
Characterization of pSer129-αSyn Pathology and Neurofilament Light-Chain Release across In Vivo, Ex Vivo, and In Vitro Models of Pre-Formed-Fibril-Induced αSyn Aggregation

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Pre-Formed Fibrils
2.3. Stereotaxic Injections
2.4. Terminal Sampling Procedures
2.5. Brain Immunohistochemistry
2.6. Organotypic Hippocampal Slice Cultures
2.7. Primary Hippocampal Cultures
2.8. Fractionation by Ultracentrifugation
2.9. Immunoblotting
2.10. Neurofilament Light-Chain Immunoassay
2.11. Homogeneous Time-Resolved Fluorescence (HTRF)
2.12. Fluidigm
2.13. Statistics
3. Results
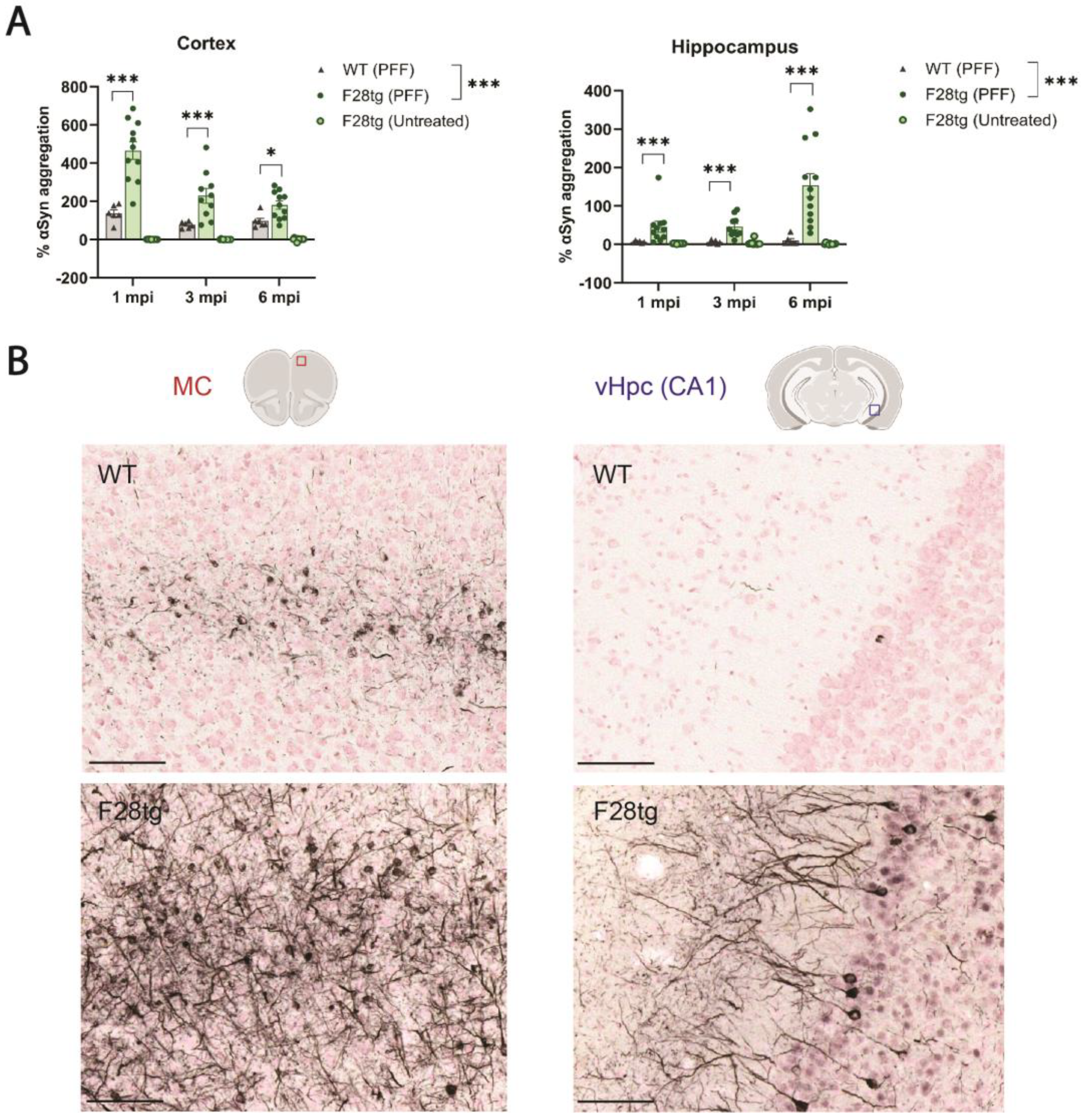
3.1. Overexpression of Human αSyn Increases αSyn Pathology upon PFF Injection In Vivo
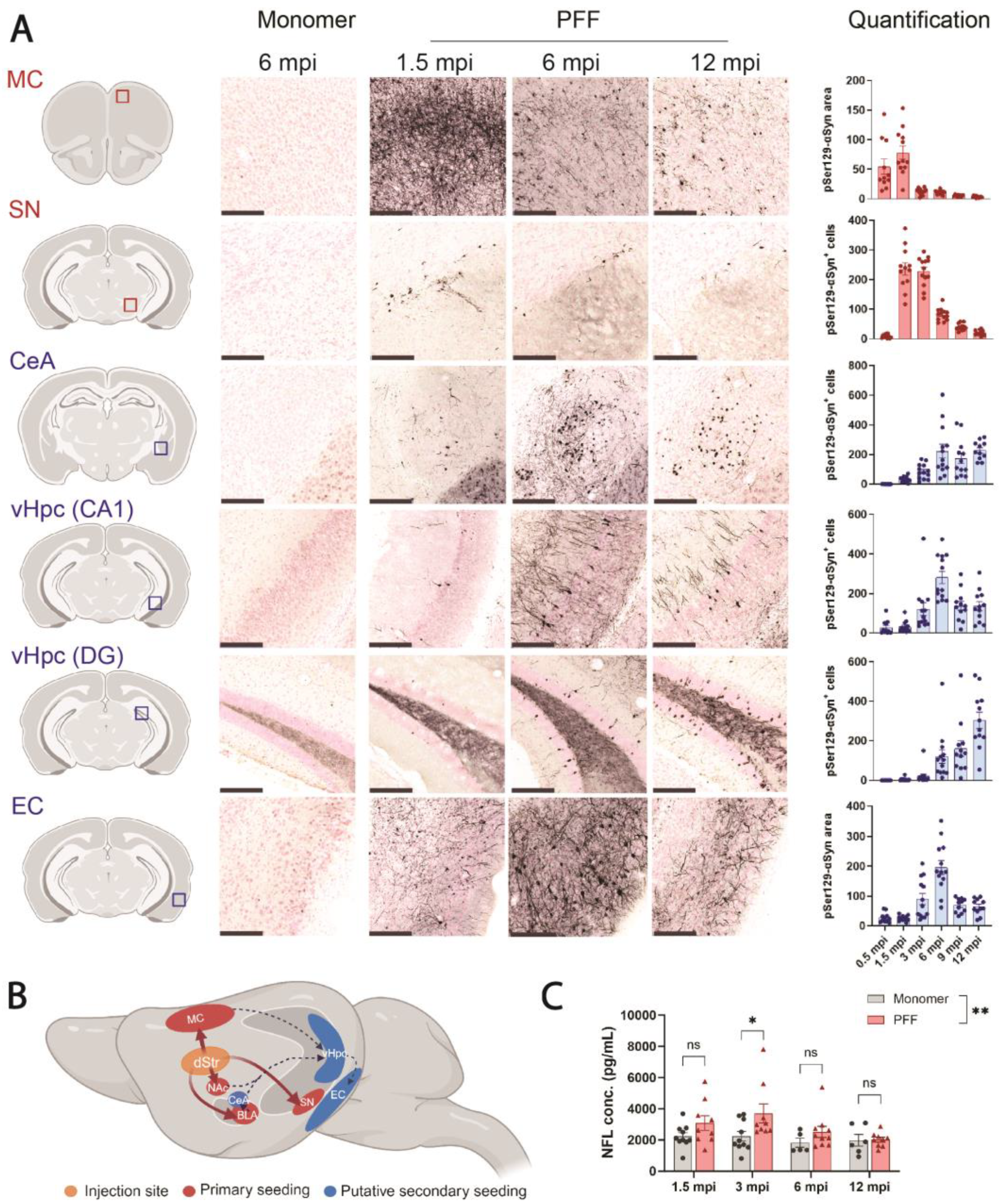
3.2. PFF Injection into F28tg Mice Induces the Spread of αSyn Pathology and Release of NFL to the CSF
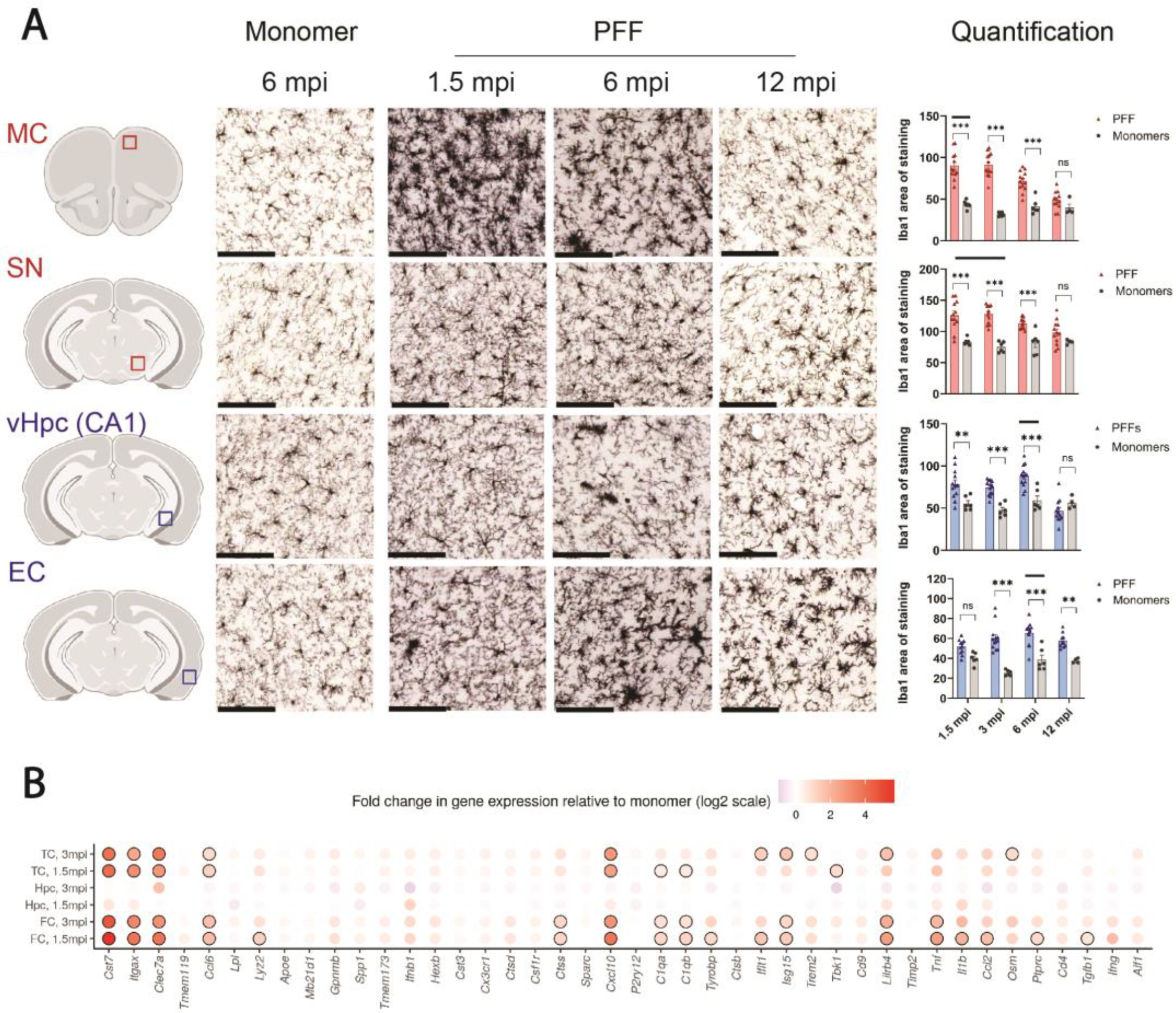
3.3. The Progressive Spread of αSyn Is Paralleled by Reactive Microgliosis In Vivo
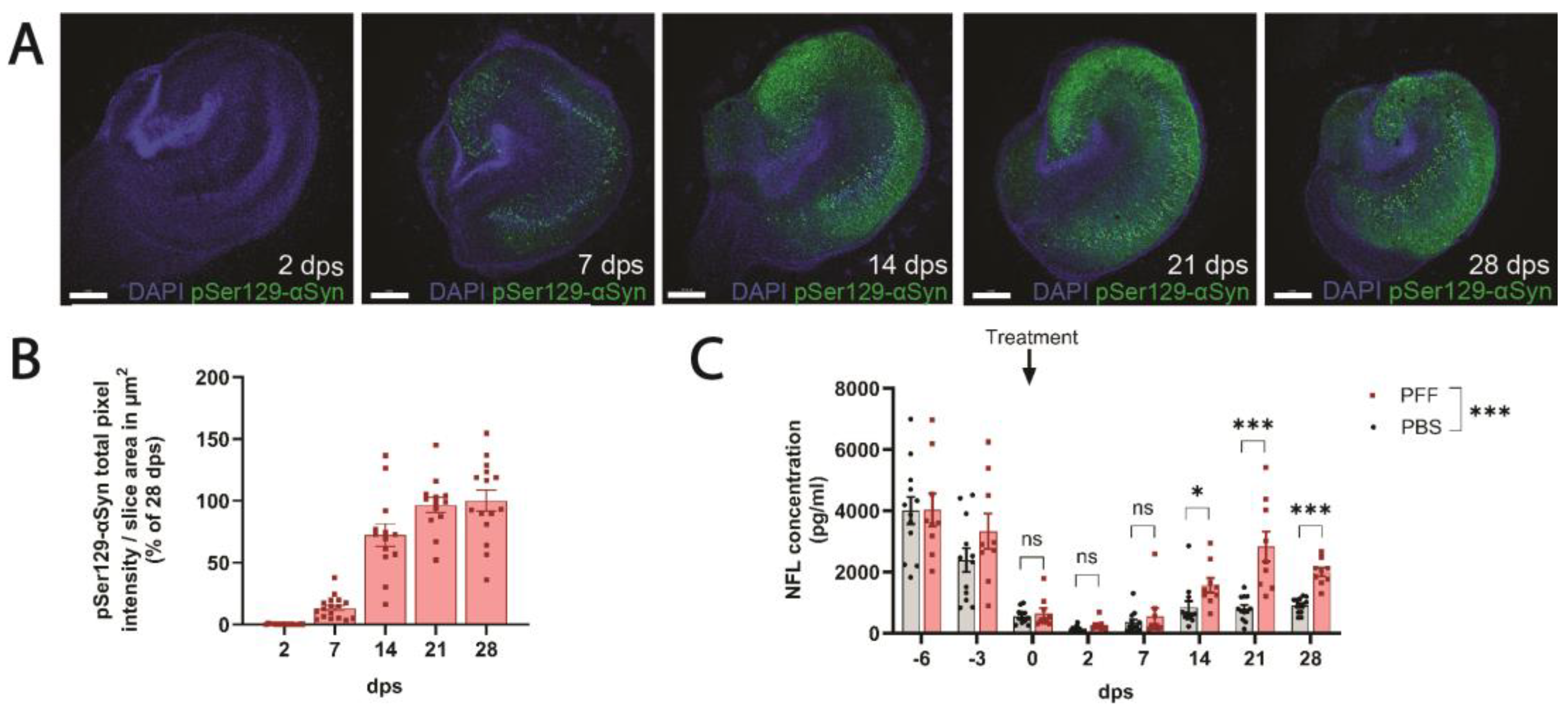
3.4. PFF-Treated Ex Vivo Organotypic Hippocampal Slices Cultures Develop pSer129-αSyn Pathology and Release NFL in a Time-Dependent Manner
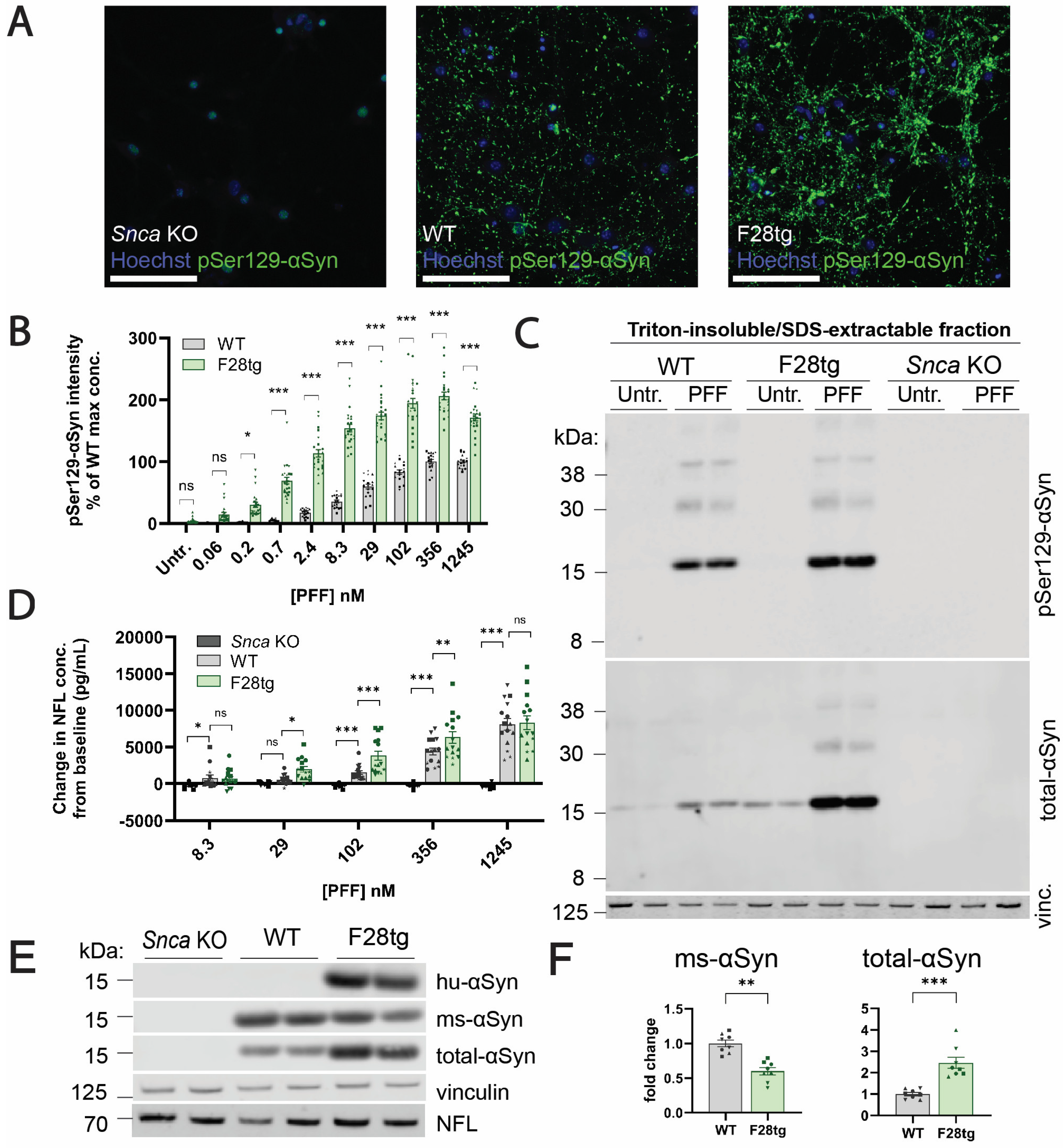
3.5. αSyn Aggregation Is a Key Determinant of NFL Release in Primary Hippocampal Cultures
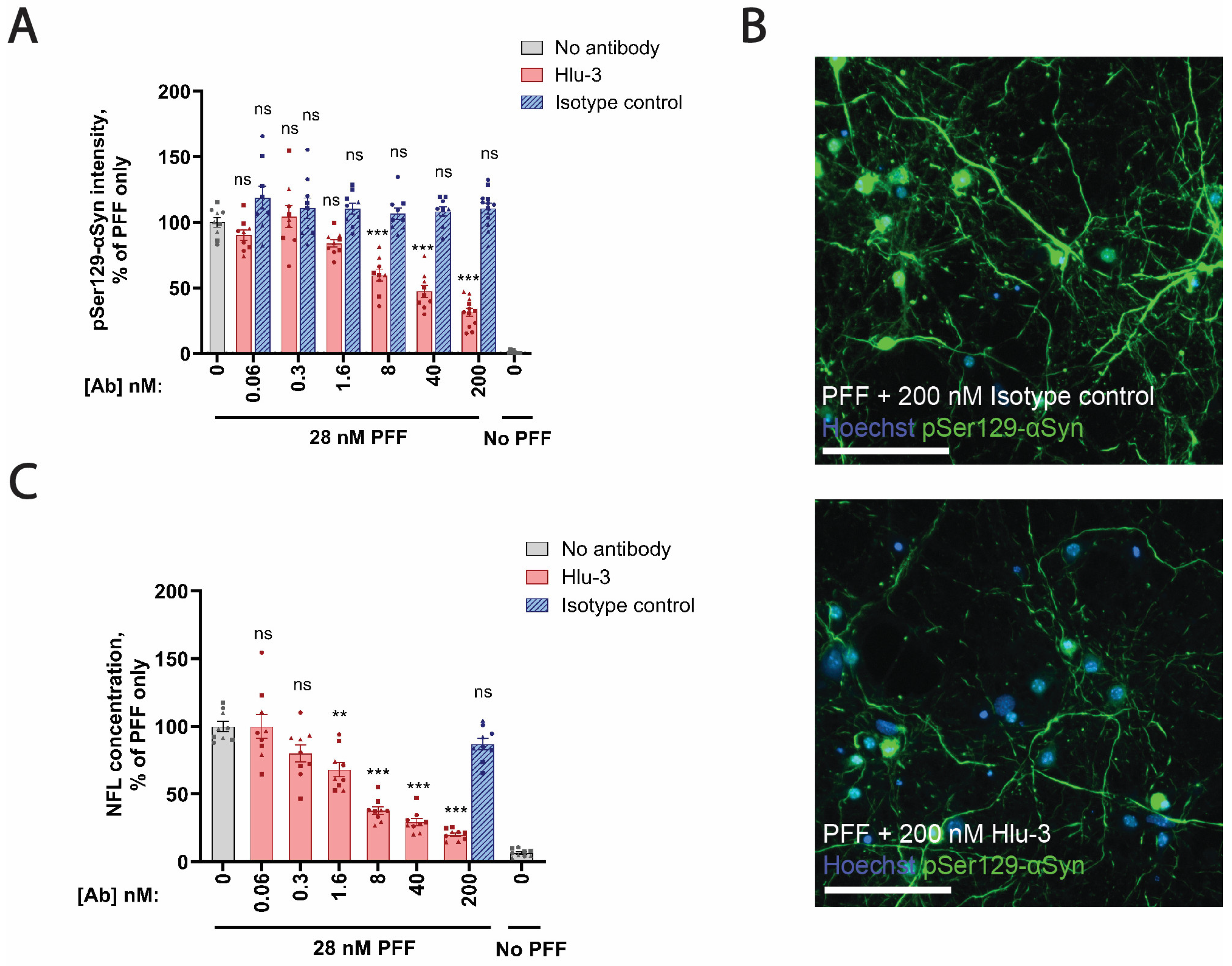
3.6. PFF-Induced Neurodegeneration In Vitro Is Inhibited by an Anti-αSyn Antibody
