
Glymphatic System Pathology and Neuroinflammation as Two Risk Factors of Neurodegeneration



Abstract
1. Introduction to Neurodegeneration
2. Neuroinflammation in Neurodegeneration
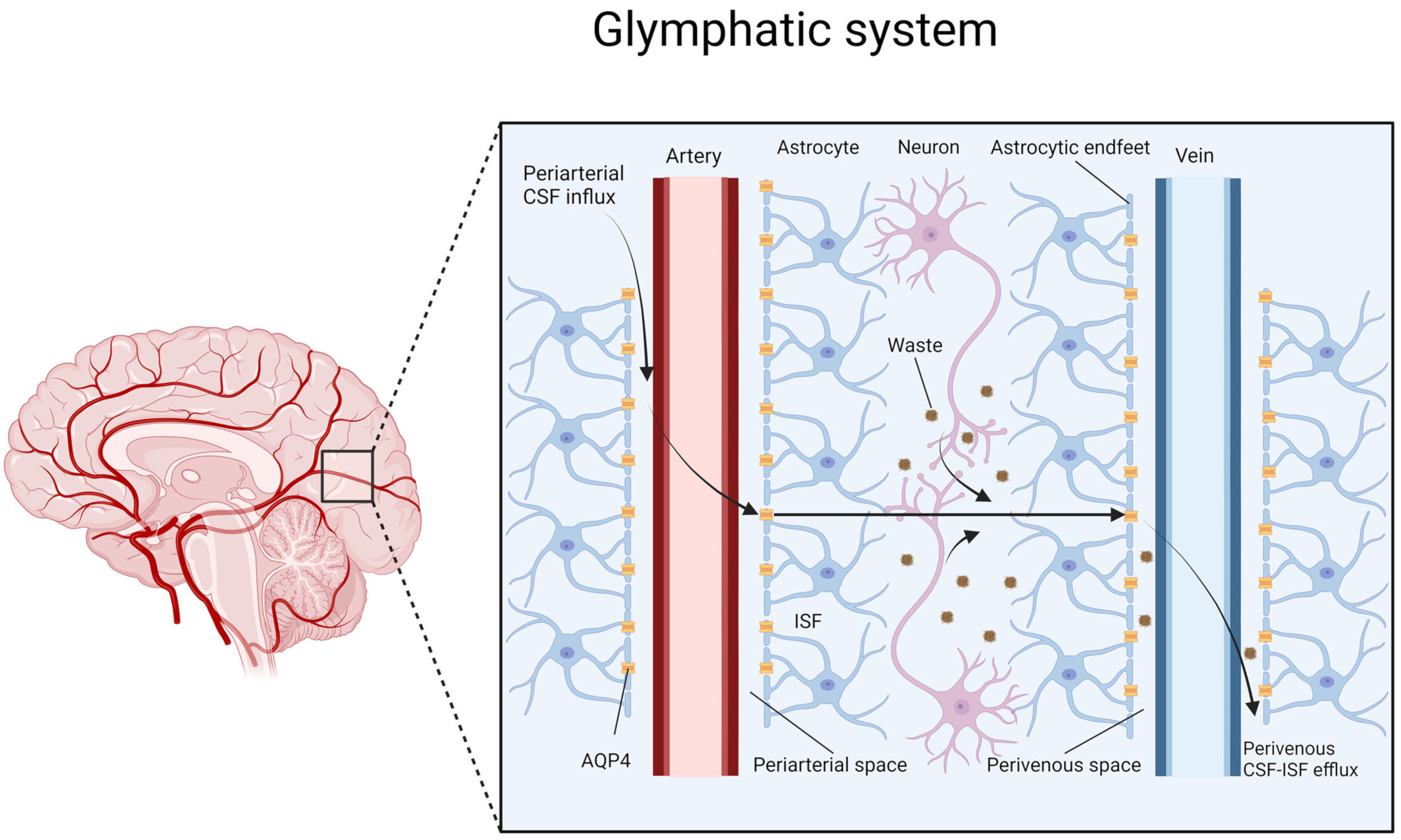
3. Glymphatic System
4. Astrocytes and the Role of AQP4

{kind=link}
{kind=link}
{kind=link}
| Multiple Roles of AQP4 in Neurodegeneration | Description | References |
|---|---|---|
| Loss of the perivascular localization of AQP4 | Results in glymphatic impairment and in an increase in Aβ level | Pedersen, T.J. et al., 2023 [42] |
| Deletion of the Aqp4 gene | Results in reduced clearance of injected α-Syn from the brain | Zhang, Y. et al., 2023 [5] |
| Significant reduction of perivascular localization of AQP4 | Observed in postmortem studies of patients with AD compared to subjects without cognitive impairment | Simon, M. et al., 2022 [41] |
| AQP4 deficiency | In an animal model accelerated the accumulation of α-Syn, facilitated the loss of dopaminergic neurons and, accelerated PD-like symptoms | Zhang, Y. et al., 2023 [5] |
| Pro-inflammatory role of AQP4 | Activation of microglia and swelling of astrocytes were observed in an animal model of PD | Prydz, A. et al., 2020 [57] |
| Involvement of AQP4 in the inflammatory response of astrocytes | Increase in the level of proinflammatory cytokines measured in astrocyte cultures was observed | Li, L. et al., 2011 [58] |
| AQP4 deletion | Results in exacerbation of cognitive deficits and was associated with an increase in Aβ accumulation in an animal model of AD | Xu, Z. et al., 2015 [39] |
5. Protein Aggregates as a Driver of the Vicious Circle of Impaired Glymphatic Clearance and Neurodegeneration
6. Peripheral Inflammatory Cells in the Brain
7. Gut–Brain Axis
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Erkkinen, M.G.; Kim, M.O.; Geschwind, M.D. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033118. [Google Scholar] [CrossRef]
- Gao, H.-M.; Hong, J.-S. Why neurodegenerative diseases are progressive: Uncontrolled inflammation drives disease progression. Trends Immunol. 2008, 29, 357–365. [Google Scholar] [CrossRef]
- Brown, R.C.; Lockwood, A.H.; Sonawane, B.R. Neurodegenerative diseases: An overview of environmental risk factors. Environ. Health Perspect. 2005, 113, 1250–1256. [Google Scholar] [CrossRef]
- Pihlstrom, L.; Wiethoff, S.; Houlden, H. Genetics of neurodegenerative diseases: An overview. Handb. Clin. Neurol. 2017, 145, 309–323. [Google Scholar]
- Zhang, Y.; Zhang, C.; He, X.-Z.; Li, Z.-H.; Meng, J.-C.; Mao, R.-T.; Li, X.; Xue, R.; Gui, Q.; Zhang, G.-X.; et al. Interaction Between the Glymphatic System and α-Synuclein in Parkinson’s Disease. Mol. Neurobiol. 2023, 60, 2209–2222. [Google Scholar] [CrossRef]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Ishida, K.; Yamada, K.; Nishiyama, R.; Hashimoto, T.; Nishida, I.; Abe, Y.; Yasui, M.; Iwatsubo, T. Glymphatic system clears extracellular tau and protects from tau aggregation and neurodegeneration. J. Exp. Med. 2022, 219, e20211275. [Google Scholar] [CrossRef] [PubMed]
