
Probing the Effects of Retinoblastoma Binding Protein 6 (RBBP6) Knockdown on the Sensitivity of Cisplatin in Cervical Cancer Cells



Abstract
:1. Introduction
2. Materials and Methods
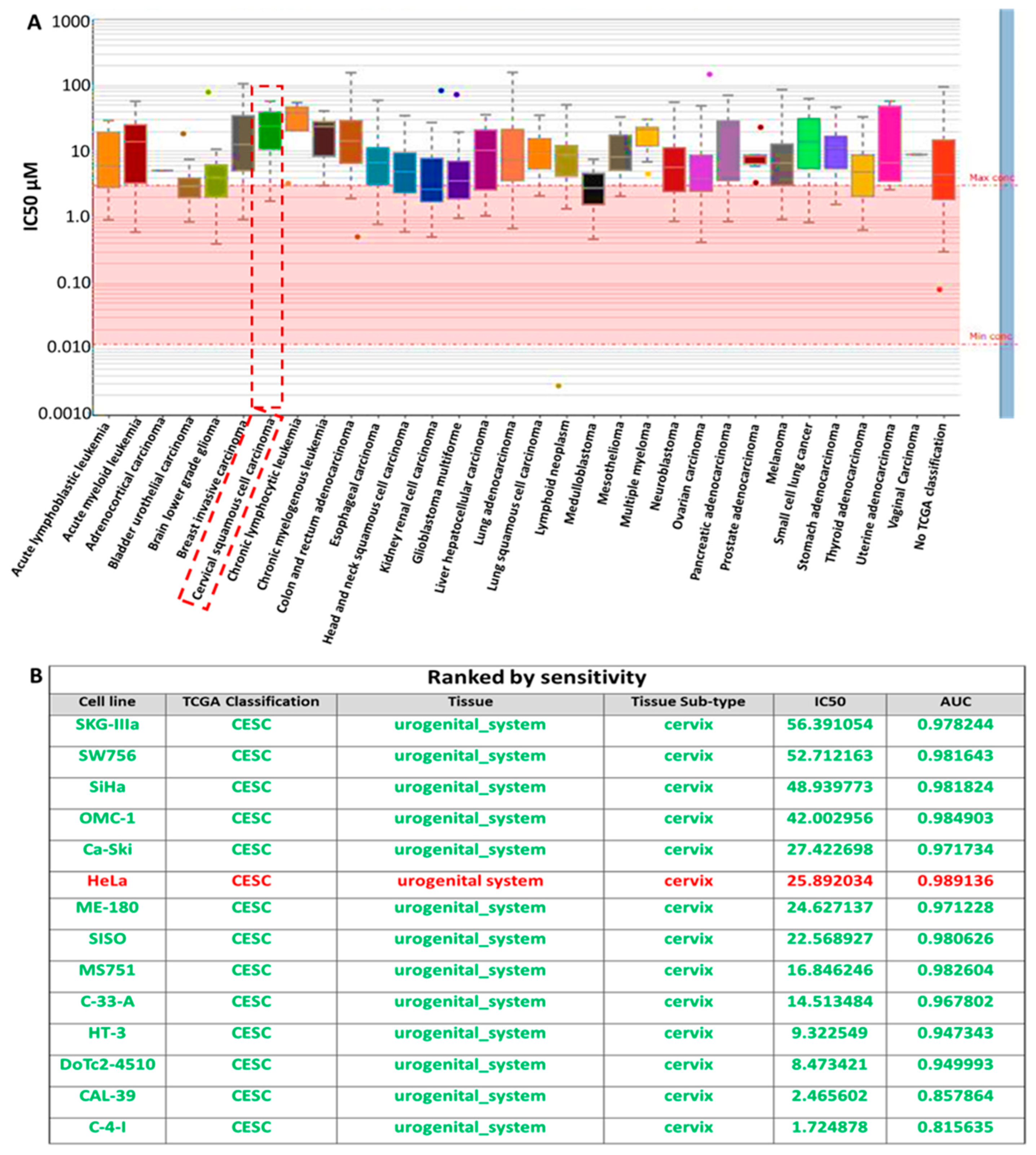
2.1. Bioinformatics Analysis
2.2. In Vitro Analysis
2.2.1. Materials
2.2.2. Cell Culture
2.2.3. Cell Viability Assay
RBBP6 Silencing and CDDP Treatment
2.2.4. RNA Extraction
2.2.5. Reverse Transcription
2.2.6. Real-Time qPCR
2.2.7. Western Blot Analysis
2.2.8. Flow Cytometry
2.2.9. xCELLigence
2.2.10. Statistical Analysis
3. Results
3.1. The Effects of RBBP6 Knockdown on p53 and Bcl-2 Gene Expression
3.2. Analysis of the Sensitivity of RBBP6/P53/BCL-2 Oncogenes to Cisplatin Treatment
3.3. RBBP6, Bcl-2, and p53 Gene Expression in Response to CDDP Treatment
3.4. Gene Expression in Response to Combined RBBP6 Knockdown and CDDP Treatment
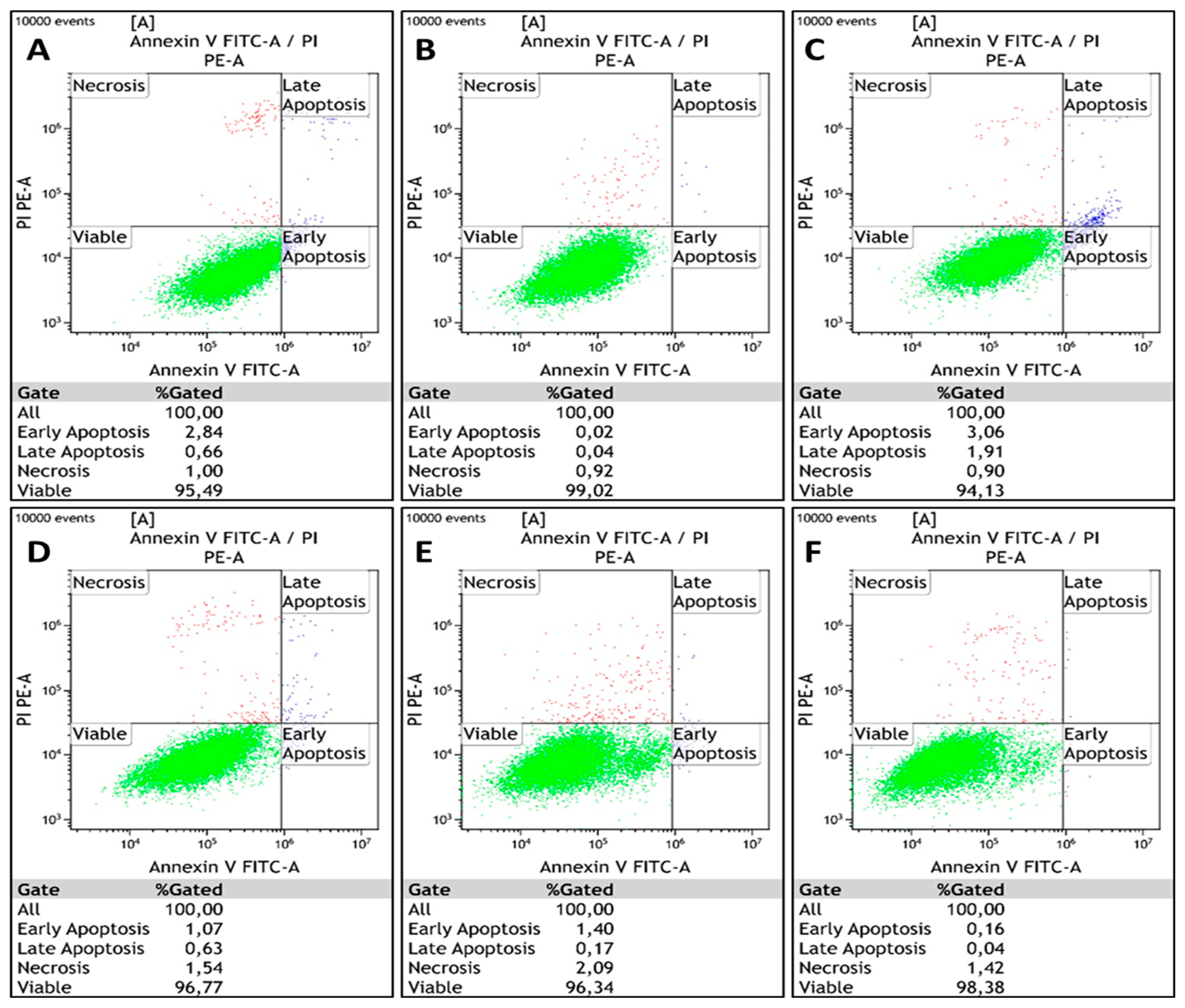
3.5. Apoptosis Detection Assay
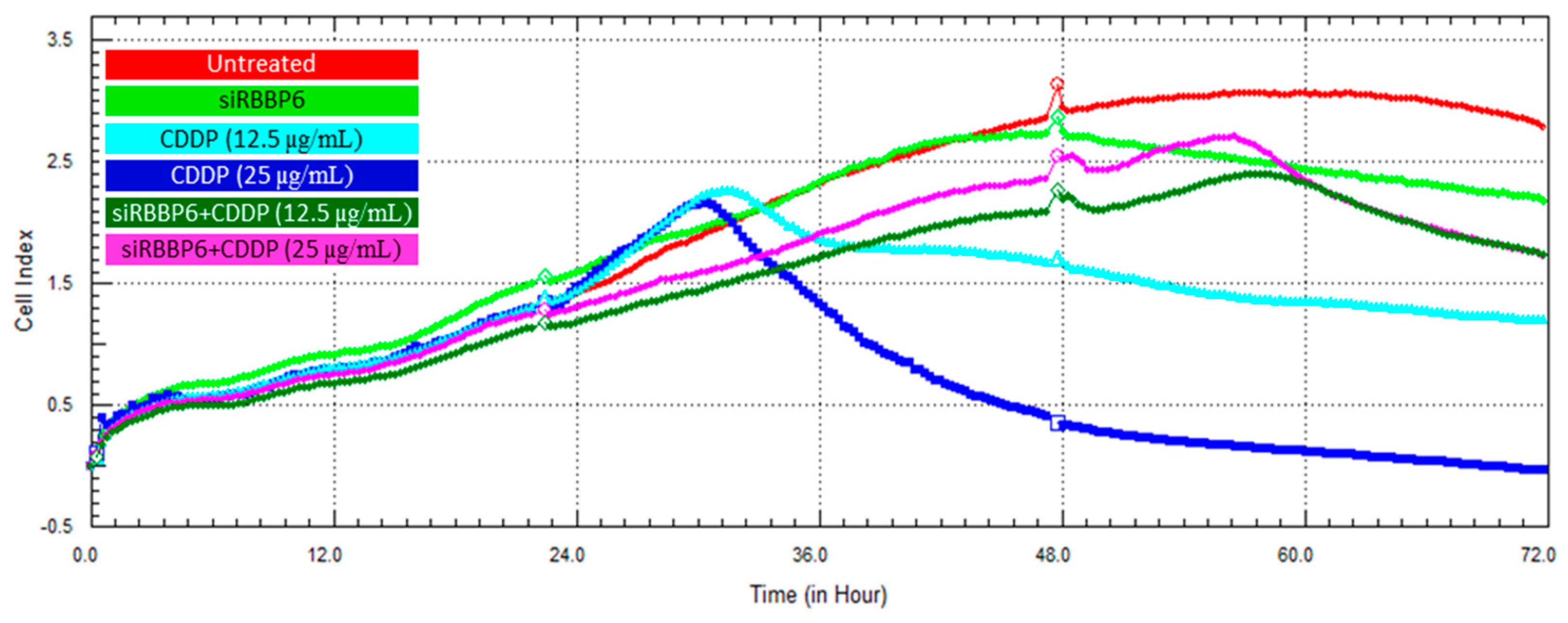
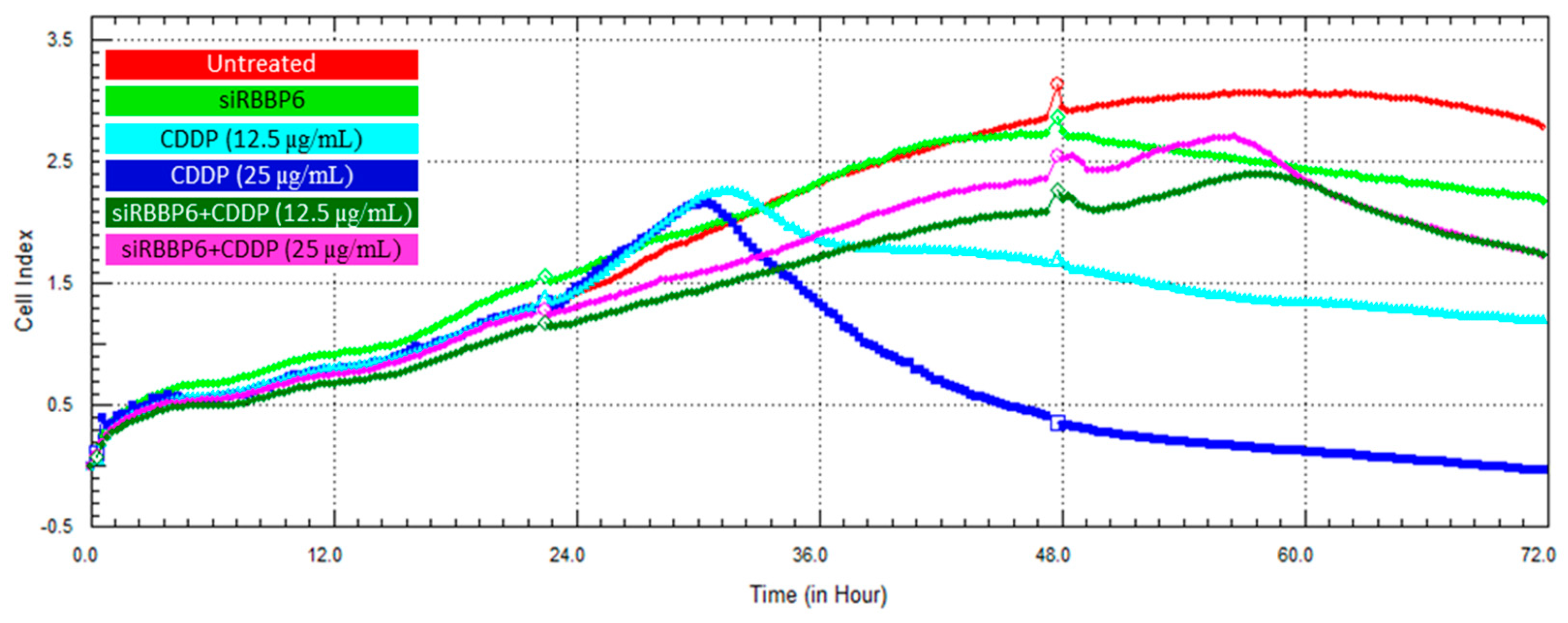
3.6. Real-Time Cell Growth Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA A Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Arbyn, M.; Weiderpass, E.; Bruni, L.; de Sanjosé, S.; Saraiya, M.; Ferlay, J.; Bray, F. Estimates of incidence and mortality of cervical cancer in 2018: A worldwide analysis. Lancet. Glob. Health 2020, 8, e191–e203. [Google Scholar] [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries and 25 major cancers in 2018. Eur. J. Cancer 2018, 103, 356–387. [Google Scholar] [CrossRef]
- van Meir, H.; Kenter, G.G.; Burggraaf, J.; Kroep, J.R.; Welters, M.J.; Melief, C.J.; van der Burg, S.H.; van Poelgeest, M.I. The need for improvement of the treatment of advanced and metastatic cervical cancer, the rationale for combined chemo-immunotherapy. Anti-Cancer Agents Med. Chem. 2014, 14, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Tchounwou, P.B.; Dasari, S.; Noubissi, F.K.; Ray, P.; Kumar, S. Advances in Our Understanding of the Molecular Mechanisms of Action of Cisplatin in Cancer Therapy. J. Exp. Pharmacol. 2021, 13, 303–328. [Google Scholar] [CrossRef] [PubMed]