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baba, M.; Nakajo, S.; Tu, P.H.; Tomita, T.; Nakaya, K.; Lee, V.M.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol. 1998, 152, 879–884. [Google Scholar] [PubMed]
- Kim, W.S.; Kågedal, K.; Halliday, G.M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar]
- Li, J.Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Björklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef]
- Desplats, P.; Lee, H.J.; Bae, E.J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef]
- Goedert, M.; Masuda-Suzukake, M.; Falcon, B. Like prions: The propagation of aggregated tau and α-synuclein in neurodegeneration. Brain 2017, 140, 266–278. [Google Scholar] [CrossRef]
- Goedert, M. NEURODEGENERATION. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 2015, 349, 1255555. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef]
- Brundin, P.; Melki, R. Prying into the Prion Hypothesis for Parkinson’s Disease. J. Neurosci. 2017, 37, 9808–9818. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef]
- Karpowicz, R.J.; Haney, C.M.; Mihaila, T.S.; Sandler, R.M.; Petersson, E.J.; Lee, V.M.Y. Selective imaging of internalized proteopathic α-synuclein seeds in primary neurons reveals mechanistic insight into transmission of synucleinopathies. J. Biol. Chem. 2017, 292, 13482–13497. [Google Scholar] [CrossRef]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef]
