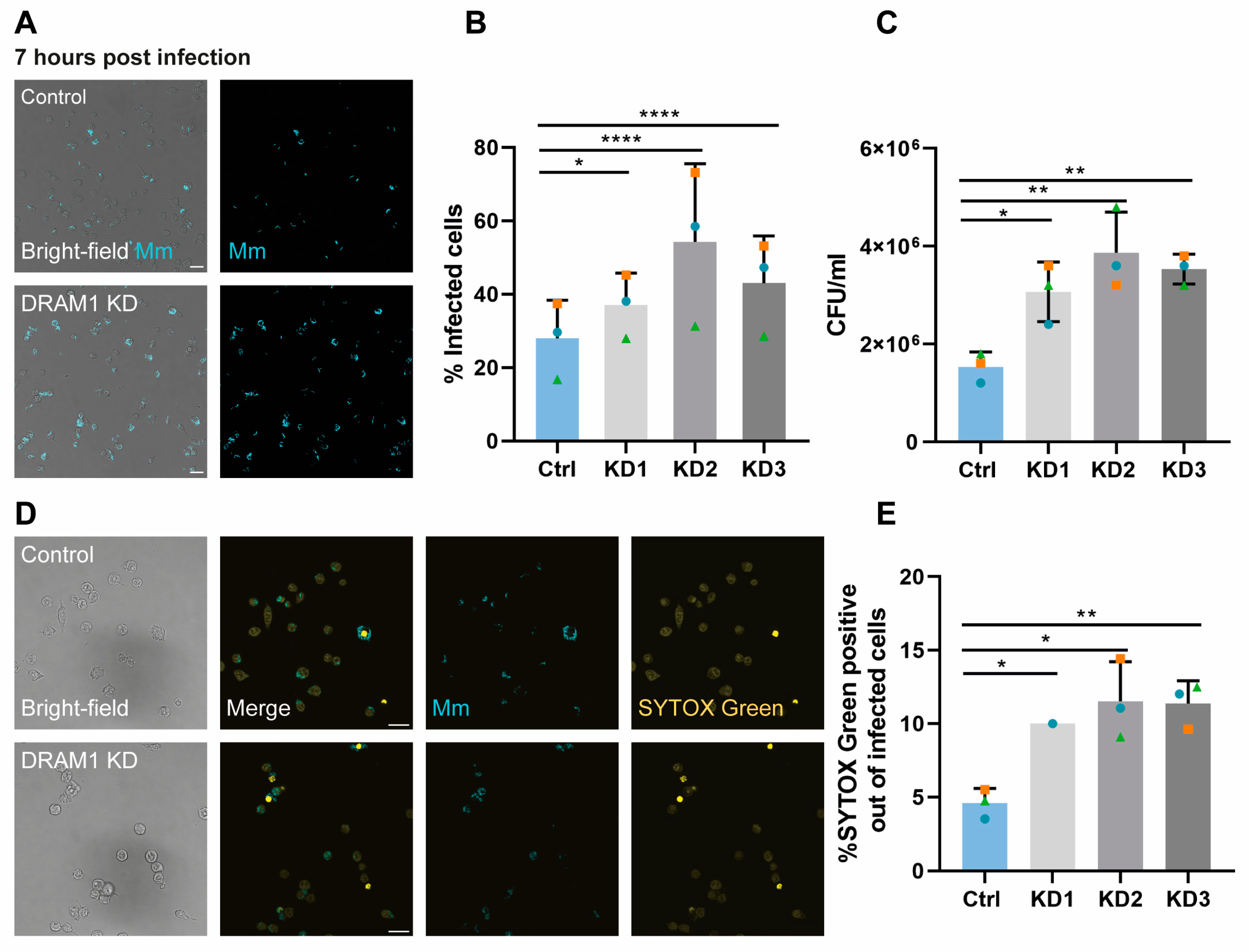
3.6. DRAM1 Is Required for Macrophage Defence against Mm
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fountain, A.; Inpanathan, S.; Alves, P.; Verdawala, M.B.; Botelho, R.J. Phagosome maturation in macrophages: Eat, digest, adapt, and repeat. Adv. Biol. Regul. 2021, 82, 100832. [Google Scholar] [CrossRef]
- Flannagan, R.S.; Cosío, G.; Grinstein, S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat. Rev. Microbiol. 2009, 7, 355–366. [Google Scholar] [CrossRef]
- Deretic, V. Autophagy in inflammation, infection, and immunometabolism. Immunity 2021, 54, 437–453. [Google Scholar] [CrossRef]
- Boyle, K.B.; Randow, F. The role of ‘eat-me’signals and autophagy cargo receptors in innate immunity. Curr. Opin. Microbiol. 2013, 16, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Peña-Martinez, C.; Rickman, A.D.; Heckmann, B.L. Beyond autophagy: LC3-associated phagocytosis and endocytosis. Sci. Adv. 2022, 8, eabn1702. [Google Scholar] [CrossRef]
- Riebisch, A.K.; Mühlen, S.; Beer, Y.Y.; Schmitz, I. Autophagy—A Story of Bacteria Interfering with the Host Cell Degradation Machinery. Pathogens 2021, 10, 110. [Google Scholar] [CrossRef] [PubMed]
- Grijmans, B.J.; van der Kooij, S.B.; Varela, M.; Meijer, A.H. LAPped in proof: LC3-associated phagocytosis and the arms race against bacterial pathogens. Front. Cell. Infect. Microbiol. 2022, 11, 809121. [Google Scholar] [CrossRef]
- Deretic, V.; Singh, S.; Master, S.; Harris, J.; Roberts, E.; Kyei, G.; Davis, A.; De Haro, S.; Naylor, J.; Lee, H.H. Mycobacterium tuberculosis inhibition of phagolysosome biogenesis and autophagy as a host defence mechanism. Cell. Microbiol. 2006, 8, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Alipoor, S.D.; Adcock, I.M.; Tabarsi, P.; Folkerts, G.; Mortaz, E. MiRNAs in tuberculosis: Their decisive role in the fate of TB. Eur. J. Pharmacol. 2020, 886, 173529. [Google Scholar] [CrossRef]
- Songane, M.; Kleinnijenhuis, J.; Netea, M.G.; van Crevel, R. The role of autophagy in host defence against Mycobacterium tuberculosis infection. Tuberculosis 2012, 92, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Simeone, R.; Sayes, F.; Lawarée, E.; Brosch, R. Breaching the phagosome, the case of the tuberculosis agent. Cell. Microbiol. 2021, 23, e13344. [Google Scholar] [CrossRef]
- Russell, D.G. Mycobacterium tuberculosis and the intimate discourse of a chronic infection. Immunol. Rev. 2011, 240, 252–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, S.; Spaink, H.P.; Forn-Cuní, G. Drug resistance in nontuberculous mycobacteria: Mechanisms and models. Biology 2021, 10, 96. [Google Scholar] [CrossRef] [PubMed]
- Silwal, P.; Kim, I.S.; Jo, E.-K. Autophagy and host defense in nontuberculous mycobacterial infection. Front. Immunol. 2021, 12, 728742. [Google Scholar] [CrossRef] [PubMed]
- Tobin, D.M.; Ramakrishnan, L. Comparative pathogenesis of Mycobacterium marinum and Mycobacterium tuberculosis. Cell. Microbiol. 2008, 10, 1027–1039. [Google Scholar] [CrossRef]
