
Chemogenetic Manipulation of Amygdala Kappa Opioid Receptor Neurons Modulates Amygdala Neuronal Activity and Neuropathic Pain Behaviors


 , and
, and 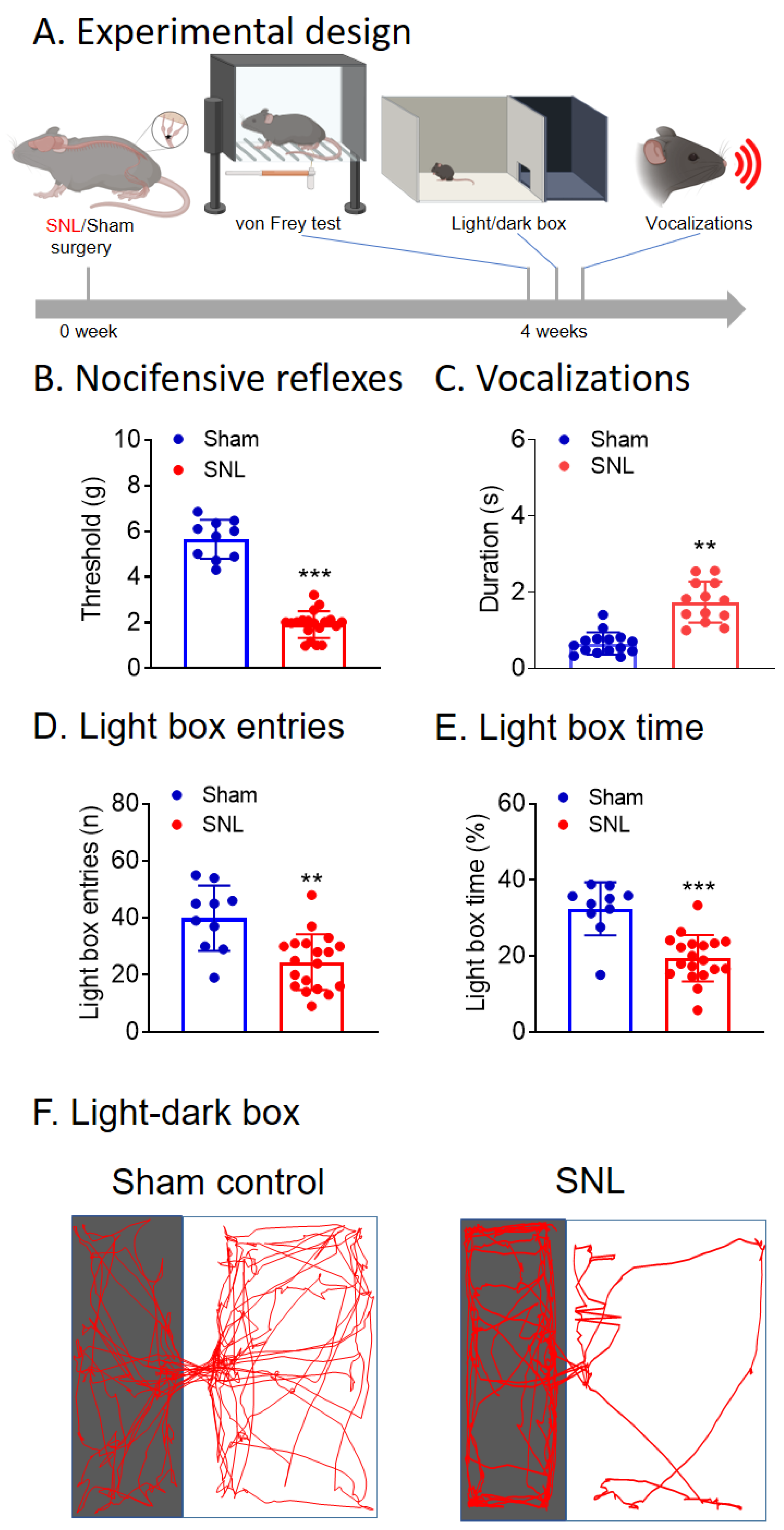{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Neuropathic Pain Model
2.3. Chemogenetic Manipulations
2.4. Experimental Protocol
2.5. Pain-Related Behaviors
2.6. Brain Slice Electrophysiology
2.7. Histological Verification of Injection and Recording Sites
2.8. Statistical Analysis
3. Results
3.1. Chronic Neuropathic Pain (SNL Model) Behaviors of KOR-Cre Mice
3.2. Chemogenetic Inhibition of CeA-KOR Neurons Induces Pain-Like Behaviors in Naïve Mice


3.3. Chemogenetic Activation of CeA-KOR Neurons Decreases Neuropathic Pain Behaviors
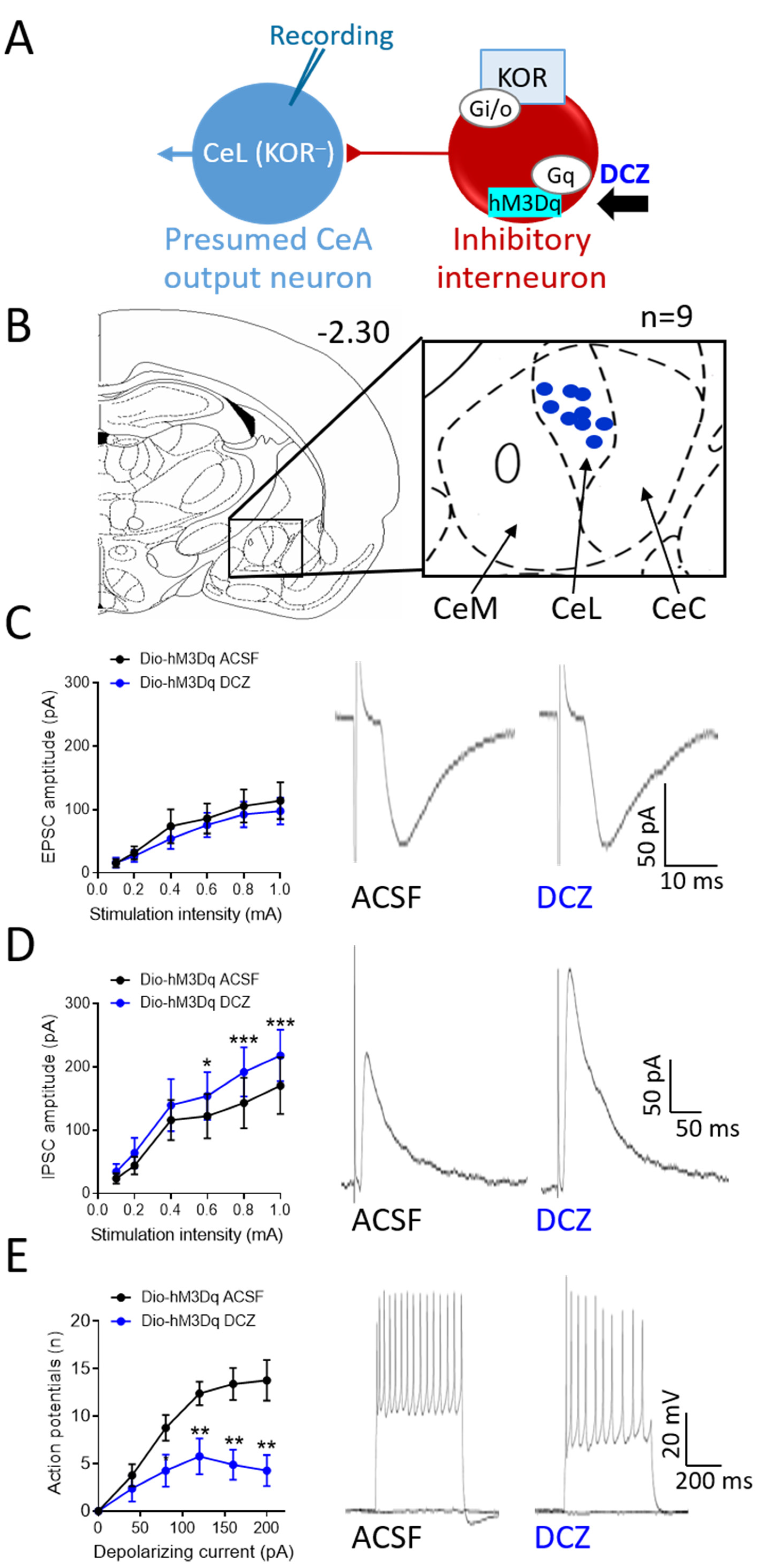
3.4. Validation of Chemogenetic Manipulations of CeA-KOR Neurons


3.5. Effects of Chemogenetic Modulation of CeA-KOR Neurons on Downstream CeL Neurons
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Dahlhamer, J.; Lucas, J.; Zelaya, C.; Nahin, R.; Mackey, S.; DeBar, L.; Kerns, R.; Von Korff, M.; Porter, L.; Helmick, C. Prevalence of Chronic Pain and High-Impact Chronic Pain among Adults—United States, 2016. Morb. Mortal. Wkly. Rep. 2018, 67, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.; Carsten Schultheis, B.; Hanes, M.C.; Jolly, S.M.; Chakravarthy, K.V.; Deer, T.R.; Levy, R.M.; Hunter, C.W. A Comprehensive Algorithm for Management of Neuropathic Pain. Pain Med. Off. J. Am. Acad. Pain Med. 2019, 20, S2. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Kuner, T.; Kuner, R. Synaptic Plasticity in Pathological Pain. Trends Neurosci. 2014, 37, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Veinante, P.; Yalcin, I.; Barrot, M. The Amygdala between Sensation and Affect: A Role in Pain. J. Mol. Psychiatry 2013, 1, 9. [Google Scholar] [CrossRef] [PubMed]
- Neugebauer, V. Amygdala Physiology in Pain. Handb. Behav. Neurosci. 2020, 26, 101. [Google Scholar] [CrossRef] [PubMed]
- Neugebauer, V.; Mazzitelli, M.; Cragg, B.; Ji, G.; Navratilova, E.; Porreca, F. Amygdala, Neuropeptides, and Chronic Pain-Related Affective Behaviors. Neuropharmacology 2020, 170, 108052. [Google Scholar] [CrossRef] [PubMed]