- Mahul-Mellier, A.L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Lashuel, H.A. The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 4971–4982. [Google Scholar] [CrossRef] [PubMed]
- Rahayel, S.; Mišić, B.; Zheng, Y.-Q.; Liu, Z.-Q.; Abdelgawad, A.; Abbasi, N.; Caputo, A.; Zhang, B.; Lo, A.; Kehm, V.; et al. Differentially targeted seeding reveals unique pathological alpha-synuclein propagation patterns. Brain 2022, 145, 1743–1756. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, M.B.; Ferreira, S.A.; Schacht, A.C.; Jacobsen, J.; Simonsen, M.; Betzer, C.; Jensen, P.H.; Brooks, D.J.; Landau, A.M.; Romero-Ramos, M. PET imaging reveals early and progressive dopaminergic deficits after intra-striatal injection of preformed alpha-synuclein fibrils in rats. Neurobiol. Dis. 2021, 149, 105229. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Takano, H.; Riddle, D.M.; Trojanowski, J.Q.; Coulter, D.A.; Lee, V.M. α-Synuclein (αSyn) Preformed Fibrils Induce Endogenous αSyn Aggregation, Compromise Synaptic Activity and Enhance Synapse Loss in Cultured Excitatory Hippocampal Neurons. J. Neurosci. 2019, 39, 5080–5094. [Google Scholar] [CrossRef] [PubMed]
- Moudio, S.; Rodin, F.; Albargothy, N.J.; Karlsson, U.; Reyes, J.F.; Hallbeck, M. Exposure of α-Synuclein Aggregates to Organotypic Slice Cultures Recapitulates Key Molecular Features of Parkinson’s Disease. Front. Neurol. 2022, 13, 826102. [Google Scholar] [CrossRef] [PubMed]