- Makovec, T. Cisplatin and beyond: Molecular mechanisms of action and drug resistance development in cancer chemotherapy. Radiol. Oncol. 2019, 53, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef]
- Leisching, G.; Loos, B.; Botha, M.; Engelbrecht, A.-M. Bcl-2 confers survival in cisplatin treated cervical cancer cells: Circumventing cisplatin dose-dependent toxicity and resistance. J. Transl. Med. 2015, 13, 328. [Google Scholar] [CrossRef]
- Minagawa, Y.; Kigawa, J.; Itamochi, H.; Kanamori, Y.; Shimada, M.; Takahashi, M.; Terakawa, N. Cisplatin-resistant HeLa cells are resistant to apoptosis via p53 -dependent and -independent Pathways. Jpn. J. Cancer Res. 1999, 90, 1373–1379. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Duan, G.; Tang, Q.; Yan, H.; Xie, L.; Wang, Y.; Zheng, X.E.; Zhuge, Y.; Shen, S.; Zhang, B.; Zhang, X.; et al. A Strategy to Delay the Development of Cisplatin Resistance by Maintaining a Certain Amount of Cisplatin-Sensitive Cells. Sci. Rep. 2017, 7, 432. [Google Scholar] [CrossRef] [PubMed]
- Ishida, S.; McCormick, F.; Smith-McCune, K.; Hanahan, D. Enhancing tumor-specific uptake of the anticancer drug cisplatin with a copper chelator. Cancer Cell 2010, 17, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Krishnamurthy, S. Cellular responses to Cisplatin-induced DNA damage. J. Nucleic Acids 2010, 2010, 201367. [Google Scholar] [CrossRef]