- Kato, F.; Sugimura, Y.K.; Takahashi, Y. Pain-Associated Neural Plasticity in the Parabrachial to Central Amygdala Circuit. Adv. Exp. Med. Biol. 2018, 1099, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Neugebauer, V.; Li, W.; Bird, G.C.; Han, J.S. The Amygdala and Persistent Pain. Neuroscientist 2004, 10, 221–234. [Google Scholar] [CrossRef]
- Gauriau, C.; Bernard, J.F. Pain Pathways and Parabrachial Circuits in the Rat. Exp. Physiol. 2002, 87, 251–258. [Google Scholar] [CrossRef]
- Neugebauer, V.; Presto, P.; Yakhnitsa, V.; Antenucci, N.; Mendoza, B.; Ji, G. Pain-Related Cortico-Limbic Plasticity and Opioid Signaling. Neuropharmacology 2023, 231, 109510. [Google Scholar] [CrossRef]
- Becker, J.J.; Carrasquillo, Y. Projections, Where Art Thou: The State and Future of the Central Amygdala. J. Physiol. 2019, 597, 365. [Google Scholar] [CrossRef]
- Liu, J.; Hu, T.; Zhang, M.Q.; Xu, C.Y.; Yuan, M.Y.; Li, R.X. Differential Efferent Projections of GABAergic Neurons in the Basolateral and Central Nucleus of Amygdala in Mice. Neurosci. Lett. 2021, 745, 135621. [Google Scholar] [CrossRef] [PubMed]
- Raver, C.; Uddin, O.; Ji, Y.; Li, Y.; Cramer, N.; Jenne, C.; Morales, M.; Masri, R.; Keller, A. An Amygdalo-Parabrachial Pathway Regulates Pain Perception and Chronic Pain. J. Neurosci. 2020, 40, 3424–3442. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.M.; Neugebauer, V. Amygdala Plasticity and Pain. Pain Res. Manag. 2017, 2017, 8296501. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.D.; Valdivia, S.; Khan, A.; Ahn, H.S.; Adke, A.P.; Gonzalez, S.M.; Sugimura, Y.K.; Carrasquillo, Y. Dual and Opposing Functions of the Central Amygdala in the Modulation of Pain. Cell Rep. 2019, 29, 332. [Google Scholar] [CrossRef] [PubMed]
- Limoges, A.; Yarur, H.E.; Tejeda, H.A. Dynorphin/Kappa Opioid Receptor System Regulation on Amygdaloid Circuitry: Implications for Neuropsychiatric Disorders. Front. Syst. Neurosci. 2022, 16, 963691. [Google Scholar] [CrossRef] [PubMed]
- Crowley, N.A.; Bloodgood, D.W.; Hardaway, J.A.; Kendra, A.M.; McCall, J.G.; Al-Hasani, R.; McCall, N.M.; Yu, W.; Schools, Z.L.; Krashes, M.J.; et al. Dynorphin Controls the Gain of an Amygdalar Anxiety Circuit. Cell Rep. 2016, 14, 2774. [Google Scholar] [CrossRef] [PubMed]