- Feigin, V.L.; Vos, T.; Nichols, E.; O Owolabi, M.; Carroll, W.M.; Dichgans, M.; Deuschl, G.; Parmar, P.; Brainin, M.; Murray, C. The global burden of neurological disorders: Translating evidence into policy. Lancet Neurol. 2020, 19, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Farì, G.; Lunetti, P.; Pignatelli, G.; Raele, M.V.; Cera, A.; Mintrone, G.; Ranieri, M.; Megna, M.; Capobianco, L. The Effect of Physical Exercise on Cognitive Impairment in Neurodegenerative Disease: From Pathophysiology to Clinical and Rehabilitative Aspects. Int. J. Mol. Sci. 2021, 22, 11632. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, P.; Park, H.; Baumann, M.; Dunlop, J.; Frydman, J.; Kopito, R.; McCampbell, A.; Leblanc, G.; Venkateswaran, A.; Nurmi, A.; et al. Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl. Neurodegener. 2017, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Irvine, G.B.; El-Agnaf, O.M.; Shankar, G.M.; Walsh, D.M. Protein aggregation in the Brain: The molecular basis for Alzheimer’s and Parkinson’s diseases. Mol. Med. 2008, 14, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Forloni, G. Alpha Synuclein: Neurodegeneration and Inflammation. Int. J. Mol. Sci. 2023, 24, 5914. [Google Scholar] [CrossRef] [PubMed]
- Minter, M.R.; Taylor, J.M.; Crack, P.J. The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J. Neurochem. 2016, 136, 457–474. [Google Scholar] [CrossRef]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a Common Feature of Neurodegenerative Disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef] [PubMed]
- Lyman, M.; Lloyd, D.G.; Ji, X.; Vizcaychipi, M.P.; Ma, D. Neuroinflammation: The role and consequences. Neurosci. Res. 2014, 79, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Araque, A.; Navarrete, M. Glial cells in neuronal network function. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 2375–2381. [Google Scholar] [CrossRef] [PubMed]
- Franklin, H.; Clarke, B.E.; Patani, R. Astrocytes and microglia in neurodegenerative diseases: Lessons from human in vitro models. Prog. Neurobiol. 2021, 200, 101973. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Neuroinflammation Induces Neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar]
- Hablitz, L.M.; Nedergaard, M. The Glymphatic System: A Novel Component of Fundamental Neurobiology. J. Neurosci. 2021, 41, 7698–7711. [Google Scholar] [CrossRef]
- Benveniste, H.; Elkin, R.; Heerdt, P.M.; Koundal, S.; Xue, Y.; Lee, H.; Wardlaw, J.; Tannenbaum, A. The glymphatic system and its role in cerebral homeostasis. J. Appl. Physiol. 2020, 129, 1330–1340. [Google Scholar] [CrossRef]
- Padamsey, Z.; Rochefort, N.L. Paying the brain’s energy bill. Curr. Opin. Neurobiol. 2023, 78, 102668. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [PubMed]
- Mezey, É.; Palkovits, M. Neuroanatomy: Forgotten findings of brain lymphatics. Nature 2015, 524, 415. [Google Scholar] [CrossRef] [PubMed]
- Földi, M.; Csanda, E.; Simon, M.; Obál, F.; Schneider, I.; Dobranovics, I.; Zoltán, O.T.; Kozma, M.; Poberai, M. Lymphogenic haemangiopathy. “Prelymphatic” pathways in the wall of cerebral and cervical blood vessels. Angiologica 1968, 5, 250–262. [Google Scholar]
- Jessen, N.A.; Munk, A.S.F.; Lundgaard, I.; Nedergaard, M. The Glymphatic System: A Beginner’s Guide. Neurochem. Res. 2015, 40, 2583–2599. [Google Scholar] [CrossRef]
- Natale, G.; Limanaqi, F.; Busceti, C.L.; Mastroiacovo, F.; Nicoletti, F.; Puglisi-Allegra, S.; Fornai, F. Glymphatic System as a Gateway to Connect Neurodegeneration From Periphery to CNS. Front. Neurosci. 2021, 15, 639140. [Google Scholar] [CrossRef] [PubMed]
- Mestre, H.; Kostrikov, S.; Mehta, R.I.; Nedergaard, M. Perivascular spaces, glymphatic dysfunction, and small vessel disease. Clin. Sci. 2017, 131, 2257–2274. [Google Scholar] [CrossRef]