- Seol, H.J.; Ulak, R.; Ki, K.D.; Lee, J.M. Cytotoxic and targeted systemic therapy in advanced and recurrent cervical cancer: Experience from clinical trials. Tohoku J. Exp. Med. 2014, 232, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.W.; Pouliot, L.M.; Hall, M.D.; Gottesman, M.M. Cisplatin resistance: A cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol. Rev. 2012, 64, 706–721. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.G.; Bundy, B.N.; Watkins, E.B.; Thigpen, J.T.; Deppe, G.; Maiman, M.A.; Clarke-Pearson, D.L.; Insalaco, S. Concurrent cisplatin-based radiotherapy and chemotherapy for locally advanced cervical cancer. N. Engl. J. Med. 1999, 340, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Luo, H.; Zhang, W.; Shen, Z.; Hu, X.; Zhu, X. Molecular mechanisms of cisplatin resistance in cervical cancer. Drug Des. Dev. Ther. 2016, 10, 1885–1895. [Google Scholar] [CrossRef]
- Sampath, J.; Sun, D.; Kidd, V.J.; Grenet, J.; Gandhi, A.; Shapiro, L.H.; Wang, Q.; Zambetti, G.P.; Schuetz, J.D. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J. Biol. Chem. 2001, 276, 39359–39367. [Google Scholar] [CrossRef]
- Kong, L.; Hao, Q.; Wang, Y.; Zhou, P.; Zou, B.; Zhang, Y.X. Regulation of p53 expression and apoptosis by vault RNA2-1-5p in cervical cancer cells. Oncotarget 2015, 6, 28371–28388. [Google Scholar] [CrossRef]
- Lu, H.; Wu, Y.; Liu, X.; Jiang, H.; Pang, Q.; Peng, L.; Cheng, J.; Deng, S.; Gu, J.; Zhao, R.; et al. A prospective study on neoadjuvant chemoradiotherapy plus anti-EGFR monoclonal antibody followed by surgery for locally advanced cervical cancer. OncoTargets Ther. 2018, 11, 3785–3792. [Google Scholar] [CrossRef] [PubMed]
- Xi, C.; Wang, L.; Yu, J.; Ye, H.; Cao, L.; Gong, Z. Inhibition of eukaryotic translation initiation factor 4E is effective against chemo-resistance in colon and cervical cancer. Biochem. Biophys. Res. Commun. 2018, 503, 2286–2292. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, T.; Su, W.; Dou, Z.; Zhao, D.; Jin, X.; Lei, H.; Wang, J.; Xie, X.; Cheng, B.; et al. Mutant p53 in cancer: From molecular mechanism to therapeutic modulation. Cell Death Dis. 2022, 13, 974. [Google Scholar] [CrossRef] [PubMed]