- Gilpin, N.W.; Roberto, M.; Koob, G.F.; Schweitzer, P. Kappa Opioid Receptor Activation Decreases Inhibitory Transmission and Antagonizes Alcohol Effects in Rat Central Amygdala. Neuropharmacology 2014, 77, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Navratilova, E.; Qu, C.; Ji, G.; Neugebauer, V.; Guerrero, M.; Rosen, H.; Roberts, E.; Porreca, F. Opposing Effects on Descending Control of Nociception by µ and κ Opioid Receptors in the Anterior Cingulate Cortex. Anesthesiology 2024, 140, 272–283. [Google Scholar] [CrossRef]
- Cahill, C.M.; Taylor, A.M.W.; Cook, C.; Ong, E.; Moron, J.A.; Evans, C.J. Does the Kappa Opioid Receptor System Contribute to Pain Aversion? Front. Pharmacol. 2014, 5, 121563. [Google Scholar] [CrossRef]
- Peckys, D.; Landwehrmeyer, G.B. Expression of Mu, Kappa, and Delta Opioid Receptor Messenger RNA in the Human CNS: A 33P in Situ Hybridization Study. Neuroscience 1999, 88, 1093–1135. [Google Scholar] [CrossRef] [PubMed]
- Knoll, A.T.; Carlezon, W.A. Dynorphin, Stress, and Depression. Brain Res. 2010, 1314C, 56. [Google Scholar] [CrossRef] [PubMed]
- Marchant, N.J.; Densmore, V.S.; Osborne, P.B. Coexpression of Prodynorphin and Corticotrophin-Releasing Hormone in the Rat Central Amygdala: Evidence of Two Distinct Endogenous Opioid Systems in the Lateral Division. J. Comp. Neurol. 2007, 504, 702–715. [Google Scholar] [CrossRef] [PubMed]
- Pomrenze, M.B.; Millan, E.Z.; Hopf, F.W.; Keiflin, R.; Maiya, R.; Blasio, A.; Dadgar, J.; Kharazia, V.; De Guglielmo, G.; Crawford, E.; et al. A Transgenic Rat for Investigating the Anatomy and Function of Corticotrophin Releasing Factor Circuits. Front. Neurosci. 2015, 9, 487. [Google Scholar] [CrossRef] [PubMed]
- Hein, M.; Ji, G.; Tidwell, D.; D’Souza, P.; Kiritoshi, T.; Yakhnitsa, V.; Navratilova, E.; Porreca, F.; Neugebauer, V. Kappa Opioid Receptor Activation in the Amygdala Disinhibits CRF Neurons to Generate Pain-like Behaviors. Neuropharmacology 2021, 185, 108456. [Google Scholar] [CrossRef] [PubMed]
- Navratilova, E.; Ji, G.; Phelps, C.; Qu, C.; Hein, M.; Yakhnitsa, V.; Neugebauer, V.; Porreca, F. Kappa Opioid Signaling in the Central Nucleus of the Amygdala Promotes Disinhibition and Aversiveness of Chronic Neuropathic Pain. Pain 2019, 160, 824. [Google Scholar] [CrossRef]