- Rasmussen, M.K.; Mestre, H.; Nedergaard, M. The glymphatic pathway in neurological disorders. Lancet Neurol. 2018, 17, 1016–1024. [Google Scholar] [CrossRef]
- Gędek, A.; Koziorowski, D.; Szlufik, S. Assessment of factors influencing glymphatic activity and implications for clinical medicine. Front. Neurol. 2023, 14, 1232304. [Google Scholar] [CrossRef]
- Kopeć, K.; Szleszkowski, S.; Koziorowski, D.; Szlufik, S. Glymphatic System and Mitochondrial Dysfunction as Two Crucial Players in Pathophysiology of Neurodegenerative Disorders. Int. J. Mol. Sci. 2023, 24, 10366. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- Kim, Y.; Park, J.; Choi, Y.K. The Role of Astrocytes in the Central Nervous System Focused on BK Channel and Heme Oxygenase Metabolites: A Review. Antioxidants 2019, 8, 121. [Google Scholar] [CrossRef]
- Ricci, G.; Volpi, L.; Pasquali, L.; Petrozzi, L.; Siciliano, G. Astrocyte–neuron interactions in neurological disorders. J. Biol. Phys. 2009, 35, 317–336. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, F.; Quintana, F.J. The Role of Astrocytes in CNS Inflammation. Trends Immunol. 2020, 41, 805–819. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Benveniste, E.N. Immune function of astrocytes. Glia 2001, 36, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Mader, S.; Brimberg, L. Aquaporin-4 Water Channel in the Brain and Its Implication for Health and Disease. Cells 2019, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Silva, I.; Silva, J.; Ferreira, R. Glymphatic system, AQP4, and their implications in Alzheimer’s disease. Neurol. Res. Pract. 2021, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Xiao, N.; Chen, Y.; Huang, H.; Marshall, C.; Gao, J.; Cai, Z.; Wu, T.; Hu, G.; Xiao, M. Deletion of aquaporin-4 in APP/PS1 mice exacerbates brain Abeta accumulation and memory deficits. Mol. Neurodegener. 2015, 10, 58. [Google Scholar] [CrossRef]
- Zeppenfeld, D.M.; Simon, M.; Haswell, J.D.; D’abreo, D.; Murchison, C.; Quinn, J.F.; Grafe, M.R.; Woltjer, R.L.; Kaye, J.; Iliff, J.J. Association of Perivascular Localization of Aquaporin-4 With Cognition and Alzheimer Disease in Aging Brains. JAMA Neurol. 2017, 74, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; Wang, M.X.; Ismail, O.; Braun, M.; Schindler, A.G.; Reemmer, J.; Wang, Z.; Haveliwala, M.A.; O’Boyle, R.P.; Han, W.Y.; et al. Loss of perivascular aquaporin-4 localization impairs glymphatic exchange and promotes amyloid beta plaque formation in mice. Alzheimer’s Res. Ther. 2022, 14, 59. [Google Scholar] [CrossRef]
- Pedersen, T.J.; Keil, S.A.; Han, W.; Wang, M.X.; Iliff, J.J. The effect of aquaporin-4 mis-localization on Abeta deposition in mice. Neurobiol. Dis. 2023, 181, 106100. [Google Scholar] [CrossRef]
- Gundersen, G.A.; Vindedal, G.F.; Skare, O.; Nagelhus, E.A. Evidence that pericytes regulate aquaporin-4 polarization in mouse cortical astrocytes. Brain. Struct Funct. 2014, 219, 2181–2186. [Google Scholar] [CrossRef]
- Eidsvaag, V.A.; Enger, R.; Hansson, H.-A.; Eide, P.K.; Nagelhus, E.A. Human and mouse cortical astrocytes differ in aquaporin-4 polarization toward microvessels. Glia 2017, 65, 964–973. [Google Scholar] [CrossRef]
- Gomolka, R.S.; Hablitz, L.M.; Mestre, H.; Giannetto, M.; Du, T.; Hauglund, N.L.; Xie, L.; Peng, W.; Martinez, P.M.; Nedergaard, M.; et al. Loss of aquaporin-4 results in glymphatic system dysfunction via brain-wide interstitial fluid stagnation. eLife 2023, 12, e82232. [Google Scholar] [CrossRef]
