
A Possible Therapeutic Application of the Selective Inhibitor of Urate Transporter 1, Dotinurad, for Metabolic Syndrome, Chronic Kidney Disease, and Cardiovascular Disease


Abstract
:1. Introduction
2. The Association of URAT1 and Other UA Transporters with Metabolic Syndrome
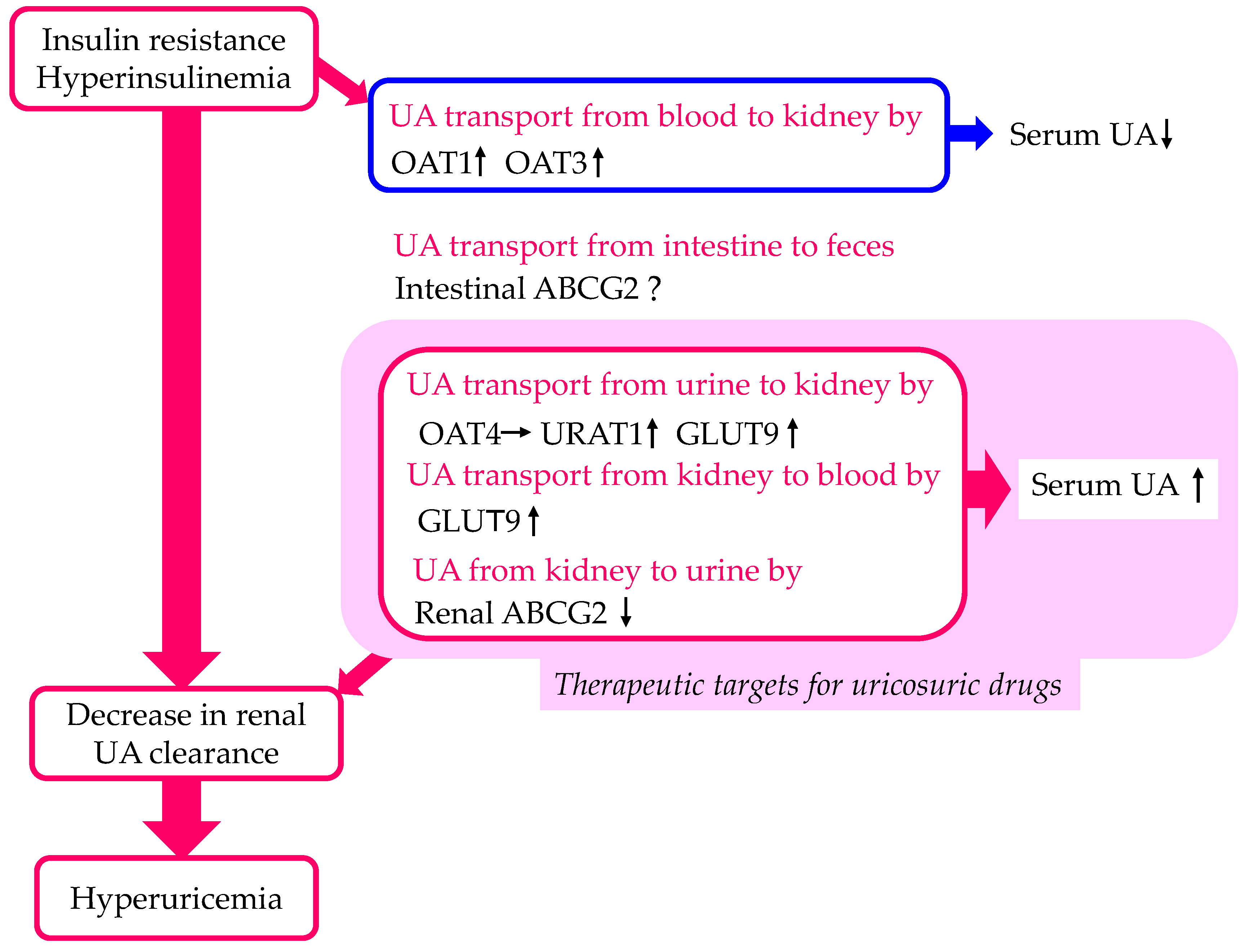
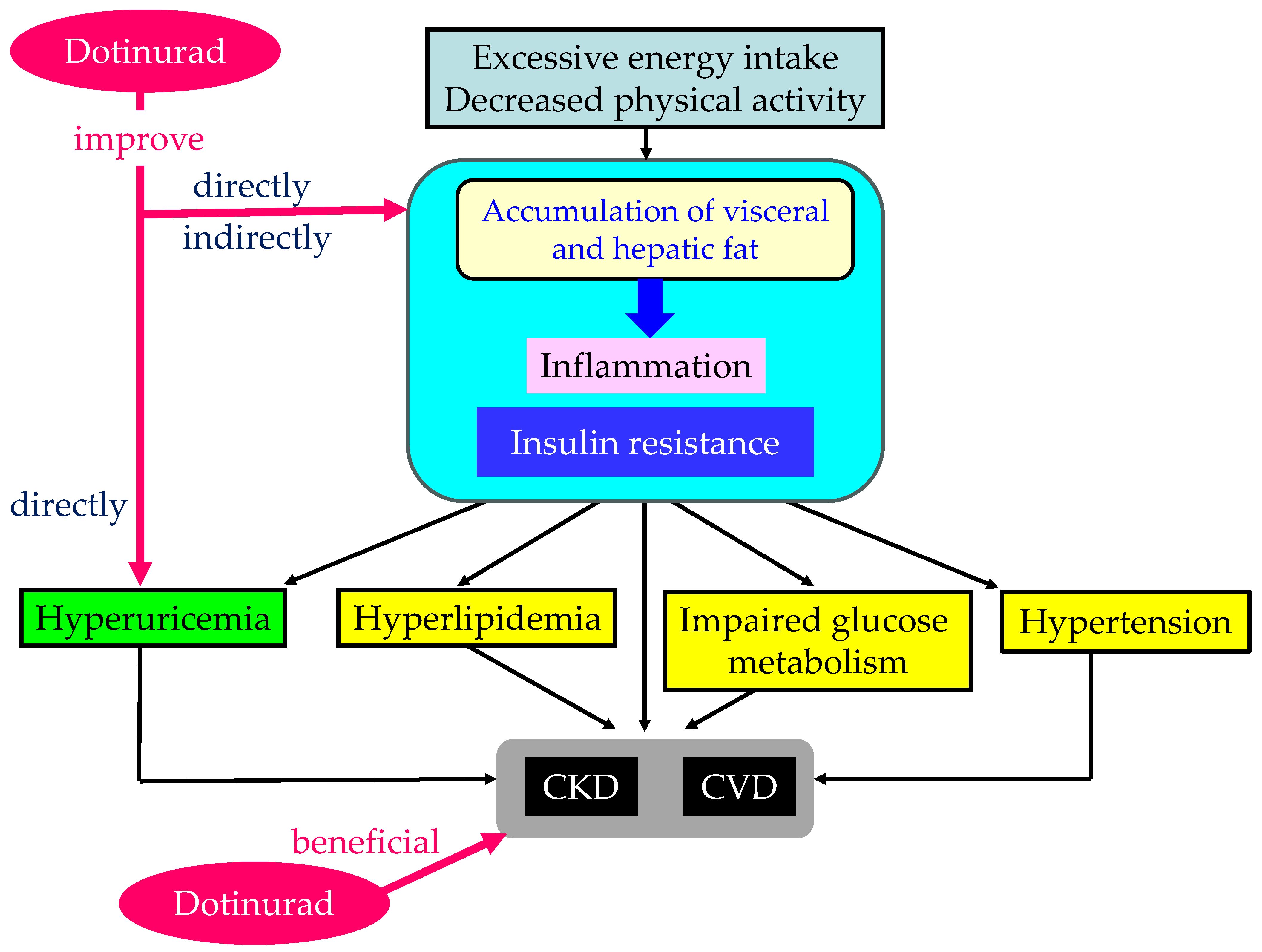
2.1. Metabolic Syndrome and Hyperuricemia
2.2. The Effect of Insulin Resistance on URAT1 Expression
2.3. The Effect of Insulin on UA Transport by Other Urate Transporters
2.4. The Effect of Inhibition of URAT1 on Metabolic Parameters in Humans
2.5. The Effect of Inhibition of URAT1 on Metabolic Parameters in Mice
2.6. The Effects of Other UA-Lowering Drugs on Metabolic Parameters
2.7. The Possible Mechanisms of an Improvement in Metabolic Parameters by Dotinurad
3. The Association of URAT1 and Other UA Transporters with CKD
3.1. CKD and Hyperuricemia
3.2. The Effect of CKD on Renal URAT1 Expression
3.3. The Effect of CKD on Other Urate Transporter Expressions
3.4. The Effect of IS on CKD
3.5. The Effect of Inhibition of URAT1 on CKD
3.6. The Effects of Other UA-Lowering Drugs on CKD
3.7. The Effects of Febuxostat and Dotinurad on Advanced CKD

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UA-Lowering Drugs | XO Inhibitors | Uricosuric Drugs | ||||
|---|---|---|---|---|---|---|
| Allopurinol | Febuxostat | Topiroxostat | Benzbromarone | Probenecid | Dotinurad | |
| Inhibition of UA Transporters | ABCG2 | ABCG2 | ABCG2 | ABCG2 | URAT1 | |
| URAT1 | URAT1 | |||||
| GLUT9 | GLUT9 | |||||
| OAT1 | OAT1 | |||||
| OAT3 | OAT3 | |||||
| Albuminuria | No data | Improved [85,87,90] | Improved [92] | No data | No data | Improved [14,79] |
| eGFR or serum creatinine | Improved [82,83] | Improved [85] | Not improved [93] and Improved [94] | No data | No data | Improved [14,79,80,81] |
| eGFR in patients with CKD stage 4 and 5 | No data | Not improved [96,97] | No data | No data | No data | Improved [79,80,81] |
| Proximal tubular impairment | No data | Improved [87] | Not improved [93] | No data | No data | No data |
| Renal outcomes | Improved [84] | Improved [91] | No data | Improved [95] | No data | No data |
4. The Association of URAT1 and Other UA Transporters with the Development of CVD
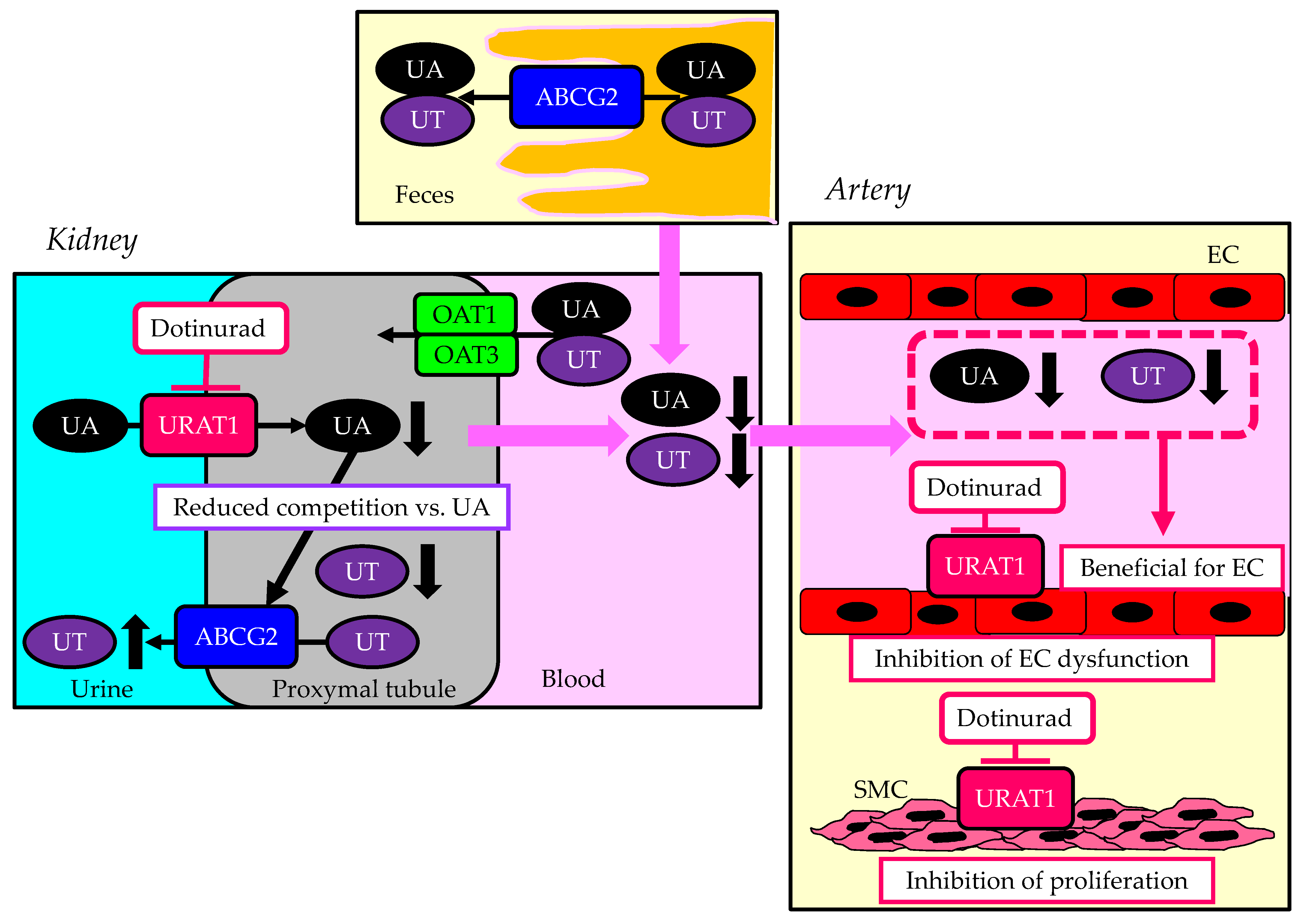
4.1. The Association of URAT1 with Atherogenesis
4.2. The Association of Other UA Transporters with Atherogenesis
4.3. The Effect of Inhibition of URAT1 on Atherosclerosis
4.4. The Effects of Other UA-Lowering Drugs on Endothelial Function
4.5. The Effects of UA-Lowering Drugs on CVD
5. A Summary of Unfavorable Effects of the Inhibition of ABCG2, OAT1, and OAT3 on the Kidneys and Vascular Endothelial Cells in CKD Patients
6. A Summary of the Beneficial Effects of Dotinurad on the Kidneys and Atherosclerosis in CKD Patients
7. Possible Beneficial Effects of Dotinurad for Heart Failure (HF)
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Enomoto, A.; Kimura, H.; Chairoungdua, A.; Shigeta, Y.; Jutabha, P.; Cha, S.H.; Hosoyamada, M.; Takeda, M.; Sekine, T.; Igarashi, T. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 2002, 417, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H. Molecular Biological and Clinical Understanding of the Pathophysiology and Treatments of Hyperuricemia and Its Association with Metabolic Syndrome, Cardiovascular Diseases and Chronic Kidney Disease. Int. J. Mol. Sci. 2021, 22, 9221. [Google Scholar] [CrossRef] [PubMed]
- Dalbeth, N.; Merriman, T. Crystal ball gazing: New therapeutic targets for hyperuricaemia and gout. Rheumatology 2009, 48, 222–226. [Google Scholar] [CrossRef]
- Merriman, T.R.; Dalbeth, N. The genetic basis of hyperuricaemia and gout. Jt. Bone Spine 2011, 78, 35–40. [Google Scholar] [CrossRef]
- Xu, L.; Shi, Y.; Zhuang, S.; Liu, N. Recent advances on uric acid transporters. Oncotarget 2017, 8, 100852–100862. [Google Scholar] [CrossRef]
- Caulfield, M.J.; Munroe, P.B.; O’Neill, D.; Witkowska, K.; Charchar, F.J.; Doblado, M.; Evans, S.; Eyheramendy, S.; Onipinla, A.; Howard, P.; et al. SLC2A9 is a high-capacity urate transporter in humans. PLoS Med. 2008, 5, e197. [Google Scholar] [CrossRef]
- Li, S.; Sanna, S.; Maschio, A.; Busonero, F.; Usala, G.; Mulas, A.; Lai, S.; Dei, M.; Orrù, M.; Albai, G. The GLUT9 gene is associated with serum uric acid levels in Sardinia and Chianti cohorts. PLoS Genet. 2007, 3, e194. [Google Scholar] [CrossRef] [PubMed]
- Vitart, V.; Rudan, I.; Hayward, C.; Gray, N.K.; Floyd, J.; Palmer, C.N.; Knott, S.A.; Kolcic, I.; Polasek, O.; Graessler, J.; et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat. Genet. 2008, 40, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Woodward, O.M.; Köttgen, A.; Coresh, J.; Boerwinkle, E.; Guggino, W.B.; Köttgen, M. Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc. Natl. Acad. Sci. USA 2009, 106, 10338–10342. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, H.; Takada, T.; Ichida, K.; Nakamura, T.; Nakayama, A.; Ikebuchi, Y.; Ito, K.; Kusanagi, Y.; Chiba, T.; Tadokoro, S. Common defects of ABCG2, a high-capacity urate exporter, cause gout: A function-based genetic analysis in a Japanese population. Sci. Transl. Med. 2009, 1, 5ra11. [Google Scholar] [CrossRef]