- Ferenczy, A.; Franco, E. Persistent human papillomavirus infection and cervical neoplasia. Lancet. Oncol. 2002, 3, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Hassin, O.; Oren, M. Drugging p53 in cancer: One protein, many targets. Nat. Rev. Drug Discov. 2023, 22, 127–144. [Google Scholar] [CrossRef] [PubMed]
- Co, N.N.; Iglesias, D.; Celestino, J.; Kwan, S.Y.; Mok, S.C.; Schmandt, R.; Lu, K.H. Loss of LKB1 in high-grade endometrial carcinoma: LKB1 is a novel transcriptional target of p53. Cancer 2014, 120, 3457–3468. [Google Scholar] [CrossRef] [PubMed]
- Russell, B.L.; Ntwasa, M. Expression, purification, and characterisation of the p53 binding domain of Retinoblastoma binding protein 6 (RBBP6). PLoS ONE 2023, 18, e0277478. [Google Scholar] [CrossRef] [PubMed]
- Motadi, L.R.; Lekganyane, M.M.; Moela, P. RBBP6 expressional effects on cell proliferation and apoptosis in breast cancer cell lines with distinct p53 statuses. Cancer Manag. Res. 2018, 10, 3357–3369. [Google Scholar] [CrossRef] [PubMed]
- Dlamini, Z.; Ledwaba, T.; Hull, R.; Naicker, S.; Mbita, Z. RBBP6 Is Abundantly Expressed in Human Cervical Carcinoma and May Be Implicated in Its Malignant Progression. Biomark. Cancer 2019, 11, 1179299X19829149. [Google Scholar] [CrossRef]
- Makgoo, L.; Laka, K.; Mbita, Z. Downregulation of RBBP6 variant 1 during arsenic trioxide-mediated cell cycle arrest and curcumin-induced apoptosis in MCF-7 breast cancer cells. Future Sci. OA 2019, 5, FSO409. [Google Scholar] [CrossRef]
- Moela, P.; Choene, M.M.; Motadi, L.R. Silencing RBBP6 (Retinoblastoma Binding Protein 6) sensitises breast cancer cells MCF7 to staurosporine and camptothecin-induced cell death. Immunobiology 2014, 219, 593–601. [Google Scholar] [CrossRef]