- Escartin, C.; Galea, E.; Lakatos, A.; O’callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrogliosis. Cold Spring Harb. Perspect. Biol. 2014, 7, a020420. [Google Scholar] [CrossRef]
- Colangelo, A.M.; Alberghina, L.; Papa, M. Astrogliosis as a therapeutic target for neurodegenerative diseases. Neurosci. Lett. 2014, 565, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Price, B.R.; Johnson, L.A.; Norris, C.M. Reactive astrocytes: The nexus of pathological and clinical hallmarks of Alzheimer’s disease. Ageing Res. Rev. 2021, 68, 101335. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.-L.; Long, C.-X.; Sun, L.; Xie, C.; Lin, X.; Caiet, H. Astrocytic expression of Parkinson’s disease-related A53T alpha-synuclein causes neurodegeneration in mice. Mol. Brain 2010, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Li, J.; Zheng, J.; Qin, S. Reactive Astrocytes in Neurodegenerative Diseases. Aging Dis. 2019, 10, 664–675. [Google Scholar] [CrossRef]
- Wolburg, H.; Wolburg-Buchholz, K.; Fallier-Becker, P.; Noell, S.; Mack, A.F. Structure and functions of aquaporin-4-based orthogonal arrays of particles. Int. Rev. Cell Mol. Biol. 2011, 287, 1–41. [Google Scholar]
- Eide, P.K.; Hansson, H.A. Astrogliosis and impaired aquaporin-4 and dystrophin systems in idiopathic normal pressure hydro-cephalus. Neuropathol. Appl. Neurobiol. 2018, 44, 474–490. [Google Scholar] [CrossRef] [PubMed]
- Kecheliev, V.; Boss, L.; Maheshwari, U.; Konietzko, U.; Keller, A.; Razansky, D.; Nitsch, R.M.; Klohs, J.; Ni, R. Aquaporin 4 is differentially increased and dislocated in association with tau and amyloid-beta. Life Sci. 2023, 321, 121593. [Google Scholar] [CrossRef]
- Si, X.; Dai, S.; Fang, Y.; Tang, J.; Wang, Z.; Li, Y.; Song, Z.; Chen, Y.; Liu, Y.; Zhao, G.; et al. Matrix metalloproteinase-9 inhibition prevents aquaporin-4 depolarization-mediated glymphatic dysfunction in Parkinson’s disease. J. Adv. Res. 2023, 56, 125–136. [Google Scholar] [CrossRef]
- Salman, M.M.; Kitchen, P.; Halsey, A.; Wang, M.X.; Törnroth-Horsefield, S.; Conner, A.C.; Badaut, J.; Iliff, J.J.; Bill, R.M. Emerging roles for dynamic aquaporin-4 subcellular relocalization in CNS water homeostasis. Brain 2021, 145, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Prydz, A.; Stahl, K.; Zahl, S.; Skauli, N.; Skare, Ø.; Ottersen, O.P.; Amiry-Moghaddam, M. Pro-Inflammatory Role of AQP4 in Mice Subjected to Intrastriatal Injections of the Parkinsonogenic Toxin MPP+. Cells 2020, 9, 2418. [Google Scholar] [CrossRef]
- Li, L.; Zhang, H.; Varrin-Doyer, M.; Zamvil, S.S.; Verkman, A. Proinflammatory role of aquaporin-4 in autoimmune neuroinflammation. FASEB J. 2011, 25, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- LaRocca, T.J.; Cavalier, A.N.; Roberts, C.M.; Lemieux, M.R.; Ramesh, P.; Garcia, M.A.; Link, C.D. Amyloid beta acts synergistically as a pro-inflammatory cytokine. Neurobiol. Dis. 2021, 159, 105493. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.M., 3rd; Cookson, M.R.; Van Den Bosch, L.; Zetterberg, H.; Holtzman, D.M.; Dewachter, I. Hallmarks of neurodegenerative diseases. Cell 2023, 186, 693–714. [Google Scholar] [CrossRef]