- Sattui, S.E.; Gaffo, A.L. Treatment of hyperuricemia in gout: Current therapeutic options, latest developments and clinical implications. Ther. Adv. Musculoskelet. Dis. 2016, 8, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Reinders, M.K.; van Roon, E.N.; Houtman, P.M.; Brouwers, J.R.; Jansen, T.L. Biochemical effectiveness of allopurinol and allopurinol-probenecid in previously benzbromarone-treated gout patients. Clin. Rheumatol. 2007, 26, 1459–1465. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Ashizawa, N.; Matsumoto, K.; Saito, R.; Motoki, K.; Sakai, M.; Chikamatsu, N.; Hagihara, C.; Hashiba, M.; Iwanaga, T. Pharmacological Evaluation of Dotinurad, a Selective Urate Reabsorption Inhibitor. J. Pharmacol. Exp. Ther. 2019, 371, 162–170. [Google Scholar] [CrossRef]
- Yanai, H.; Katsuyama, H.; Hakoshima, M.; Adachi, H. Urate Transporter 1 Can Be a Therapeutic Target Molecule for Chronic Kidney Disease and Diabetic Kidney Disease: A Retrospective Longitudinal Study. Biomedicines 2023, 11, 567. [Google Scholar] [CrossRef]
- Tanaka, A.; Taguchi, I.; Hisauchi, I.; Yoshida, H.; Shimabukuro, M.; Hongo, H.; Ishikawa, T.; Kadokami, T.; Yagi, S.; Sata, M. Clinical effects of a selective urate reabsorption inhibitor dotinurad in patients with hyperuricemia and treated hypertension: A multicenter, prospective, exploratory study (DIANA). Eur. J. Med. Res. 2023, 28, 238. [Google Scholar] [CrossRef]
- Yuan, H.; Yu, C.; Li, X.; Sun, L.; Zhu, X.; Zhao, C.; Zhang, Z.; Yang, Z. Serum Uric Acid Levels and Risk of Metabolic Syndrome: A Dose-Response Meta-Analysis of Prospective Studies. J. Clin. Endocrinol. Metab. 2015, 100, 4198–4207. [Google Scholar] [CrossRef]
- Hjortnaes, J.; Algra, A.; Olijhoek, J.; Huisman, M.; Jacobs, J.; van der Graaf, Y.; Visseren, F. Serum uric acid levels and risk for vascular diseases in patients with metabolic syndrome. J. Rheumatol. 2007, 34, 1882–1887. [Google Scholar]
- Takahashi, S.; Yamamoto, T.; Tsutsumi, Z.; Moriwaki, Y.; Yamakita, J.; Higashino, K. Close correlation between visceral fat accumulation and uric acid metabolism in healthy men. Metabolism 1997, 46, 1162–1165. [Google Scholar] [CrossRef]
- Facchini, F.; Chen, Y.D.; Hollenbeck, C.B.; Reaven, G.M. Relationship between resistance to insulin-mediated glucose uptake, urinary uric acid clearance, and plasma uric acid concentration. JAMA 1991, 266, 3008–3011. [Google Scholar] [CrossRef] [PubMed]
- Doshi, M.; Takiue, Y.; Saito, H.; Hosoyamada, M. The increased protein level of URAT1 was observed in obesity/metabolic syndrome model mice. Nucleosides Nucleotides Nucleic Acids 2011, 30, 1290–1294. [Google Scholar] [CrossRef]
- Miao, Z.; Yan, S.; Wang, J.; Wang, B.; Li, Y.; Xing, X.; Yuan, Y.; Meng, D.; Wang, L.; Gu, J. Insulin resistance acts as an independent risk factor exacerbating high-purine diet induced renal injury and knee joint gouty lesions. Inflamm. Res. 2009, 58, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, D.M.; Liu, J.H.; Hu, L.S.; Xue, Q.C.; Ding, X.Q.; Kong, L.D. Wuling San protects kidney dysfunction by inhibiting renal TLR4/MyD88 signaling and NLRP3 inflammasome activation in high fructose-induced hyperuricemic mice. J. Ethnopharmacol. 2015, 169, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Nie, Q.; Zhang, Z.; Zhao, J.; Zhang, F.; Wang, C.; Wang, X.; Song, G. Resveratrol affects the expression of uric acid transporter by improving inflammation. Mol. Med. Rep. 2021, 24, 564. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G.; Rössler, O.G. Resveratrol regulates gene transcription via activation of stimulus-responsive transcription factors. Pharmacol. Res. 2017, 117, 166–176. [Google Scholar] [CrossRef]
- Cheng, K.; Song, Z.; Chen, Y.; Li, S.; Zhang, Y.; Zhang, H.; Zhang, L.; Wang, C.; Wang, T. Resveratrol protects against renal damage via attenuation of inflammation and oxidative stress in high-fat-diet-induced obese mice. Inflammation 2019, 42, 937–945. [Google Scholar] [CrossRef]
- Saldanha, J.F.; Leal, V.O.; Stenvinkel, P.; Carraro-Eduardo, J.C.; Mafra, D. Resveratrol: Why is it a promising therapy for chronic kidney disease patients? Oxid. Med. Cell. Longev. 2013, 2013, 963217. [Google Scholar] [CrossRef]
- Quiñones Galvan, A.; Natali, A.; Baldi, S.; Frascerra, S.; Sanna, G.; Ciociaro, D.; Ferrannini, E. Effect of insulin on uric acid excretion in humans. Am. J. Physiol. 1995, 268, E1–E5. [Google Scholar] [CrossRef]
- Muscelli, E.; Natali, A.; Bianchi, S.; Bigazzi, R.; Galvan, A.Q.; Sironi, A.M.; Frascerra, S.; Ciociaro, D.; Ferrannini, E. Effect of insulin on renal sodium and uric acid handling in essential hypertension. Am. J. Hypertens. 1996, 9, 746–752. [Google Scholar] [CrossRef]
- Ter Maaten, J.C.; Voorburg, A.; Heine, R.J.; Ter Wee, P.M.; Donker, A.J.; Gans, R.O. Renal handling of urate and sodium during acute physiological hyperinsulinaemia in healthy subjects. Clin. Sci. 1997, 92, 51–58. [Google Scholar] [CrossRef]
- Toyoki, D.; Shibata, S.; Kuribayashi-Okuma, E.; Xu, N.; Ishizawa, K.; Hosoyamada, M.; Uchida, S. Insulin stimulates uric acid reabsorption via regulating urate transporter 1 and ATP-binding cassette subfamily G member 2. Am. J. Physiol. Renal. Physiol. 2017, 313, F826–F834. [Google Scholar] [CrossRef]
- Keembiyehetty, C.; Augustin, R.; Carayannopoulos, M.O.; Steer, S.; Manolescu, A.; Cheeseman, C.I.; Moley, K.H. Mouse glucose transporter 9 splice variants are expressed in adult liver and kidney and are up-regulated in diabetes. Mol. Endocrinol. 2006, 20, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.K.; Leask, M.P.; Estiverne, C.; Choi, H.K.; Merriman, T.R.; Mount, D.B. Genetic and Physiological Effects of Insulin on Human Urate Homeostasis. Front. Physiol. 2021, 12, 713710. [Google Scholar] [CrossRef]