- Betzer, C.; Lassen, L.B.; Olsen, A.; Kofoed, R.H.; Reimer, L.; Gregersen, E.; Zheng, J.; Calì, T.; Gai, W.P.; Chen, T.; et al. Alpha-synuclein aggregates activate calcium pump SERCA leading to calcium dysregulation. EMBO Rep. 2018, 19, 44617. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Gamble, K.L.; Schultheiss, C.E.; Riddle, D.M.; West, A.B.; Lee, V.M. Formation of α-synuclein Lewy neurite-like aggregates in axons impedes the transport of distinct endosomes. Mol. Biol. Cell 2014, 25, 4010–4023. [Google Scholar] [CrossRef] [PubMed]
- Chou, Q.L.; Alik, A.; Marquier, F.; Melki, R.; Treussart, F.; Simonneau, M. Impact of α-Synuclein Fibrillar Strains and β-Amyloid Assemblies on Mouse Cortical Neurons Endo-Lysosomal Logistics. eNeuro 2022, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Tanik, S.A.; Schultheiss, C.E.; Volpicelli-Daley, L.A.; Brunden, K.R.; Lee, V.M. Lewy body-like α-synuclein aggregates resist degradation and impair macroautophagy. J. Biol. Chem. 2013, 288, 15194–15210. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.C.; Minakaki, G.; Menges, S.; Salvi, R.; Savitskiy, S.; Kazman, A.; Vicente Miranda, H.; Mielenz, D.; Klucken, J.; Winkler, J.; et al. Extracellular aggregated alpha synuclein primarily triggers lysosomal dysfunction in neural cells prevented by trehalose. Sci. Rep. 2019, 9, 544. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10, 4066. [Google Scholar] [CrossRef]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; de Bernard, M. Triggering of inflammasome by aggregated α-synuclein, an inflammatory response in synucleinopathies. PLoS ONE 2013, 8, e55375. [Google Scholar] [CrossRef] [PubMed]