- El Khoury, J.B.; Moore, K.J.; Means, T.K.; Leung, J.; Terada, K.; Toft, M.; Freeman, M.W.; Luster, A.D. CD36 mediates the innate host response to beta-amyloid. J. Exp. Med. 2003, 197, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Alasmari, F.; Alshammari, M.A.; Alasmari, A.F.; Alanazi, W.A.; Alhazzani, K. Neuroinflammatory Cytokines Induce Amyloid Beta Neurotoxicity through Modulating Amyloid Precursor Protein Levels/Metabolism. BioMed. Res. Int. 2018, 2018, 3087475. [Google Scholar] [CrossRef] [PubMed]
- Webers; Heneka, M.T.; Gleeson, P.A. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol. Cell Biol. 2020, 98, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.-K.; Tao, K.-X.; Wang, X.-B.; Yao, X.-Y.; Pang, M.-Z.; Liu, J.-Y.; Wang, F.; Liu, C.-F. Role of α-synuclein in microglia: Autophagy and phagocytosis balance neuroinflammation in Parkinson’s disease. Inflamm. Res. 2023, 72, 443–462. [Google Scholar] [CrossRef] [PubMed]
- Jewell, S.; Herath, A.M.; Gordon, R. Inflammasome Activation in Parkinson’s Disease. J. Park. Dis. 2022, 12, S113–S128. [Google Scholar] [CrossRef] [PubMed]
- Chung, L.Y.-R.; Lin, Y.-T.; Liu, C.; Tai, Y.-C.; Lin, H.-Y.; Lin, C.-H.; Chen, C.-C. Neuroinflammation Upregulated Neuronal Toll-Like Receptors 2 and 4 to Drive Synucleinopathy in Neurodegen-eration. Front. Pharmacol. 2022, 13, 845930. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Rey, N.L.; Tyson, T.; Esquibel, C.; Meyerdirk, L.; Schulz, E.; Pierce, S.; Burmeister, A.R.; Madaj, Z.; Steiner, J.A.; et al. Microglia affect α-synuclein cell-to-cell transfer in a mouse model of Parkinson’s disease. Mol. Neurodegener. 2019, 14, 34. [Google Scholar] [CrossRef] [PubMed]
- Kress, B.T.; Iliff, J.J.; Xia, M.; Wang, M.; Wei, H.S.; Zeppenfeld, D.; Xie, L.; Kang, H.; Xu, Q.; Liew, J.A.; et al. Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol. 2014, 76, 845–861. [Google Scholar] [CrossRef]
- Lope-Piedrafita, S. Diffusion Tensor Imaging (DTI). Methods Mol. Biol. 2018, 1718, 103–116. [Google Scholar]
- Ota, M.; Sato, N.; Nakaya, M.; Shigemoto, Y.; Kimura, Y.; Chiba, E.; Yokoi, Y.; Tsukamoto, T.; Matsuda, H. Relationships Between the Deposition of Amyloid-beta and Tau Protein and Glymphatic System Activity in Alzheimer’s Disease: Diffusion Tensor Image Study. J. Alzheimer’s Dis. 2022, 90, 295–303. [Google Scholar] [CrossRef]
- Cui, H.; Wang, W.; Zheng, X.; Xia, D.; Liu, H.; Qin, C.; Tian, H.; Teng, J. Decreased AQP4 Expression Aggravates a-Synuclein Pathology in Parkinson’s Disease Mice, Possibly via Impaired Glymphatic Clearance. J. Mol. Neurosci. 2021, 71, 2500–2513. [Google Scholar] [CrossRef]
- Alliot, F.; Godin, I.; Pessac, B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res. Dev. Brain Res. 1999, 117, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Pessac, B.; Godin, I.; Alliot, F. Microglia: Origin and development. Bull Acad. Natl. Med. 2001, 185, 337; discussion 346–347. [Google Scholar] [PubMed]
- Lopes Pinheiro, M.A.; Kooij, G.; Mizee, M.R.; Kamermans, A.; Enzmann, G.; Lyck, R.; Schwaninger, M.; Engelhardt, B.; de Vries, H.E. Immune cell trafficking across the barriers of the central nervous system in multiple sclerosis and stroke. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2016, 1862, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Laaker, C.; Baenen, C.; Kovács, K.G.; Sandor, M.; Fabry, Z. Immune cells as messengers from the CNS to the periphery: The role of the meningeal lymphatic system in immune cell migration from the CNS. Front. Immunol. 2023, 14, 1233908. [Google Scholar] [CrossRef] [PubMed]