- Tanaka, Y.; Nagoshi, T.; Takahashi, H.; Oi, Y.; Yoshii, A.; Kimura, H.; Ito, K.; Kashiwagi, Y.; Tanaka, T.D.; Yoshimura, M. URAT1-selective inhibition ameliorates insulin resistance by attenuating diet-induced hepatic steatosis and brown adipose tissue whitening in mice. Mol. Metab. 2022, 55, 101411. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Spiga, R.; Marini, M.A.; Mancuso, E.; Di Fatta, C.; Fuoco, A.; Perticone, F.; Andreozzi, F.; Mannino, G.C.; Sesti, G. Uric Acid Is Associated With Inflammatory Biomarkers and Induces Inflammation Via Activating the NF-κB Signaling Pathway in HepG2 Cells. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1241–1249. [Google Scholar] [CrossRef] [PubMed]
- Czech, M.P. Mechanisms of insulin resistance related to white, beige, and brown adipocytes. Mol. Metab. 2020, 34, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Sun, L.; Li, W.; Liu, H.; Liu, Y.; Wei, Y.; Yuan, Y.; Zheng, L.; Yin, S.; Dai, C.; et al. Metformin alleviates hyperuricaemia-induced serum FFA elevation and insulin resistance by inhibiting adipocyte hypertrophy and reversing suppressed white adipose tissue beiging. Clin. Sci. 2020, 134, 1537–1553. [Google Scholar] [CrossRef]
- Baldwin, W.; McRae, S.; Marek, G.; Wymer, D.; Pannu, V.; Baylis, C.; Johnson, R.J.; Sautin, Y.Y. Hyperuricemia as a mediator of the proinflammatory endocrine imbalance in the adipose tissue in a murine model of the metabolic syndrome. Diabetes 2011, 60, 1258–1269. [Google Scholar] [CrossRef]
- Kwon, M.M.; O’Dwyer, S.M.; Baker, R.K.; Covey, S.D.; Kieffer, T.J. FGF21-mediated improvements in glucose clearance require uncoupling protein 1. Cell Rep. 2015, 13, 1521–1527. [Google Scholar] [CrossRef]
- Sautin, Y.Y.; Nakagawa, T.; Zharikov, S.; Johnson, R.J. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am. J. Physiol. Cell Physiol. 2007, 293, C584–C596. [Google Scholar] [CrossRef]
- Nadwa, E.H.; Morcos, G.N.B.; Salama, N.M.; Shafik, A.N. Comparing the Effects of Febuxostat and Allopurinol in an Animal Model of Metabolic Syndrome. Pharmacology 2021, 106, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Cho, I.J.; Oh, D.H.; Yoo, J.; Hwang, Y.C.; Ahn, K.J.; Chung, H.Y.; Jeong, S.W.; Moon, J.Y.; Lee, S.H.; Lim, S.J.; et al. Allopurinol ameliorates high fructose diet induced hepatic steatosis in diabetic rats through modulation of lipid metabolism, inflammation, and ER stress pathway. Sci. Rep. 2021, 11, 9894. [Google Scholar] [CrossRef] [PubMed]
- Avramoglu, R.K.; Basciano, H.; Adeli, K. Lipid and lipoprotein dysregulation in insulin resistant states. Clin. Chim. Acta 2006, 368, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Hinds, T.D., Jr.; Hosick, P.A.; Chen, S.; Tukey, R.H.; Hankins, M.W.; Nestor-Kalinoski, A.; Stec, D.E. Mice with hyperbilirubinemia due to Gilbert’s syndrome polymorphism are resistant to hepatic steatosis by decreased serine 73 phosphorylation of PPARα. Am. J. Physiol. Endocrinol. Metab. 2017, 312, E244–E252. [Google Scholar] [CrossRef] [PubMed]
- Stec, D.E.; John, K.; Trabbic, C.J.; Luniwal, A.; Hankins, M.W.; Baum, J.; Hinds, T.D., Jr. Bilirubin Binding to PPARα Inhibits Lipid Accumulation. PLoS ONE 2016, 11, e0153427. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Adeosun, S.O.; Alamodi, A.A.; Stec, D.E. Does bilirubin prevent hepatic steatosis through activation of the PPARα nuclear receptor? Med. Hypotheses 2016, 95, 54–57. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Burns, K.A.; Hosick, P.A.; McBeth, L.; Nestor-Kalinoski, A.; Drummond, H.A.; AlAmodi, A.A.; Hankins, M.W.; Heuvel, J.P.V.; Stec, D.E. Biliverdin Reductase A Attenuates Hepatic Steatosis by Inhibition of Glycogen Synthase Kinase (GSK) 3β Phosphorylation of Serine 73 of Peroxisome Proliferator-activated Receptor (PPAR) α. J. Biol. Chem. 2016, 291, 25179–25191. [Google Scholar] [CrossRef]
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.R.; Van Hul, W.; Mertens, I.; et al. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 2015, 63, 164–173. [Google Scholar] [CrossRef]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int. J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Ntambi, J.M.; Friedman, J.M. Stearoyl-CoA desaturase-1 and the metabolic syndrome. Curr. Drug Targets-Immune Endocr. Metab. Disord. 2003, 3, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Soriguer, F.; Rojo-Martínez, G.; de Fonseca, F.R.; García-Escobar, E.; Fuentes, E.G.; Olveira, G. Obesity and the metabolic syndrome in Mediterranean countries: A hypothesis related to olive oil. Mol. Nutr. Food. Res. 2007, 51, 1260–1267. [Google Scholar] [CrossRef] [PubMed]
- Dobrzyn, A.; Ntambi, J.M. The role of stearoyl-CoA desaturase in body weight regulation. Trends Cardiovasc. Med. 2004, 14, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.M.; Dobrzyn, A.; Lee, S.H.; Dobrzyn, P.; Miyazaki, M.; Ntambi, J.M. Stearoyl-CoA desaturase 1 deficiency increases insulin signalling and glycogen accumulation in brown adipose tissue. Am. J. Physiol. Endocrinol. Metab. 2005, 288, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Ran, Z.; Xue, X.; Han, L.; Terkeltaub, R.; Merriman, T.R.; Zhao, T.; He, Y.; Wang, C.; Li, X.; Liu, Z.; et al. Decrease in Serum Urate Level Is Associated With Loss of Visceral Fat in Male Gout Patients. Front. Endocrinol. 2021, 12, 724822. [Google Scholar] [CrossRef]