- Stinear, T.P.; Seemann, T.; Harrison, P.F.; Jenkin, G.A.; Davies, J.K.; Johnson, P.D.; Abdellah, Z.; Arrowsmith, C.; Chillingworth, T.; Churcher, C. Insights from the complete genome sequence of Mycobacterium marinum on the evolution of Mycobacterium tuberculosis. Genome Res. 2008, 18, 729–741. [Google Scholar] [CrossRef] [Green Version]
- Gröschel, M.I.; Sayes, F.; Simeone, R.; Majlessi, L.; Brosch, R. ESX secretion systems: Mycobacterial evolution to counter host immunity. Nat. Rev. Microbiol. 2016, 14, 677–691. [Google Scholar] [CrossRef]
- Houben, D.; Demangel, C.; Van Ingen, J.; Perez, J.; Baldeón, L.; Abdallah, A.M.; Caleechurn, L.; Bottai, D.; Van Zon, M.; De Punder, K. ESX-1-mediated translocation to the cytosol controls virulence of mycobacteria. Cell. Microbiol. 2012, 14, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- Bernard, E.M.; Fearns, A.; Bussi, C.; Santucci, P.; Peddie, C.J.; Lai, R.J.; Collinson, L.M.; Gutierrez, M.G.M. tuberculosis infection of human iPSC-derived macrophages reveals complex membrane dynamics during xenophagy evasion. J. Cell Sci. 2021, 134, jcs252973. [Google Scholar]
- van der Vaart, M.; Korbee, C.J.; Lamers, G.E.; Tengeler, A.C.; Hosseini, R.; Haks, M.C.; Ottenhoff, T.H.; Spaink, H.P.; Meijer, A.H. The DNA damage-regulated autophagy modulator DRAM1 links mycobacterial recognition via TLR-MYD88 to autophagic defense. Cell Host Microbe 2014, 15, 753–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi-Giesgen, I.; Behrends, C.; Alpi, A.F. The ubiquitin ligation machinery in the defense against bacterial pathogens. EMBO Rep. 2021, 22, e52864. [Google Scholar] [CrossRef]
- Kimmey, J.M.; Stallings, C.L. Bacterial pathogens versus autophagy: Implications for therapeutic interventions. Trends Mol. Med. 2016, 22, 1060–1076. [Google Scholar] [CrossRef] [PubMed]
- Köster, S.; Upadhyay, S.; Chandra, P.; Papavinasasundaram, K.; Yang, G.; Hassan, A.; Grigsby, S.J.; Mittal, E.; Park, H.S.; Jones, V. Mycobacterium tuberculosis is protected from NADPH oxidase and LC3-associated phagocytosis by the LCP protein CpsA. Proc. Natl. Acad. Sci. USA 2017, 114, E8711–E8720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [Green Version]
- O’Prey, J.; Skommer, J.; Wilkinson, S.; Ryan, K.M. Analysis of DRAM-related proteins reveals evolutionarily conserved and divergent roles in the control of autophagy. Cell Cycle 2009, 8, 2260–2265. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.K.; Lee, H.-M.; Park, K.-S.; Shin, D.-M.; Kim, T.S.; Kim, Y.S.; Suh, H.-W.; Kim, S.Y.; Kim, I.S.; Kim, J.-M. MIR144* inhibits antimicrobial responses against Mycobacterium tuberculosis in human monocytes and macrophages by targeting the autophagy protein DRAM2. Autophagy 2017, 13, 423–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrschtik, M.; O’prey, J.; Lao, L.; Long, J.; Beaumatin, F.; Strachan, D.; O’prey, M.; Skommer, J.; Ryan, K. DRAM-3 modulates autophagy and promotes cell survival in the absence of glucose. Cell Death Differ. 2015, 22, 1714–1726. [Google Scholar] [CrossRef]