- Alam, A.; Thelin, E.P.; Tajsic, T.; Khan, D.Z.; Khellaf, A.; Patani, R.; Helmy, A. Cellular infiltration in traumatic brain injury. J. Neuroinflamm. 2020, 17, 328. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-W.; Zhang, X.; Huang, W.-J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef] [PubMed]
- Rezai-Zadeh, K.; Gate, D.; Town, T. CNS infiltration of peripheral immune cells: D-Day for neurodegenerative disease? J. Neuroimmune Pharmacol. 2009, 4, 462–475. [Google Scholar] [CrossRef]
- Brochard, V.; Combadière, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.-M.; et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Investig. 2009, 119, 182–192. [Google Scholar] [CrossRef]
- Bas, J.; Calopa, M.; Mestre, M.; Molleví, D.G.; Cutillas, B.; Ambrosio, S.; Buendia, E. Lymphocyte populations in Parkinson’s disease and in rat models of parkinsonism. J. Neuroimmunol. 2001, 113, 146–152. [Google Scholar] [CrossRef]
- Sulzer, D.; Alcalay, R.N.; Garretti, F.; Cote, L.; Kanter, E.; Agin-Liebes, J.; Liong, C.; McMurtrey, C.; Hildebrand, W.H.; Mao, X.; et al. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 2017, 546, 656–661. [Google Scholar] [CrossRef]
- Gelders, G.; Baekelandt, V.; Van der Perren, A. Linking Neuroinflammation and Neurodegeneration in Parkinson’s Disease. J. Immunol. Res. 2018, 2018, 4784268. [Google Scholar] [CrossRef]
- Hernández, J.C.C.; Bracko, O.; Kersbergen, C.J.; Muse, V.; Haft-Javaherian, M.; Berg, M.; Park, L.; Vinarcsik, L.K.; Ivasyk, I.; Rivera, D.A.; et al. Neutrophil adhesion in brain capillaries reduces cortical blood flow and impairs memory function in Alz-heimer’s disease mouse models. Nat. Neurosci. 2019, 22, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Zenaro, E.; Pietronigro, E.; Della Bianca, V.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 2015, 21, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Baik, S.H.; Cha, M.-Y.; Hyun, Y.-M.; Cho, H.; Hamza, B.; Kim, D.K.; Han, S.-H.; Choi, H.; Kim, K.H.; Moon, M.; et al. Migration of neutrophils targeting amyloid plaques in Alzheimer’s disease mouse model. Neurobiol. Aging 2014, 35, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Smyth, L.C.D.; Murray, H.C.; Hill, M.; van Leeuwen, E.; Highet, B.; Magon, N.J.; Osanlouy, M.; Mathiesen, S.N.; Mockett, B.; Singh-Bains, M.K.; et al. Neutrophil-vascular interactions drive myeloperoxidase accumulation in the brain in Alzheimer’s disease. Acta Neuropathol. Commun. 2022, 10, 38. [Google Scholar] [CrossRef]
- Song, L.; Yang, Y.T.; Guo, Q.; Zhao, X.-M. the ZIB Consortium Cellular transcriptional alterations of peripheral blood in Alzheimer’s disease. BMC Med. 2022, 20, 266. [Google Scholar] [CrossRef]
- Pham, C.T.N. Neutrophil serine proteases: Specific regulators of inflammation. Nat. Rev. Immunol. 2006, 6, 541–550. [Google Scholar] [CrossRef]
- Korkmaz, B.; Horwitz, M.S.; Jenne, D.E.; Gauthier, F. Neutrophil elastase, proteinase 3, and cathepsin gas therapeutic targets in human diseases. Pharmacol. Rev. 2010, 62, 726–759. [Google Scholar] [CrossRef]
- Lee, T.D.; Gonzalez, M.L.; Kumar, P.; Chary-Reddy, S.; Grammas, P.; Pereira, H.A. CAP37, a novel inflammatory mediator: Its expression in endothelial cells and localization to atherosclerotic lesions. Am. J. Pathol. 2002, 160, 841–848. [Google Scholar] [CrossRef]