- Pike, A.F.; Varanita, T.; Herrebout, M.A.C.; Plug, B.C.; Kole, J.; Musters, R.J.P.; Teunissen, C.E.; Hoozemans, J.J.M.; Bubacco, L.; Veerhuis, R. α-Synuclein evokes NLRP3 inflammasome-mediated IL-1β secretion from primary human microglia. Glia 2021, 69, 1413–1428. [Google Scholar] [CrossRef]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef]
- Stoll, A.C.; Sortwell, C.E. Leveraging the preformed fibril model to distinguish between alpha-synuclein inclusion- and nigrostriatal degeneration-associated immunogenicity. Neurobiol. Dis. 2022, 171, 105804. [Google Scholar] [CrossRef]
- Duffy, M.F.; Collier, T.J.; Patterson, J.R.; Kemp, C.J.; Luk, K.C.; Tansey, M.G.; Paumier, K.L.; Kanaan, N.M.; Fischer, D.L.; Polinski, N.K.; et al. Lewy body-like alpha-synuclein inclusions trigger reactive microgliosis prior to nigral degeneration. J. Neuroinflamm. 2018, 15, 129. [Google Scholar] [CrossRef]
- Iba, M.; McDevitt, R.A.; Kim, C.; Roy, R.; Sarantopoulou, D.; Tommer, E.; Siegars, B.; Sallin, M.; Kwon, S.; Sen, J.M.; et al. Aging exacerbates the brain inflammatory micro-environment contributing to α-synuclein pathology and functional deficits in a mouse model of DLB/PD. Mol. Neurodegener. 2022, 17, 60. [Google Scholar] [CrossRef]