- Mosweu, M.; Motadi, L.; Moela, P. Investigating the Effects of RBBP6 Gene Expression on Telomerase Activity in Cervical Cancer Cells. Cancer Manag. Res. 2020, 12, 10725–10734. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Wang, Y.; Zheng, M.; Chen, J.; Song, G.; Zhou, Z.; Zhou, C.; Sun, X.; Zhong, L.; Ding, E.; et al. RBBP6 increases radioresistance and serves as a therapeutic target for preoperative radiotherapy in colorectal cancer. Cancer Sci. 2018, 109, 1075–1087. [Google Scholar] [CrossRef] [PubMed]
- Mbita, Z.; Meyer, M.; Skepu, A.; Hosie, M.; Rees, J.; Dlamini, Z. De-regulation of the RBBP6 isoform 3/DWNN in human cancers. Mol. Cell. Biochem. 2012, 362, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Yoshitake, Y.; Nakatsura, T.; Monji, M.; Senju, S.; Matsuyoshi, H.; Tsukamoto, H.; Hosaka, S.; Komori, H.; Fukuma, D.; Ikuta, Y.; et al. Proliferation potential-related protein, an ideal esophageal cancer antigen for immunotherapy, identified using complementary DNA microarray analysis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 6437–6448. [Google Scholar] [CrossRef] [PubMed]
- Chibi, M.; Meyer, M.; Skepu, A.; Rees, D.J.G.; Moolman-Smook, J.C.; Pugh, D.J.R. RBBP6 interacts with multifunctional protein YB-1 through its RING finger domain, leading to ubiquitination and proteosomal degradation of YB-1. J. Mol. Biol. 2008, 384, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Tang, H.; Wu, Z.; Zhou, C.; Jiang, T.; Xue, Y.; Huang, G.; Yan, D.; Peng, Z. Overexpression of RBBP6, alone or combined with mutant TP53, is predictive of poor prognosis in colon cancer. PLoS ONE 2013, 8, e66524. [Google Scholar] [CrossRef] [PubMed]
- Motadi, L.R.; Bhoola, K.D.; Dlamini, Z. Expression and function of retinoblastoma binding protein 6 (RBBP6) in human lung cancer. Immunobiology 2011, 216, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Mbita, Z.; Hull, R.; Mbele, M.; Makhafola, T.; Dlamini, Z. Expression Analysis of RbBP6 in human cancers: A Prospective biomarker. Anti-Cancer Drugs 2019, 30, 767–773. [Google Scholar] [CrossRef]