- Yakhnitsa, V.; Ji, G.; Hein, M.; Presto, P.; Griffin, Z.; Ponomareva, O.; Navratilova, E.; Porreca, F.; Neugebauer, V. Kappa Opioid Receptor Blockade in the Amygdala Mitigates Pain Like-Behaviors by Inhibiting Corticotropin Releasing Factor Neurons in a Rat Model of Functional Pain. Front. Pharmacol. 2022, 13, 903978. [Google Scholar] [CrossRef] [PubMed]
- Roth, B.L. DREADDs for Neuroscientists. Neuron 2016, 89, 683–694. [Google Scholar] [CrossRef]
- Ho Kim, S.; Mo Chung, J. An Experimental Model for Peripheral Neuropathy Produced by Segmental Spinal Nerve Ligation in the Rat. Pain 1992, 50, 355–363. [Google Scholar] [CrossRef]
- Ji, G.; Neugebauer, V. Hemispheric Lateralization of Pain Processing by Amygdala Neurons. J. Neurophysiol. 2009, 102, 2253. [Google Scholar] [CrossRef]
- Nation, K.M.; De Felice, M.; Hernandez, P.I.; Dodick, D.W.; Neugebauer, V.; Navratilova, E.; Porreca, F. Lateralized Kappa Opioid Receptor Signaling from the Amygdala Central Nucleus Promotes Stress-Induced Functional Pain. Pain 2018, 159, 919. [Google Scholar] [CrossRef] [PubMed]
- Phelps, C.E.; Navratilova, E.; Dickenson, A.H.; Porreca, F.; Bannister, K. Kappa Opioid Signaling in the Right Central Amygdala Causes Hind Paw Specific Loss of Diffuse Noxious Inhibitory Controls in Experimental Neuropathic Pain. Pain 2019, 160, 1614–1621. [Google Scholar] [CrossRef] [PubMed]
- Carrasquillo, Y.; Gereau IV, R.W. Hemispheric Lateralization of a Molecular Signal for Pain Modulation in the Amygdala. Mol. Pain 2008, 4, 24. [Google Scholar] [CrossRef] [PubMed]
- Allen, H.N.; Bobnar, H.J.; Kolber, B.J. Left and Right Hemispheric Lateralization of the Amygdala in Pain. Prog. Neurobiol. 2021, 196, 101891. [Google Scholar] [CrossRef] [PubMed]
- Brackley, A.D.; Gomez, R.; Guerrero, K.A.; Akopian, A.N.; Glucksman, M.J.; Du, J.; Carlton, S.M.; Jeske, N.A. A-Kinase Anchoring Protein 79/150 Scaffolds Transient Receptor Potential A 1 Phosphorylation and Sensitization by Metabotropic Glutamate Receptor Activation. Sci. Rep. 2017, 7, 1842. [Google Scholar] [CrossRef] [PubMed]
- Calvino, B.; Besson, J.M.; Boehrer, A.; Depaulis, A. Ultrasonic Vocalization (22–28 KHz) in a Model of Chronic Pain, the Arthritic Rat: Effects of Analgesic Drugs. Neuroreport 1996, 7, 581–584. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, D.; Ardid, D.; Chapuy, E.; Le Bars, D.; Eschalier, A. Effect of Analgesics on Audible and Ultrasonic Pain-Induced Vocalization in the Rat. Life Sci. 1998, 63, 1761–1768. [Google Scholar] [CrossRef] [PubMed]