- Tran, H.T.; Chung, C.H.; Iba, M.; Zhang, B.; Trojanowski, J.Q.; Luk, K.C.; Lee, V.M. A-synuclein immunotherapy blocks uptake and templated propagation of misfolded α-synuclein and neurodegeneration. Cell Rep. 2014, 7, 2054–2065. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A.; et al. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Covell, D.J.; Kehm, V.M.; Zhang, B.; Song, I.Y.; Byrne, M.D.; Pitkin, R.M.; Decker, S.C.; Trojanowski, J.Q.; Lee, V.M.Y. Molecular and Biological Compatibility with Host Alpha-Synuclein Influences Fibril Pathogenicity. Cell Rep. 2016, 16, 3373–3387. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.; Canaslan, S.; Espinosa, J.C.; Fernández-Borges, N.; Villar-Piqué, A.; Llorens, F.; Varges, D.; Maass, F.; Torres, J.M.; Hermann, P.; et al. Validation of Plasma and CSF Neurofilament Light Chain as an Early Marker for Sporadic Creutzfeldt-Jakob Disease. Mol. Neurobiol. 2022, 59, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Soylu-Kucharz, R.; Sandelius, Å.; Sjögren, M.; Blennow, K.; Wild, E.J.; Zetterberg, H.; Björkqvist, M. Neurofilament light protein in CSF and blood is associated with neurodegeneration and disease severity in Huntington’s disease R6/2 mice. Sci. Rep. 2017, 7, 14114. [Google Scholar] [CrossRef] [PubMed]
- Clement, A.; Mitchelmore, C.; Andersson, D.R.; Asuni, A.A. Cerebrospinal fluid neurofilament light chain as a biomarker of neurodegeneration in the Tg4510 and MitoPark mouse models. Neuroscience 2017, 354, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Bacioglu, M.; Maia, L.F.; Preische, O.; Schelle, J.; Apel, A.; Kaeser, S.A.; Schweighauser, M.; Eninger, T.; Lambert, M.; Pilotto, A.; et al. Neurofilament Light Chain in Blood and CSF as Marker of Disease Progression in Mouse Models and in Neurodegenerative Diseases. Neuron 2016, 91, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Specht, C.G.; Schoepfer, R. Deletion of the alpha-synuclein locus in a subpopulation of C57BL/6J inbred mice. BMC Neurosci. 2001, 2, 11. [Google Scholar] [CrossRef]
- Westerlund, M.; Ran, C.; Borgkvist, A.; Sterky, F.H.; Lindqvist, E.; Lundströmer, K.; Pernold, K.; Brené, S.; Kallunki, P.; Fisone, G.; et al. Lrrk2 and α-synuclein are co-regulated in rodent striatum. Mol. Cell. Neurosci. 2008, 39, 586–591. [Google Scholar] [CrossRef]
- Polinski, N.K.; Volpicelli-Daley, L.A.; Sortwell, C.E.; Luk, K.C.; Cremades, N.; Gottler, L.M.; Froula, J.; Duffy, M.F.; Lee, V.M.Y.; Martinez, T.N.; et al. Best Practices for Generating and Using Alpha-Synuclein Pre-Formed Fibrils to Model Parkinson’s Disease in Rodents. J. Park. Dis. 2018, 8, 303–322. [Google Scholar] [CrossRef]
- Liu, M.; Kuhel, D.G.; Shen, L.; Hui, D.Y.; Woods, S.C. Apolipoprotein E does not cross the blood-cerebrospinal fluid barrier, as revealed by an improved technique for sampling CSF from mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R903–R908. [Google Scholar] [CrossRef]
- Zurhellen, C. (NeuroScience Associates, Inc., Knoxville, TN, USA). Personal communication, 2024.