- Nakamura, T.; Nampei, M.; Murase, T.; Satoh, E.; Akari, S.; Katoh, N.; Mizukami, H. Influence of xanthine oxidoreductase inhibitor, topiroxostat, on body weight of diabetic obese mice. Nutr. Diabetes 2021, 11, 12. [Google Scholar] [CrossRef]
- Soletsky, B.; Feig, D.I. Uric acid reduction rectifies prehypertension in obese adolescents. Hypertension 2012, 60, 1148–1156. [Google Scholar] [CrossRef]
- Johnson, R.J.; Kang, D.H.; Feig, D.; Kivlighn, S.; Kanellis, J.; Watanabe, S.; Tuttle, K.R.; Rodriguez-Iturbe, B.; Herrera-Acosta, J.; Mazzali, M. Is there a pathogenetic role for uric acid in hypertension and cardiovascular and renal disease? Hypertension 2003, 41, 1183–1190. [Google Scholar] [CrossRef]
- Berger, L.; Yü, T.F. Renal function in gout. IV. An analysis of 524 gouty subjects including long-term follow-up studies. Am. J. Med. 1975, 59, 605–613. [Google Scholar] [CrossRef]
- Feig, D.I.; Madero, M.; Jalal, D.I.; Sanchez-Lozada, L.G.; Johnson, R.J. Uric acid and the origins of hypertension. J. Pediatr. 2013, 162, 896–902. [Google Scholar] [CrossRef]
- Mazzali, M.; Hughes, J.; Kim, Y.G.; Jefferson, J.A.; Kang, D.H.; Gordon, K.L.; Lan, H.Y.; Kivlighn, S.; Johnson, R.J. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension 2001, 38, 1101–1106. [Google Scholar] [CrossRef]
- Uedono, H.; Tsuda, A.; Ishimura, E.; Yasumoto, M.; Ichii, M.; Ochi, A.; Ohno, Y.; Nakatani, S.; Mori, K.; Uchida, J.; et al. Relationship between serum uric acid levels and intrarenal hemodynamic parameters. Kidney Blood Press. Res. 2015, 40, 315–322. [Google Scholar] [CrossRef]
- Mallat, S.G.; Al Kattar, S.; Tanios, B.Y.; Jurjus, A. Hyperuricemia, Hypertension, and Chronic Kidney Disease: An Emerging Association. Curr. Hypertens. Rep. 2016, 18, 74. [Google Scholar] [CrossRef]
- Takae, K.; Nagata, M.; Hata, J.; Mukai, N.; Hirakawa, Y.; Yoshida, D.; Kishimoto, H.; Tsuruya, K.; Kitazono, T.; Kiyohara, Y.; et al. Serum Uric Acid as a Risk Factor for Chronic Kidney Disease in a Japanese Community—The Hisayama Study. Circ. J. 2016, 80, 1857–1862. [Google Scholar] [CrossRef]
- Iseki, K.; Ikemiya, Y.; Inoue, T.; Iseki, C.; Kinjo, K.; Takishita, S. Significance of hyperuricemia as a risk factor for developing ESRD in a screened cohort. Am. J. Kidney Dis. 2004, 44, 642–650. [Google Scholar] [CrossRef]
- Nagura, M.; Tamura, Y.; Kumagai, T.; Hosoyamada, M.; Uchida, S. Uric acid metabolism of kidney and intestine in a rat model of chronic kidney disease. Nucleosides Nucleotides Nucleic Acids 2016, 35, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Takada, T.; Yamamoto, T.; Matsuo, H.; Tan, J.K.; Ooyama, K.; Sakiyama, M.; Miyata, H.; Yamanashi, Y.; Toyoda, Y.; Higashino, T.; et al. Identification of ABCG2 as an Exporter of Uremic Toxin Indoxyl Sulfate in Mice and as a Crucial Factor Influencing CKD Progression. Sci. Rep. 2018, 8, 11147. [Google Scholar] [CrossRef]
- Uchida, S.; Kumagai, T.; Chang, W.X.; Tamura, Y.; Shibata, S. Time to Target Uric Acid to Retard Chronic Kidney Disease Progression. Uric Acid Chronic Kidney Dis. 2018, 192, 56–68. [Google Scholar]
- Tan, S.P.F.; Scotcher, D.; Rostami-Hodjegan, A.; Galetin, A. Effect of Chronic Kidney Disease on the Renal Secretion via Organic Anion Transporters 1/3: Implications for Physiologically-Based Pharmacokinetic Modeling and Dose Adjustment. Clin. Pharmacol. Ther. 2022, 112, 643–652. [Google Scholar] [CrossRef]
- Shen, H.; Nelson, D.M.; Oliveira, R.V.; Zhang, Y.; Mcnaney, C.A.; Gu, X.; Chen, W.; Su, C.; Reily, M.D.; Shipkova, P.A.; et al. Discovery and validation of pyridoxic acid and homovanillic acid as novel endogenous plasma biomarkers of organic anion transporter (OAT) 1 and OAT3 in cynomolgus monkeys. Drug Metab. Dispos. 2018, 46, 178–188. [Google Scholar] [CrossRef]
- Shen, H.; Holenarsipur, V.K.; Mariappan, T.T.; Drexler, D.M.; Cantone, J.L.; Rajanna, P.; Singh Gautam, S.; Zhang, Y.; Gan, J.; Shipkova, P.A.; et al. Evidence for the validity of pyridoxic acid (PDA) as a plasma-based endogenous probe for OAT1 and OAT3 function in healthy subjects. J. Pharmacol. Exp. Ther. 2019, 368, 136–145. [Google Scholar] [CrossRef]
- Willemin, M.E.; Van Der Made, T.K.; Pijpers, I.; Dillen, L.; Kunze, A.; Jonkers, S.; Steemans, K.; Tuytelaars, A.; Jacobs, F.; Monshouwer, M.; et al. Clinical investigation on endogenous biomarkers to predict strong OAT-mediated drug–drug interactions. Clin. Pharmacokinet. 2021, 60, 1187–1199. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zelnick, L.R.; Wang, K.; Hoofnagle, A.N.; Becker, J.O.; Hsu, C.Y.; Feldman, H.I.; Mehta, R.C.; Lash, J.P.; Waikar, S.S.; et al. Kidney clearance of secretory solutes is associated with progression of CKD: The CRIC study. J. Am. Soc. Nephrol. 2020, 31, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zelnick, L.R.; Chen, Y.; Hoofnagle, A.N.; Watnick, T.; Seliger, S.; Kestenbaum, B. Alterations of proximal tubular secretion in autosomal dominant polycystic kidney disease. Clin. J. Am. Soc. Nephrol. 2020, 15, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Bush, K.T.; Nigam, S.K. Key Role for the Organic Anion Transporters, OAT1 and OAT3, in the in vivo Handling of Uremic Toxins and Solutes. Sci. Rep. 2017, 7, 4939. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, C.H.; Yoshida, K.; Zhao, P.; Meyer, T.W.; Zhang, L.; Huang, S.M.; Giacomini, K.M. Identification and Quantitative Assessment of Uremic Solutes as Inhibitors of Renal Organic Anion Transporters, OAT1 and OAT3. Mol. Pharm. 2016, 13, 3130–3140. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T. Uremic toxicity of indoxyl sulfate. Nagoya J. Med. Sci. 2010, 72, 1–11. [Google Scholar] [PubMed]