- Wang, Q.S.; Wei, S.R.; Xiao, H.L. RBBP6 induces non-small cell lung cancer cell proliferation and high expression is associated with poor prognosis. Oncol. Lett. 2020, 19, 2895–2901. [Google Scholar] [CrossRef]
- Ouyang, Y.W.; Pan, X.L.; Peng, Z.L.; Qu, Y.; Zhang, H.Y. [Expression of metastasis suppressor gene KAI1 in cervical carcinoma and infections of HPV16 E6, E7 and HPV18 E6/E7]. Sichuan Da Xue Xue Bao Yi Xue Ban J. Sichuan Univ. Med. Sci. Ed. 2008, 39, 410–413. [Google Scholar]
- Kaloni, D.; Diepstraten, S.T.; Strasser, A.; Kelly, G.L. BCL-2 protein family: Attractive targets for cancer therapy. Apoptosis Int. J. Program. Cell Death 2023, 28, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.N.; Shi, H.R.; Zhao, X.L.; Zhang, R.T.; Liu, G.Z.; Zhang, J.X. The TLR3, PI3K, survivin, FasL, and Fas genes as major risk factors of occurrence and development of cervical cancer disease. Gene 2014, 550, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Pugh, D.J.R.; Ab, E.; Faro, A.; Lutya, P.T.; Hoffmann, E.; Rees, D.J.G. DWNN, a novel ubiquitin-like domain, implicates RBBP6 in mRNA processing and ubiquitin-like pathways. BMC Struct. Biol. 2006, 6, 1. [Google Scholar] [CrossRef]
- Yu, S.; Garcia, A.A. Advancements in Recurrent and Metastatic Cervical Cancer. Am. J. Hematol. Oncol. 2015, 11, 26–31. [Google Scholar]
- Brozovic, A.; Fritz, G.; Christmann, M.; Zisowsky, J.; Jaehde, U.; Osmak, M.; Kaina, B. Long-term activation of SAPK/JNK, p38 kinase and fas-L expression by cisplatin is attenuated in human carcinoma cells that acquired drug resistance. Int. J. Cancer 2004, 112, 974–985. [Google Scholar] [CrossRef]