- Vivian, J.A.; Miczek, K.A. Morphine Attenuates Ultrasonic Vocalization during Agonistic Encounters in Adult Male Rats. Psychopharmacology 1993, 111, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Han, J.S.; Bird, G.C.; Li, W.; Jones, J.; Neugebauer, V. Computerized Analysis of Audible and Ultrasonic Vocalizations of Rats as a Standardized Measure of Pain-Related Behavior. J. Neurosci. Methods 2005, 141, 261–269. [Google Scholar] [CrossRef]
- Presto, P.; Ji, G.; Junell, R.; Griffin, Z.; Neugebauer, V. Fear Extinction-Based Inter-Individual and Sex Differences in Pain-Related Vocalizations and Anxiety-like Behaviors but Not Nocifensive Reflexes. Brain Sci. 2021, 11, 1339. [Google Scholar] [CrossRef]
- Ashraf-Uz-Zaman, M.; Ji, G.; Tidwell, D.; Yin, L.; Thakolwiboon, S.; Pan, J.; Junell, R.; Griffin, Z.; Shahi, S.; Barthels, D.; et al. Evaluation of Urea-Based Inhibitors of the Dopamine Transporter Using the Experimental Autoimmune Encephalomyelitis Model of Multiple Sclerosis. ACS Chem. Neurosci. 2022, 13, 217–228. [Google Scholar] [CrossRef]
- Crawley, J.; Goodwin, F.K. Preliminary Report of a Simple Animal Behavior Model for the Anxiolytic Effects of Benzodiazepines. Pharmacol. Biochem. Behav. 1980, 13, 167–170. [Google Scholar] [CrossRef]
- Gandhi, P.J.; Gawande, D.Y.; Shelkar, G.P.; Gakare, S.G.; Kiritoshi, T.; Ji, G.; Misra, B.; Pavuluri, R.; Liu, J.; Neugebauer, V.; et al. Dysfunction of Glutamate Delta-1 Receptor-Cerebellin 1 Trans-Synaptic Signaling in the Central Amygdala in Chronic Pain. Cells 2021, 10, 2644. [Google Scholar] [CrossRef] [PubMed]
- Tappe-Theodor, A.; Fu, Y.; Kuner, R.; Neugebauer, V. Homer1a Signaling in the Amygdala Counteracts Pain-Related Synaptic Plasticity, MGluR1 Function and Pain Behaviors. Mol. Pain 2011, 7, 1744–8069. [Google Scholar] [CrossRef] [PubMed]
- Ji, G.; Zhang, W.; Mahimainathan, L.; Narasimhan, M.; Kiritoshi, T.; Fan, X.; Wang, J.; Green, T.A.; Neugebauer, V. 5-HT2C Receptor Knockdown in the Amygdala Inhibits Neuropathic-Pain-Related Plasticity and Behaviors. J. Neurosci. 2017, 37, 1378. [Google Scholar] [CrossRef]
- Corder, G.; Ahanonu, B.; Grewe, B.F.; Wang, D.; Schnitzer, M.J.; Scherrer, G. An Amygdalar Neural Ensemble That Encodes the Unpleasantness of Pain. Science 2019, 363, 276. [Google Scholar] [CrossRef] [PubMed]