- Lin, C.J.; Chen, H.H.; Pan, C.F.; Chuang, C.K.; Wang, T.J.; Sun, F.J.; Wu, C.J. p-Cresylsulfate and indoxyl sulfate level at different stages of chronic kidney disease. J. Clin. Lab. Anal. 2011, 25, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Holle, J.; Kirchner, M.; Okun, J.; Bayazit, A.K.; Obrycki, L.; Canpolat, N.; Bulut, I.K.; Azukaitis, K.; Duzova, A.; Ranchin, B.; et al. Serum indoxyl sulfate concentrations associate with progression of chronic kidney disease in children. PLoS ONE 2020, 15, e0240446. [Google Scholar] [CrossRef]
- Yanai, H.; Yamaguchi, N.; Adachi, H. Chronic Kidney Disease Stage G4 in a Diabetic Patient Improved by Multi-Disciplinary Treatments Based Upon Literature Search for Therapeutic Evidence. Cardiol. Res. 2022, 13, 309–314. [Google Scholar] [CrossRef]
- Kurihara, O.; Yamada, T.; Kato, K.; Miyauchi, Y. Efficacy of dotinurad in patients with severe renal dysfunction. Clin. Exp. Nephrol. 2023. online ahead of print. [Google Scholar] [CrossRef]
- Yanai, K.; Hirai, K.; Kaneko, S.; Mutsuyoshi, Y.; Kitano, T.; Miyazawa, H.; Ito, K.; Ueda, Y.; Ookawara, S.; Morishita, Y. The Efficacy and Safety of Dotinurad on Uric Acid and Renal Function in Patients with Hyperuricemia and Advanced Chronic Kidney Disease: A Single Center, Retrospective Analysis. Drug Des. Devel. Ther. 2023, 17, 3233–3248. [Google Scholar] [CrossRef]
- Siu, Y.P.; Leung, K.T.; Tong, M.K.; Kwan, T.H. Use of allopurinol in slowing the progression of renal disease through its ability to lower serum uric acid level. Am. J. Kidney Dis. 2006, 47, 51–59. [Google Scholar] [CrossRef]
- Goicoechea, M.; de Vinuesa, S.G.; Verdalles, U.; Ruiz-Caro, C.; Ampuero, J.; Rincón, A.; Arroyo, D.; Luño, J. Effect of allopurinol in chronic kidney disease progression and cardiovascular risk. Clin. J. Am. Soc. Nephrol. 2010, 5, 1388–1393. [Google Scholar] [CrossRef] [PubMed]
- Goicoechea, M.; Garcia de Vinuesa, S.; Verdalles, U.; Verde, E.; Macias, N.; Santos, A.; Pérez de Jose, A.; Cedeño, S.; Linares, T.; Luño, J. Allopurinol and progression of CKD and cardiovascular events: Long-term follow-up of a randomized clinical trial. Am. J. Kidney Dis. 2015, 65, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Shibagaki, Y.; Ohno, I.; Hosoya, T.; Kimura, K. Safety, efficacy and renal effect of febuxostat in patients with moderate-to-severe kidney dysfunction. Hypertens. Res. 2014, 37, 919–925. [Google Scholar] [CrossRef] [PubMed]
- Tsuruta, Y.; Mochizuki, T.; Moriyama, T.; Itabashi, M.; Takei, T.; Tsuchiya, K.; Nitta, K. Switching from allopurinol to febuxostat for the treatment of hyperuricemia and renal function in patients with chronic kidney disease. Clin. Rheumatol. 2014, 33, 1643–1648. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Nakayama, M.; Kanno, M.; Kimura, H.; Watanabe, K.; Tani, Y.; Hayashi, Y.; Asahi, K.; Terawaki, H.; Watanabe, T. Renoprotective effects of febuxostat in hyperuricemic patients with chronic kidney disease: A parallel-group, randomized, controlled trial. Clin. Exp. Nephrol. 2015, 19, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- Kamijo-Ikemori, A.; Sugaya, T.; Ichikawa, D.; Hoshino, S.; Matsui, K.; Yokoyama, T.; Yasuda, T.; Hirata, K.; Kimura, K. Urinary liver type fatty acid binding protein in diabetic nephropathy. Clin. Chim. Acta 2013, 424, 104–108. [Google Scholar] [CrossRef]
- Câmara, N.O.; Williams, W.W., Jr.; Pacheco-Silva, A. Proximal tubular dysfunction as an indicator of chronic graft dysfunction. Braz. J. Med. Biol. Res. 2009, 42, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, H.J.; Ahn, H.S.; Oh, S.W.; Han, K.H.; Um, T.H.; Cho, C.R.; Han, S.Y. Renoprotective effects of febuxostat compared with allopurinol in patients with hyperuricemia: A systematic review and meta-analysis. Kidney Res. Clin. Pract. 2017, 36, 274–281. [Google Scholar] [CrossRef]
- Hsu, Y.O.; Wu, I.W.; Chang, S.H.; Lee, C.C.; Tsai, C.Y.; Lin, C.Y.; Lin, W.T.; Huang, Y.T.; Wu, C.Y.; Kuo, G.; et al. Comparative Renoprotective Effect of Febuxostat and Allopurinol in Predialysis Stage 5 Chronic Kidney Disease Patients: A Nationwide Database Analysis. Clin. Pharmacol. Ther. 2020, 107, 1159–1169. [Google Scholar] [CrossRef]
- Horino, T.; Hatakeyama, Y.; Ichii, O.; Matsumoto, T.; Shimamura, Y.; Inoue, K.; Terada, Y.; Okuhara, Y. Effects of topiroxostat in hyperuricemic patients with chronic kidney disease. Clin. Exp. Nephrol. 2018, 22, 337–345. [Google Scholar] [CrossRef]
- Skálová, S. The diagnostic role of urinary N-acetyl-beta-D-glucosaminidase (NAG) activity in the detection of renal tubular impairment. Acta Medica 2005, 48, 75–80. [Google Scholar]