- Jung, H.S.; Erkin, O.C.; Kwon, M.J.; Kim, S.H.; Jung, J.I.; Oh, Y.-K.; Her, S.W.; Ju, W.; Choi, Y.-L.; Song, S.Y.; et al. The synergistic therapeutic effect of cisplatin with Human papillomavirus E6/E7 short interfering RNA on cervical cancer cell lines in vitro and in vivo. Int. J. Cancer 2012, 130, 1925–1936. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Deng, B.; Xing, G.; Teng, Y.; Tian, C.; Cheng, X.; Yin, X.; Yang, J.; Gao, X.; Zhu, Y.; et al. PACT is a negative regulator of p53 and essential for cell growth and embryonic development. Proc. Natl. Acad. Sci. USA 2007, 104, 7951. [Google Scholar] [CrossRef] [PubMed]
- Midgley, C.A.; Lane, D.P. p53 protein stability in tumour cells is not determined by mutation but is dependent on Mdm2 binding. Oncogene 1997, 15, 1179–1189. [Google Scholar] [CrossRef]
- Di Giammartino, D.C.; Li, W.; Ogami, K.; Yashinskie, J.J.; Hoque, M.; Tian, B.; Manley, J.L. RBBP6 isoforms regulate the human polyadenylation machinery and modulate expression of mRNAs with AU-rich 3’ UTRs. Genes Dev. 2014, 28, 2248–2260. [Google Scholar] [CrossRef]
- Dasgupta, A.; Nomura, M.; Shuck, R.; Yustein, J. Cancer’s Achilles’ Heel: Apoptosis and Necroptosis to the Rescue. Int. J. Mol. Sci. 2016, 18, 23. [Google Scholar] [CrossRef] [PubMed]
- Putral, L.N.; Bywater, M.J.; Gu, W.; Saunders, N.A.; Gabrielli, B.G.; Leggatt, G.R.; McMillan, N.A. RNA interference against human papillomavirus oncogenes in cervical cancer cells results in increased sensitivity to cisplatin. Mol. Pharmacol. 2005, 68, 1311–1319. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sense Strand | Antisense Strand | |
|---|---|---|
| Sequence | 5′-GCGAUGGCAACUACAAAAGtt-3′ | 5′-CUUUUGUAGUUGCCAUCGCtg-3′ |
| Forward Primer Sequence | Reverse Primer Sequence | |
|---|---|---|
| RBBP6 | 5′-CAGCG ACGACTAAAAGAAGAG-3′ | 5′-GAGCGGCTGAATGATCGAGA-3′ |
| p53 | 5′-GACGCTAGGATCTGACTGC-3′ | 5′-GACACGCTTCCCTGGATTG-3′ |
| Bcl-2 | 5′-AGCCAGGAGAAATCAAACAGAC-3′ | 5′-GATGACTGAGTACCTGAACCG-3′ |
| GAPDH | 5′-CAGCCGCATCTTCTTTTGCG-3′ | 5′-TGGAATTTGCCATGGGTGGA-3′ |