- Nakajima, K.; Shimojo, M.; Hamanoue, M.; Ishiura, S.; Sugita, H.; Kohsaka, S. Identification of elastase as a secretory protease from cultured rat microglia. J. Neurochem. 1992, 58, 1401–1408. [Google Scholar] [CrossRef]
- Burster, T.; Beck, A.; Poeschel, S.; Øren, A.; Baechle, D.; Reich, M.; Roetzschke, O.; Falk, K.; Boehm, B.O.; Youssef, S.; et al. Interferon-gamma regulates cathepsin G activity in microglia-derived lysosomes and controls the proteolytic processing of myelin basic protein in vitro. Immunology 2007, 121, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Pereira, H.A.; Ruan, X.; Kumar, P. Activation of microglia: A neuroinflammatory role for CAP37. Glia 2003, 41, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shui, X.; Sun, R.; Wan, L.; Zhang, B.; Xiao, B.; Luo, Z. Microglial Phenotypic Transition: Signaling Pathways and Influencing Modulators Involved in Regulation in Central Nervous System Diseases. Front. Cell. Neurosci. 2021, 15, 736310. [Google Scholar] [CrossRef] [PubMed]
- Rada, B. Neutrophil Extracellular Traps. Methods Mol. Biol. 2019, 1982, 517–528. [Google Scholar]
- Dömer, D.; Walther, T.; Möller, S.; Behnen, M.; Laskay, T. Neutrophil Extracellular Traps Activate Proinflammatory Functions of Human Neutrophils. Front. Immunol. 2021, 12, 636954. [Google Scholar] [CrossRef] [PubMed]
- García-Cabrerizo, R.; Carbia, C.; Riordan, K.J.O.; Schellekens, H.; Cryan, J.F. Microbiota-gut-brain axis as a regulator of reward processes. J. Neurochem. 2021, 157, 1495–1524. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; O’Riordan, K.J.; Cowan, C.S.M.; Sandhu, K.V.; Bastiaanssen, T.F.S.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The Microbiota-Gut-Brain Axis. Physiol. Rev. 2019, 99, 1877–2013. [Google Scholar] [CrossRef] [PubMed]
- Kasarello, K.; Cudnoch-Jedrzejewska, A.; Czarzasta, K. Communication of gut microbiota and brain via immune and neuroen-docrine signaling. Front Microbiol. 2023, 14, 1118529. [Google Scholar] [CrossRef]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef]
- DeGruttola, A.K.; Low, D.; Mizoguchi, A.; Mizoguchi, E. Current Understanding of Dysbiosis in Disease in Human and Animal Models. Inflamm. Bowel Dis. 2016, 22, 1137–1150. [Google Scholar] [CrossRef] [PubMed]
- Bairamian, D.; Sha, S.; Rolhion, N.; Sokol, H.; Dorothée, G.; Lemere, C.A.; Krantic, S. Microbiota in neuroinflammation and synaptic dysfunction: A focus on Alzheimer’s disease. Mol. Neurodegener. 2022, 17, 19. [Google Scholar] [CrossRef] [PubMed]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. [Google Scholar] [CrossRef]
- Sheng, J.G.; Bora, S.H.; Xu, G.; Borchelt, D.R.; Price, D.L.; Koliatsos, V.E. Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPswe transgenic mice. Neurobiol. Dis. 2003, 14, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wang, Y.; Zhou, R.; Li, Y.; Gao, Y.; Tu, D.; Wilson, B.; Song, S.; Feng, J.; Hong, J.-S.; et al. A novel role of NLRP3-generated IL-1beta in the acute-chronic transition of peripheral lipopolysaccharide-elicited neu-roinflammation: Implications for sepsis-associated neurodegeneration. J. Neuroinflamm. 2020, 17, 64. [Google Scholar] [CrossRef]
- Pellegrini, C.; Antonioli, L.; Calderone, V.; Colucci, R.; Fornai, M.; Blandizzi, C. Microbiota-gut-brain axis in health and disease: Is NLRP3 inflammasome at the crossroads of microbiota-gut-brain communications? Prog. Neurobiol. 2020, 191, 101806. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Stamova, B.; Jin, L.-W.; DeCarli, C.; Phinney, B.; Sharp, F.R. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology 2016, 87, 2324–2332. [Google Scholar] [CrossRef] [PubMed]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480.e12. [Google Scholar] [CrossRef]