- Tsukamoto, S.; Okami, N.; Yamada, T.; Azushima, K.; Yamaji, T.; Kinguchi, S.; Uneda, K.; Kanaoka, T.; Wakui, H.; Tamura, K. Prevention of kidney function decline using uric acid-lowering therapy in chronic kidney disease patients: A systematic review and network meta-analysis. Clin. Rheumatol. 2022, 41, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Chou, H.W.; Chiu, H.T.; Tsai, C.W.; Ting, I.W.; Yeh, H.C.; Huang, H.C.; Kuo, C.C.; CMUH Kidney Research Group. Comparative effectiveness of allopurinol, febuxostat and benzbromarone on renal function in chronic kidney disease patients with hyperuricemia: A 13-year inception cohort study. Nephrol. Dial. Transplant. 2018, 33, 1620–1627. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.E.; Ahn, S.M.; Oh, J.S.; Kim, Y.G.; Lee, C.K.; Yoo, B.; Hong, S. Febuxostat dose requirement according to renal function in patients who achieve target serum urate levels: A retrospective cohort study. Jt. Bone Spine 2024, 91, 105668. [Google Scholar] [CrossRef]
- Kim, S.H.; Lee, S.Y.; Kim, J.M.; Son, C.N. Renal safety and urate-lowering efficacy of febuxostat in gout patients with stage 4-5 chronic kidney disease not yet on dialysis. Korean J. Intern. Med. 2020, 35, 998–1003. [Google Scholar] [CrossRef]
- Miyata, H.; Takada, T.; Toyoda, Y.; Matsuo, H.; Ichida, K.; Suzuki, H. Identification of Febuxostat as a New Strong ABCG2 Inhibitor: Potential Applications and Risks in Clinical Situations. Front. Pharmacol. 2016, 7, 518. [Google Scholar] [CrossRef]
- Taniguchi, T.; Omura, K.; Motoki, K.; Sakai, M.; Chikamatsu, N.; Ashizawa, N.; Takada, T.; Iwanaga, T. Hypouricemic agents reduce indoxyl sulfate excretion by inhibiting the renal transporters OAT1/3 and ABCG2. Sci. Rep. 2021, 11, 7232. [Google Scholar] [CrossRef] [PubMed]
- Maddux, B.A.; Sbraccia, P.; Kumakura, S.; Sasson, S.; Youngren, J.; Fisher, A.; Spencer, S.; Grupe, A.; Henzel, W.; Stewart, T.A.; et al. Membrane glycoprotein PC-1 and insulin resistance in non-insulin-dependent diabetes mellitus. Nature 1995, 373, 448–451. [Google Scholar] [CrossRef]
- Tassone, E.J.; Cimellaro, A.; Perticone, M.; Hribal, M.L.; Sciacqua, A.; Andreozzi, F.; Sesti, G.; Perticone, F. Uric Acid Impairs Insulin Signaling by Promoting Enpp1 Binding to Insulin Receptor in Human Umbilical Vein Endothelial Cells. Front. Endocrinol. 2018, 9, 98. [Google Scholar] [CrossRef]
- Muniyappa, R.; Sowers, J.R. Role of insulin resistance in endothelial dysfunction. Rev. Endocr. Metab. Disord. 2013, 14, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Price, K.L.; Sautin, Y.Y.; Long, D.A.; Zhang, L.; Miyazaki, H.; Mu, W.; Endou, H.; Johnson, R.J. Human vascular smooth muscle cells express a urate transporter. J. Am. Soc. Nephrol. 2006, 17, 1791–1795. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.H.; Park, S.K.; Lee, I.K.; Johnson, R.J. Uric acid-induced C-reactive protein expression: Implication on cell proliferation and nitric oxide production of human vascular cells. J. Am. Soc. Nephrol. 2005, 16, 3553–3562. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Notsu, T.; Kurata, Y.; Ninomiya, H.; Taufiq, F.; Komatsu, K.; Miake, J.; Sawano, T.; Tsuneto, M.; Shirayoshi, Y.; Hisatome, I. Inhibition of the uric acid efflux transporter ABCG2 enhances stimulating effect of soluble uric acid on IL-1β production in murine macrophage-like J774.1 cells. Hypertens. Res. 2023, 46, 2368–2377. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, T.; Ito, H. Arterial stiffness in health and disease: The role of cardio-ankle vascular index. J. Cardiol. 2021, 78, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, T.; Ito, H.; Shirai, K.; Horinaka, S.; Higaki, J.; Yamamura, S.; Saiki, A.; Takahashi, M.; Masaki, M.; Okura, T.; et al. Predictive Value of the Cardio-Ankle Vascular Index for Cardiovascular Events in Patients at Cardiovascular Risk. J. Am. Heart. Assoc. 2021, 10, e020103. [Google Scholar] [CrossRef]
- Yilmaz, M.I.; Saglam, M.; Caglar, K.; Cakir, E.; Sonmez, A.; Ozgurtas, T.; Aydin, A.; Eyileten, T.; Ozcan, O.; Acikel, C.; et al. The determinants of endothelial dysfunction in CKD: Oxidative stress and asymmetric dimethylarginine. Am. J. Kidney Dis. 2006, 47, 42–50. [Google Scholar] [CrossRef]
- Alem, M.M. Allopurinol and endothelial function: A systematic review with meta-analysis of randomized controlled trials. Cardiovasc. Ther. 2018, 36, e12432. [Google Scholar] [CrossRef]
- Xin, W.; Mi, S.; Lin, Z. Allopurinol therapy improves vascular endothelial function in subjects at risk for cardiovascular diseases: A meta-analysis of randomized controlled trials. Cardiovasc. Ther. 2016, 34, 441–449. [Google Scholar] [CrossRef]
- Vallance, P.; Leone, A.; Calver, A.; Collier, J.; Moncada, S. Accumulation of an endogenous inhibitor of nitric oxidesynthesis in chronic renal failure. Lancet 1992, 339, 572–575. [Google Scholar] [PubMed]
- Achan, V.; Broadhead, M.; Malaki, M.; Whitley, G.; Leiper, J.; MacAllister, R.; Vallance, P. Asymmetric dimethy-larginine causes hypertension and cardiac dysfunction in humans and is actively metabolized by dimethyl arginine dimethyl amino hydrolase. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1455–1459. [Google Scholar] [CrossRef] [PubMed]
- Kielstein, J.T.; Impraim, B.; Simmel, S.; Bode-Böger, S.M.; Tsikas, D.; Frölich, J.C.; Hoeper, M.M.; Haller, H.; Fliser, D. Cardiovascular effectsof systemic nitric oxide synthase inhibition with asymmetrical dimethylarginine in humans. Circulation 2004, 109, 172–177. [Google Scholar] [CrossRef]
- Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H. Significance of Endothelial Dysfunction Amelioration for Sodium-Glucose Cotransporter 2 Inhibitor-Induced Improvements in Heart Failure and Chronic Kidney Disease in Diabetic Patients. Metabolites 2023, 13, 736. [Google Scholar] [CrossRef] [PubMed]