- Liu, G.; Wan, Q.; Li, J.; Hu, X.; Gu, X.; Xu, S. Silencing miR-125b-5p attenuates inflammatory response and apoptosis inhibition in mycobacterium tuberculosis-infected human macrophages by targeting DNA damage-regulated autophagy modulator 2 (DRAM2). Cell Cycle 2020, 19, 3182–3194. [Google Scholar] [CrossRef]
- Barthet, V.J.; Mrschtik, M.; Kania, E.; McEwan, D.G.; Croft, D.; O’Prey, J.; Long, J.S.; Ryan, K.M. DRAM-4 and DRAM-5 are compensatory regulators of autophagy and cell survival in nutrient-deprived conditions. FEBS J. 2022, 289, 3752–3769. [Google Scholar] [CrossRef]
- Zhang, R.; Varela, M.; Forn-Cuní, G.; Torraca, V.; van der Vaart, M.; Meijer, A.H. Deficiency in the autophagy modulator Dram1 exacerbates pyroptotic cell death of Mycobacteria-infected macrophages. Cell Death Dis. 2020, 11, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Jagannath, C.; Liu, X.-D.; Sharafkhaneh, A.; Kolodziejska, K.E.; Eissa, N.T. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 2007, 27, 135–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Jing, W.; Runpeng, Z.; Xuewei, X.; Min, M.; Ru, C.; Yingru, X.; Shengfa, N.; Rongbo, Z. ESAT6 inhibits autophagy flux and promotes BCG proliferation through MTOR. Biochem. Biophys. Res. Commun. 2016, 477, 195–201. [Google Scholar] [CrossRef]
- Wang, J.; Wang, R.; Wang, H.; Yang, X.; Yang, J.; Xiong, W.; Wen, Q.; Ma, L. Glucocorticoids suppress antimicrobial autophagy and nitric oxide production and facilitate mycobacterial survival in macrophages. Sci. Rep. 2017, 7, 982. [Google Scholar] [CrossRef]
- Varela, M.; van der Vaart, M.; Groenewoud, A.; Meijer, A.H. Extracellular mycobacterial DNA drives disease progression by triggering Caspase-11-dependent pyroptosis of infected macrophages. bioRxiv, 2019; preprint. [Google Scholar] [CrossRef] [Green Version]
- Takaki, K.; Davis, J.; Winglee, K.; Ramakrishnan, L. Evaluation of the pathogenesis and treatment of Mycobacterium marinum infection in zebrafish. Nat. Protoc. 2013, 8, 1114–1124. [Google Scholar] [CrossRef] [Green Version]
- Benard, E.L.; van der Sar, A.M.; Ellett, F.; Lieschke, G.J.; Spaink, H.P.; Meijer, A.H. Infection of zebrafish embryos with intracellular bacterial pathogens. JoVE J. Vis. Exp. 2012, 61, e3781. [Google Scholar]
- Stamm, L.M.; Morisaki, J.H.; Gao, L.-Y.; Jeng, R.L.; McDonald, K.L.; Roth, R.; Takeshita, S.; Heuser, J.; Welch, M.D.; Brown, E.J. Mycobacterium marinum escapes from phagosomes and is propelled by actin-based motility. J. Exp. Med. 2003, 198, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Chikte, S.; Panchal, N.; Warnes, G. Use of LysoTracker dyes: A flow cytometric study of autophagy. Cytom. Part A 2014, 85, 169–178. [Google Scholar] [CrossRef]
- Green, M.V.; Pengo, T.; Raybuck, J.D.; Naqvi, T.; McMullan, H.M.; Hawkinson, J.E.; Marron Fernandez de Velasco, E.; Muntean, B.S.; Martemyanov, K.A.; Satterfield, R. Automated live-cell imaging of synapses in rat and human neuronal cultures. Front. Cell. Neurosci. 2019, 13, 467. [Google Scholar] [CrossRef] [Green Version]