- Gogolla, N.; Galimberti, I.; DePaola, V.; Caroni, P. Staining protocol for organotypic hippocampal slice cultures. Nat. Protoc. 2006, 1, 2452–2456. [Google Scholar] [CrossRef]
- Barth, M.; Bacioglu, M.; Schwarz, N.; Novotny, R.; Brandes, J.; Welzer, M.; Mazzitelli, S.; Häsler, L.M.; Schweighauser, M.; Wuttke, T.V.; et al. Microglial inclusions and neurofilament light chain release follow neuronal α-synuclein lesions in long-term brain slice cultures. Mol. Neurodegener. 2021, 16, 54. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.C.; Nadeau, K.; Abbasi, M.; Lachance, C.; Nguyen, M.; Fenrich, J. The Ultimate qPCR Experiment: Producing Publication Quality, Reproducible Data the First Time. Trends Biotechnol. 2019, 37, 761–774. [Google Scholar] [CrossRef]
- Lee, B.R.; Matsuo, Y.; Cashikar, A.G.; Kamitani, T. Role of Ser129 phosphorylation of α-synuclein in melanoma cells. J. Cell Sci. 2013, 126, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Scheiblich, H.; Dansokho, C.; Mercan, D.; Schmidt, S.V.; Bousset, L.; Wischhof, L.; Eikens, F.; Odainic, A.; Spitzer, J.; Griep, A.; et al. Microglia jointly degrade fibrillar alpha-synuclein cargo by distribution through tunneling nanotubes. Cell 2021, 184, 5089–5106.e5021. [Google Scholar] [CrossRef] [PubMed]
- Rostami, J.; Mothes, T.; Kolahdouzan, M.; Eriksson, O.; Moslem, M.; Bergström, J.; Ingelsson, M.; O’Callaghan, P.; Healy, L.M.; Falk, A.; et al. Crosstalk between astrocytes and microglia results in increased degradation of α-synuclein and amyloid-β aggregates. J. Neuroinflamm. 2021, 18, 124. [Google Scholar] [CrossRef] [PubMed]
- Damisah, E.C.; Hill, R.A.; Rai, A.; Chen, F.; Rothlin, C.V.; Ghosh, S.; Grutzendler, J. Astrocytes and microglia play orchestrated roles and respect phagocytic territories during neuronal corpse removal in vivo. Sci. Adv. 2020, 6, eaba3239. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Rasmussen, J.; Ewing, A.D.; Bodea, L.G.; Bodea, G.O.; Gearing, M.; Faulkner, G.J. An early proinflammatory transcriptional response to tau pathology is age-specific and foreshadows reduced tau burden. Brain Pathol. 2022, 32, e13018. [Google Scholar] [CrossRef] [PubMed]
- Benmamar-Badel, A.; Owens, T.; Wlodarczyk, A. Protective Microglial Subset in Development, Aging, and Disease: Lessons From Transcriptomic Studies. Front. Immunol. 2020, 11, 430. [Google Scholar] [CrossRef]
- Chen, X.-Y.; Feng, S.-N.; Bao, Y.; Zhou, Y.-X.; Ba, F. Identification of Clec7a as the therapeutic target of rTMS in alleviating Parkinson’s disease: Targeting neuroinflammation. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2023, 1869, 166814. [Google Scholar] [CrossRef]
- Szpakowski, P.; Ksiazek-Winiarek, D.; Turniak-Kusy, M.; Pacan, I.; Glabinski, A. Human Primary Astrocytes Differently Respond to Pro- and Anti-Inflammatory Stimuli. Biomedicines 2022, 10, 1769. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Gaetani, L.; Blennow, K.; Calabresi, P.; Di Filippo, M.; Parnetti, L.; Zetterberg, H. Neurofilament light chain as a biomarker in neurological disorders. J. Neurol. Neurosurg. Psychiatry 2019, 90, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.P.; Ejlerskov, P.; Rasmussen, I.; Vilhardt, F. Reciprocal signals between microglia and neurons regulate α-synuclein secretion by exophagy through a neuronal cJUN-N-terminal kinase-signaling axis. J. Neuroinflamm. 2016, 13, 59. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hansen, M.L.; Ambjørn, M.; Harndahl, M.N.; Benned-Jensen, T.; Fog, K.; Bjerregaard-Andersen, K.; Sotty, F. Characterization of pSer129-αSyn Pathology and Neurofilament Light-Chain Release across In Vivo, Ex Vivo, and In Vitro Models of Pre-Formed-Fibril-Induced αSyn Aggregation. Cells 2024, 13, 253. https://doi.org/10.3390/cells13030253
Hansen ML, Ambjørn M, Harndahl MN, Benned-Jensen T, Fog K, Bjerregaard-Andersen K, Sotty F. Characterization of pSer129-αSyn Pathology and Neurofilament Light-Chain Release across In Vivo, Ex Vivo, and In Vitro Models of Pre-Formed-Fibril-Induced αSyn Aggregation. Cells. 2024; 13(3):253. https://doi.org/10.3390/cells13030253
Chicago/Turabian StyleHansen, Maja L., Malene Ambjørn, Mikkel N. Harndahl, Tau Benned-Jensen, Karina Fog, Kaare Bjerregaard-Andersen, and Florence Sotty. 2024. "Characterization of pSer129-αSyn Pathology and Neurofilament Light-Chain Release across In Vivo, Ex Vivo, and In Vitro Models of Pre-Formed-Fibril-Induced αSyn Aggregation" Cells 13, no. 3: 253. https://doi.org/10.3390/cells13030253
APA StyleHansen, M. L., Ambjørn, M., Harndahl, M. N., Benned-Jensen, T., Fog, K., Bjerregaard-Andersen, K., & Sotty, F. (2024). Characterization of pSer129-αSyn Pathology and Neurofilament Light-Chain Release across In Vivo, Ex Vivo, and In Vitro Models of Pre-Formed-Fibril-Induced αSyn Aggregation. Cells, 13(3), 253. https://doi.org/10.3390/cells13030253