- Nata, N.; Ninwisut, N.; Inkong, P.; Supasyndh, O.; Satirapoj, B. Effects of febuxostat on markers of endothelial dysfunction and renal progression in patients with chronic kidney disease. Sci. Rep. 2023, 13, 13494. [Google Scholar] [CrossRef]
- Maruhashi, T.; Higashi, Y.; Yoshida, H.; Tanaka, A.; Eguchi, K.; Tomiyama, H.; Kario, K.; Kato, T.; Oda, N.; Tahara, N.; et al. Long-Term Effect of Febuxostat on Endothelial Function in Patients With Asymptomatic Hyperuricemia: A Sub-Analysis of the PRIZE Study. Front. Cardiovasc. Med. 2022, 9, 882821. [Google Scholar] [CrossRef]
- Kario, K.; Nishizawa, M.; Kiuchi, M.; Kiyosue, A.; Tomita, F.; Ohtani, H.; Abe, Y.; Kuga, H.; Miyazaki, S.; Kasai, T.; et al. Comparative effects of topiroxostat and febuxostat on arterial properties in hypertensive patients with hyperuricemia. J. Clin. Hypertens. 2021, 23, 334–344. [Google Scholar] [CrossRef]
- Nakata, T.; Ikeda, S.; Koga, S.; Yonekura, T.; Tsuneto, A.; Doi, Y.; Fukae, S.; Minami, T.; Kawano, H.; Maemura, K. Randomized, Open-Label, Cross-Over Comparison of the Effects of Benzbromarone and Febuxostat on Endothelial Function in Patients with Hyperuricemia. Int. Heart J. 2020, 61, 984–992. [Google Scholar] [CrossRef]
- Barrientos-Regala, M.; Macabeo, R.A.; Ramirez-Ragasa, R.; Pestaño, N.S.; Punzalan, F.E.R.; Tumanan-Mendoza, B.; Castillo, R.R. The Association of Febuxostat Compared With Allopurinol on Blood Pressure and Major Adverse Cardiac Events Among Adult Patients With Hyperuricemia: A Meta-analysis. J. Cardiovasc. Pharmacol. 2020, 76, 461–471. [Google Scholar] [CrossRef]
- Hashimoto, H.; Takeuchi, M.; Kawakami, K. Association between urate-lowering therapy and cardiovascular events in patients with asymptomatic hyperuricemia. Clin. Rheumatol. 2023, 42, 3075–3082. [Google Scholar] [CrossRef]
- Guan, X.; Zhang, S.; Liu, J.; Wu, F.; Zhou, L.; Liu, Y.; Su, N. Cardiovascular safety of febuxostat and allopurinol in patients with gout: A meta-analysis. Front. Pharmacol. 2022, 13, 998441. [Google Scholar] [CrossRef]
- Kang, E.H.; Park, E.H.; Shin, A.; Song, J.S.; Kim, S.C. Cardiovascular risk associated with allopurinol vs. benzbromarone in patients with gout. Eur. Heart J. 2021, 42, 4578–4588. [Google Scholar] [CrossRef]
- Kim, S.C.; Neogi, T.; Kang, E.H.; Liu, J.; Desai, R.J.; Zhang, M.; Solomon, D.H. Cardiovascular Risks of Probenecid Versus Allopurinol in Older Patients With Gout. J. Am. Coll. Cardiol. 2018, 71, 994–1004. [Google Scholar] [CrossRef]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; European Uremic Toxin Work Group (EUTox). Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef]
- Sung, S.H.; Chuang, S.Y.; Liu, W.L.; Cheng, H.M.; Hsu, P.F.; Pan, W.H. Hyperuricemia and pulse pressure are predictive of incident heart failure in an elderly population. Int. J. Cardiol. 2020, 300, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Cao, Y.; Han, X.; Di, H.; Yin, Y.; Wu, J.; Zhang, Y.; Zeng, X. Hyperuricemia and gout increased the risk of long-term mortality in patients with heart failure: Insights from the National Health and Nutrition Examination Survey. J. Transl. Med. 2023, 21, 463. [Google Scholar] [CrossRef]
- Carnicelli, A.P.; Sun, J.L.; Alhanti, B.; Bjursell, M.; Perl, S.; Lytle, B.; Roe, M.T.; Mentz, R.J. Elevated Uric Acid Prevalence and Clinical Outcomes in Patients with Heart Failure with Preserved Ejection Fraction: Insights from RELAX. Am. J. Med. 2020, 133, e716–e721. [Google Scholar] [CrossRef] [PubMed]
- Yılmaz Öztekin, G.M.; Genç, A.; Çağırcı, G.; Arslan, Ş. Prognostic value of the combination of uric acid and NT-proBNP in patients with chronic heart failure. Hell. J. Cardiol. 2022, 65, 35–41. [Google Scholar] [CrossRef]
- Roubille, C.; Eduin, B.; Breuker, C.; Zerkowski, L.; Letertre, S.; Mercuzot, C.; Bigot, J.; Du Cailar, G.; Roubille, F.; Fesler, P. Predictive risk factors for death in elderly patients after hospitalization for acute heart failure in an internal medicine unit. Intern. Emerg. Med. 2022, 17, 1661–1668. [Google Scholar] [CrossRef]
- Wang, X.; Fan, X.; Wu, Q.; Liu, J.; Wei, L.; Yang, D.; Bu, X.; Liu, X.; Ma, A.; Hayashi, T.; et al. Uric Acid Predicts Recovery of Left Ventricular Function and Adverse Events in Heart Failure With Reduced Ejection Fraction: Potential Mechanistic Insight From Network Analyses. Front. Cardiovasc. Med. 2022, 9, 853870. [Google Scholar] [CrossRef]
- Wang, C.; Che, H.; Zhou, Y.; Wang, R.; Zhu, D.; Cheng, L.; Rao, C.; Zhong, Q.; Li, Z.; Duan, Y.; et al. Joint association of hyperuricemia and chronic kidney disease with mortality in patients with chronic heart failure. Front. Endocrinol. 2023, 14, 1131566. [Google Scholar] [CrossRef]
- Liu, X.; Huang, G.; You, Y.; Zhang, Y.; Wang, T.; Zhu, Y.; He, Y.; Li, J.; Zhang, Z.; Xu, J. Hyperuricemia is associated with heart failure readmission in patients with heart failure and preserved ejection fraction-an observational study in Chinese. Nutr. Metab. Cardiovasc. Dis. 2023. online ahead of print. [Google Scholar]
- Sn, V.P.; Jaramillo, A.P.; Yasir, M.; Hussein, S.; Singareddy, S.; Iyer, N.; Nath, T.S. Hyperuricemia and Its Association with the Severity and Complications of Congestive Heart Failure: A Systematic Review. Cureus 2023, 15, e45246. [Google Scholar] [CrossRef]
- Packer, M. Uric Acid Is a Biomarker of Oxidative Stress in the Failing Heart: Lessons Learned from Trials with Allopurinol and SGLT2 Inhibitors. J. Card. Fail. 2020, 26, 977–984. [Google Scholar] [CrossRef]