- Brooks, M.E.; Kristensen, K.; Van Benthem, K.J.; Magnusson, A.; Berg, C.W.; Nielsen, A.; Skaug, H.J.; Machler, M.; Bolker, B.M. glmmTMB balances speed and flexibility among packages for zero-inflated generalized linear mixed modeling. R J. 2017, 9, 378–400. [Google Scholar] [CrossRef] [Green Version]
- Russell, V.; Lenth, P.B.; Giné-Vázquez, I.; Herve, M.; Jung, M.; Love, J.; Miguez, F.; Riebl, H.; Singmann, H. emmeans: Estimated Marginal Means, aka Least-Squares Means, R Package Version 1.8.4-1. 2023. Available online: https://CRAN.R-project.org/package=emmeans (accessed on 1 October 2022).
- Mah, L.Y.; O’Prey, J.; Baudot, A.D.; Hoekstra, A.; Ryan, K.M. DRAM-1 encodes multiple isoforms that regulate autophagy. Autophagy 2012, 8, 18–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilinç, G.; Saris, A.; Ottenhoff, T.H.; Haks, M.C. Host-directed therapy to combat mycobacterial infections. Immunol. Rev. 2021, 301, 62–83. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Zhang, J.; Wang, M.; Wang, Y.; Dong, M.; Ma, X.; Sun, B.; Liu, S.; Zhao, Z.; Chen, L. DRAM1 increases the secretion of PKM2-enriched EVs from hepatocytes to promote macrophage activation and disease progression in ALD. Mol. Ther.-Nucleic Acids 2022, 27, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Piper, R.C.; Katzmann, D.J. Biogenesis and Function of Multivesicular Bodies. Annu. Rev. Cell Dev. Biol. 2006, 23, 519–547. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.D.; Qi, L.; Wu, J.C.; Qin, Z.H. DRAM1 regulates autophagy flux through lysosomes. PLoS ONE 2013, 8, e63245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masud, S.; Zhang, R.; Prajsnar, T.K.; Meijer, A.H. Dram1 confers resistance to Salmonella infection. bioRxiv, 2021; preprint. [Google Scholar] [CrossRef]
- Forn-Cuní, G.; Welvaarts, L.; Stel, F.M.; van den Hondel, C.J.; Arentshorst, M.; Ram, A.F.J.; Meijer, A.H. Stimulating the autophagic-lysosomal axis enhances host defense against fungal infection in a zebrafish model of invasive Aspergillosis. Autophagy 2022, 19, 324–337. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banducci-Karp, A.; Xie, J.; Engels, S.A.G.; Sarantaris, C.; van Hage, P.; Varela, M.; Meijer, A.H.; van der Vaart, M. DRAM1 Promotes Lysosomal Delivery of Mycobacterium marinum in Macrophages. Cells 2023, 12, 828. https://doi.org/10.3390/cells12060828
Banducci-Karp A, Xie J, Engels SAG, Sarantaris C, van Hage P, Varela M, Meijer AH, van der Vaart M. DRAM1 Promotes Lysosomal Delivery of Mycobacterium marinum in Macrophages. Cells. 2023; 12(6):828. https://doi.org/10.3390/cells12060828
Chicago/Turabian StyleBanducci-Karp, Adrianna, Jiajun Xie, Sem A. G. Engels, Christos Sarantaris, Patrick van Hage, Monica Varela, Annemarie H. Meijer, and Michiel van der Vaart. 2023. "DRAM1 Promotes Lysosomal Delivery of Mycobacterium marinum in Macrophages" Cells 12, no. 6: 828. https://doi.org/10.3390/cells12060828