| Target | Dilution | Company and Catalog No. | Predicted MW (kDa) |
|---|---|---|---|
| GAPDH | 1:5000 | ABCAM, GAPDH, Rabbit mAb, ab181602 | 36 |
| RBBP6 | 1:1000 | ABCAM, RBBP6 (63) Rabbit mAb, ab237514 | 250 |
| BCL-2 | 1:1000 | Cell Signaling, BCL-2, Rabbit mAb, #2872 | 26–28 |
| P53 | 1:1000 | Cell Signaling, P53, Rabbit mAb, #9282 | 53 |
| 2nd Antibodies | 1:5000 | Cell Signaling, Anti-Rabbit IgG HPR-Licked, #7074 | |
| Untreated | siRBBP6 | CDDP 24 h | CDDP 48 h | siRBBP6 + CDDP 24 h | siRBBP6 + CDDP 48 h | |
|---|---|---|---|---|---|---|
| Viable cells (%) | 93.2 | 64.2 | 62.3 | 9.4 | 65.3 | 0.3 |
| Apoptotic (early/late) cells (%) | 6.4 | 25.7 | 32.4 | 77.0 | 23.0 | 59.4 |
| Necrotic cells (%) | 0.3 | 10.1 | 5.3 | 13.6 | 11.8 | 40.3 |
| Untreated | siRBBP6 | CDDP 24 h | CDDP 48 h | siRBBP6 + CDDP 24 h | siRBBP6 + CDDP 48 h | |
|---|---|---|---|---|---|---|
| Viable cells (%) | 95.5 | 99.0 | 94.1 | 96.8 | 96.3 | 98.4 |
| Apoptotic (early/late) cells (%) | 3.5 | 0.1 | 5.0 | 1.7 | 1.6 | 0.2 |
| Necrotic cells (%) | 1.0 | 0.9 | 0.9 | 1.5 | 2.1 | 1.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehta, H.; Ambele, M.A.; Mokgautsi, N.; Moela, P. Probing the Effects of Retinoblastoma Binding Protein 6 (RBBP6) Knockdown on the Sensitivity of Cisplatin in Cervical Cancer Cells. Cells 2024, 13, 700. https://doi.org/10.3390/cells13080700
Mehta H, Ambele MA, Mokgautsi N, Moela P. Probing the Effects of Retinoblastoma Binding Protein 6 (RBBP6) Knockdown on the Sensitivity of Cisplatin in Cervical Cancer Cells. Cells. 2024; 13(8):700. https://doi.org/10.3390/cells13080700
Chicago/Turabian StyleMehta, Harshini, Melvin Anyasi Ambele, Ntlotlang Mokgautsi, and Pontsho Moela. 2024. "Probing the Effects of Retinoblastoma Binding Protein 6 (RBBP6) Knockdown on the Sensitivity of Cisplatin in Cervical Cancer Cells" Cells 13, no. 8: 700. https://doi.org/10.3390/cells13080700