- Deis, T.; Rossing, K.; Ersbøll, M.K.; Wolsk, E.; Gustafsson, F. Uric acid in advanced heart failure: Relation to central haemodynamics and outcome. Open Heart 2022, 9, e002092. [Google Scholar] [CrossRef]
- Deng, X.L.; Yi, H.W.; Xiao, J.; Zhang, X.F.; Zhao, J.; Sun, M.; Wen, X.S.; Liu, Z.Q.; Gao, L.; Li, Z.Y.; et al. Serum uric acid: A risk factor for right ventricular dysfunction and prognosis in heart failure with preserved ejection fraction. Front. Endocrinol. 2023, 14, 1143458. [Google Scholar] [CrossRef]
- Sanikidze, Q.; Mamacashvili, I.; Petriashvili, S. PREVALENCE OF HYPERURICEMIA IN PATIENTS WITH CHRONIC HEART FAILURE. Georgian Med. News 2021, 311, 85–88. [Google Scholar]
- Doehner, W.; Anker, S.D.; Butler, J.; Zannad, F.; Filippatos, G.; Ferreira, J.P.; Salsali, A.; Kaempfer, C.; Brueckmann, M.; Pocock, S.J.; et al. Uric acid and sodium-glucose cotransporter-2 inhibition with empagliflozin in heart failure with reduced ejection fraction: The EMPEROR-reduced trial. Eur. Heart J. 2022, 43, 3435–3446. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.A.; Lin, H.; Wang, C.Q.; Zhang, J.F.; Gu, J. Association between long-term prescription of febuxostat and the progression of heart failure with preserved ejection fraction in patients with hypertension and asymptomatic hyperuricemia. Heart Vessel. 2020, 35, 1446–1453. [Google Scholar] [CrossRef] [PubMed]
- Ke, J.; Pan, J.; Lin, H.; Han, Z.; Gu, J. Uric acid-lowering therapy with benzbromarone in hypertension with asymptomatic hyperuricemia: A randomized study focusing left ventricular diastolic function. Curr. Med. Res. Opin. 2023, 39, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ma, Y.; Cao, R.; Wang, G.; Li, S.; Cao, Y.; Zhang, H.; Liu, M.; Liu, G.; Zhang, J.; et al. Soluble uric acid induces myocardial damage through activating the NLRP3 inflammasome. J. Cell Mol. Med. 2020, 24, 8849–8861. [Google Scholar] [CrossRef]
- Zhang, X.J.; Liu, D.M.; Sun, Y.; Li, Y.S.; Ma, L.L.; Kong, X.F.; Cui, X.M.; Chen, R.Y.; Zhang, Z.J.; Jiang, L.D. Potential risk of hyperuricemia: Leading cardiomyocyte hypertrophy by inducing autophagy. Am. J. Transl. Res. 2020, 12, 1894–1903. [Google Scholar] [PubMed]
- Wu, H.; Dai, R.; Wang, M.; Chen, C. Uric acid promotes myocardial infarction injury via activating pyrin domain-containing 3 inflammasome and reactive oxygen species/transient receptor potential melastatin 2/Ca2+ pathway. BMC Cardiovasc. Disord. 2023, 23, 10. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Schreckenberg, R.; Schlüter, K.D. Uric Acid Deteriorates Load-Free Cell Shortening of Cultured Adult Rat Ventricular Cardiomyocytes via Stimulation of Arginine Turnover. Biology 2022, 12, 4. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Nagoshi, T.; Takahashi, H.; Oi, Y.; Yasutake, R.; Yoshii, A.; Kimura, H.; Kashiwagi, Y.; Tanaka, T.D.; Shimoda, M.; et al. URAT1 is expressed in cardiomyocytes and dotinurad attenuates the development of diet-induced metabolic heart disease. iScience 2023, 26, 107730. [Google Scholar] [CrossRef]




| UA-Lowering Drugs | XO Inhibitors | Uricosuric Drugs | ||||
|---|---|---|---|---|---|---|
| Allopurinol | Febuxostat | Topiroxostat | Benzbromarone | Probenecid | Dotinurad | |
| Inhibition of UA Transporters | ABCG2 | ABCG2 | ABCG2 | ABCG2 | URAT1 | |
| URAT1 | URAT1 | |||||
| GLUT9 | GLUT9 | |||||
| OAT1 | OAT1 | |||||
| OAT3 | OAT3 | |||||
| Body weight | Reduced | Reduced | Suppressed weight gain | Reduced | No data | Reduced |
| (animal) [41] | (animal and human) [41,54] | (animal) [55] | (human) [54] | (animal and human) [14,33] | ||
| Visceral fat | Reduced | Reduced | No change | Reduced | No data | Reduced |
| (animal) [42,56] | (human) [54] | (animal) [55] | (human) [54] | (animal) [33] | ||
| Blood pressure | Reduced | Reduced | No data | Reduced | Reduced [56] (human) | Reduced |
| (animal and human) [41,56] | (animal and human) [41,54] | (human) [54] | (animal and human) [14,33] | |||
| Glucose metabolism | Improved | Improved | No data | No change | No data | Improved |
| (animal) [41] | (animal) [41] | (human) [54] | (animal and human) [14,33] | |||
| Serum lipids | Improved | Improved | No data | Improved | No data | Improved |
| (animal) [41] | (animal and human) [41,54] | (human) [54] | (animal and human) [14,33] | |||
| Hepatic steatosis | Improved | Improved | No change | Improved | No data | Improved |
| (animal) [42] | (human) [54] | (animal) [55] | (human) [54] | (animal) [33] | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yanai, H.; Adachi, H.; Hakoshima, M.; Iida, S.; Katsuyama, H. A Possible Therapeutic Application of the Selective Inhibitor of Urate Transporter 1, Dotinurad, for Metabolic Syndrome, Chronic Kidney Disease, and Cardiovascular Disease. Cells 2024, 13, 450. https://doi.org/10.3390/cells13050450
Yanai H, Adachi H, Hakoshima M, Iida S, Katsuyama H. A Possible Therapeutic Application of the Selective Inhibitor of Urate Transporter 1, Dotinurad, for Metabolic Syndrome, Chronic Kidney Disease, and Cardiovascular Disease. Cells. 2024; 13(5):450. https://doi.org/10.3390/cells13050450
Chicago/Turabian StyleYanai, Hidekatsu, Hiroki Adachi, Mariko Hakoshima, Sakura Iida, and Hisayuki Katsuyama. 2024. "A Possible Therapeutic Application of the Selective Inhibitor of Urate Transporter 1, Dotinurad, for Metabolic Syndrome, Chronic Kidney Disease, and Cardiovascular Disease" Cells 13, no. 5: 450. https://doi.org/10.3390/cells13050450