- Wang, Q.; Luo, Y.; Chaudhuri, K.R.; Reynolds, R.; Tan, E.-K.; Pettersson, S. The role of gut dysbiosis in Parkinson’s disease: Mechanistic insights and therapeutic options. Brain 2021, 144, 2571–2593. [Google Scholar] [CrossRef]
- Bostick, J.W.; Schonhoff, A.M.; Mazmanian, S.K. Gut microbiome-mediated regulation of neuroinflammation. Curr. Opin. Immunol. 2022, 76, 102177. [Google Scholar] [CrossRef]
- Bhattacharjee; Lukiw, W.J. Alzheimer’s disease and the microbiome. Front Cell Neurosci. 2013, 7, 153. [Google Scholar] [CrossRef]
- Romano, S.; Savva, G.M.; Bedarf, J.R.; Charles, I.G.; Hildebrand, F.; Narbad, A. Meta-analysis of the Parkinson’s disease gut microbiome suggests alterations linked to intestinal inflammation. NPJ Park. Dis. 2021, 7, 27. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef]
- Verheggen, I.; Van Boxtel, M.; Verhey, F.; Jansen, J.; Backes, W. Interaction between blood-brain barrier and glymphatic system in solute clearance. Neurosci. Biobehav. Rev. 2018, 90, 26–33. [Google Scholar] [CrossRef]
- A Erickson, M.; E Hartvigson, P.; Morofuji, Y.; Owen, J.B.; Butterfield, D.A.; A Banks, W. Lipopolysaccharide impairs amyloid beta efflux from brain: Altered vascular sequestration, cerebrospinal fluid reabsorption, peripheral clearance and transporter function at the blood–brain barrier. J. Neuroinflamm. 2012, 9, 150. [Google Scholar] [CrossRef] [PubMed]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Tóth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 6, 263ra158. [Google Scholar] [CrossRef]
- Portincasa, P.; Bonfrate, L.; Vacca, M.; De Angelis, M.; Farella, I.; Lanza, E.; Khalil, M.; Wang, D.Q.-H.; Sperandio, M.; Di Ciaula, A. Gut Microbiota and Short Chain Fatty Acids: Implications in Glucose Homeostasis. Int. J. Mol. Sci. 2022, 23, 1105. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.; Fonseca, S.; Carding, S.R. Gut microbes and metabolites as modulators of blood-brain barrier integrity and brain health. Gut Microbes 2020, 11, 135–157. [Google Scholar] [CrossRef] [PubMed]
- Matt, S.M.; Allen, J.M.; Lawson, M.A.; Mailing, L.J.; Woods, J.A.; Johnson, R.W. Butyrate and Dietary Soluble Fiber Improve Neuroinflammation Associated With Aging in Mice. Front. Immunol. 2018, 9, 1832. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szlufik, S.; Kopeć, K.; Szleszkowski, S.; Koziorowski, D. Glymphatic System Pathology and Neuroinflammation as Two Risk Factors of Neurodegeneration. Cells 2024, 13, 286. https://doi.org/10.3390/cells13030286
Szlufik S, Kopeć K, Szleszkowski S, Koziorowski D. Glymphatic System Pathology and Neuroinflammation as Two Risk Factors of Neurodegeneration. Cells. 2024; 13(3):286. https://doi.org/10.3390/cells13030286
Chicago/Turabian StyleSzlufik, Stanisław, Kamila Kopeć, Stanisław Szleszkowski, and Dariusz Koziorowski. 2024. "Glymphatic System Pathology and Neuroinflammation as Two Risk Factors of Neurodegeneration" Cells 13, no. 3: 286. https://doi.org/10.3390/cells13030286
APA StyleSzlufik, S., Kopeć, K., Szleszkowski, S., & Koziorowski, D. (2024). Glymphatic System Pathology and Neuroinflammation as Two Risk Factors of Neurodegeneration. Cells, 13(3), 286. https://doi.org/10.3390/cells13030286