- Bruchas, M.R.; Land, B.B.; Lemos, J.C.; Chavkin, C. CRF1-R Activation of the Dynorphin/Kappa Opioid System in the Mouse Basolateral Amygdala Mediates Anxiety-Like Behavior. PLoS ONE 2009, 4, 8528. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.Y.; De Felice, M.; Kopruszinski, C.M.; Eyde, N.; Lavigne, J.; Remeniuk, B.; Hernandez, P.; Yue, X.; Goshima, N.; Ossipov, M.; et al. Kappa Opioid Receptor Antagonists: A Possible New Class of Therapeutics for Migraine Prevention. Cephalalgia 2017, 37, 780. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.J.; Marchant, N.J. The Use of Chemogenetics in Behavioural Neuroscience: Receptor Variants, Targeting Approaches and Caveats. Br. J. Pharmacol. 2018, 175, 994. [Google Scholar] [CrossRef]
- Ozawa, A.; Arakawa, H. Chemogenetics Drives Paradigm Change in the Investigation of Behavioral Circuits and Neural Mechanisms Underlying Drug Action. Behav. Brain Res. 2021, 406, 113234. [Google Scholar] [CrossRef]
- Standifer, K.M.; Pasternak, G.W. G Proteins and Opioid Receptor-Mediated Signalling. Cell Signal. 1997, 9, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Ji, G.; Neugebauer, V. Kappa Opioid Receptors in the Central Amygdala Modulate Spinal Nociceptive Processing through an Action on Amygdala CRF Neurons. Mol. Brain 2020, 13, 128. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Kaneko, C.; Miyoshi, K.; Nagumo, Y.; Kuzumaki, N.; Nakajima, M.; Nanjo, K.; Matsuzawa, K.; Yamazaki, M.; Suzuki, T. Chronic Pain Induces Anxiety with Concomitant Changes in Opioidergic Function in the Amygdala. Neuropsychopharmacology 2006, 31, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Turnes, J.d.M.; Araya, E.I.; Barroso, A.R.; Baggio, D.F.; Koren, L.d.O.; Zanoveli, J.M.; Chichorro, J.G. Blockade of Kappa Opioid Receptors Reduces Mechanical Hyperalgesia and Anxiety-like Behavior in a Rat Model of Trigeminal Neuropathic Pain. Behav. Brain Res. 2022, 417, 113595. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.S.; Pickens, S.; Burma, N.E.; Ibarra-Lecue, I.; Yang, H.; Xue, L.; Cook, C.; Hakimian, J.K.; Severino, A.L.; Lueptow, L.; et al. Kappa Opioid Receptors Drive a Tonic Aversive Component of Chronic Pain. J. Neurosci. 2019, 39, 4162. [Google Scholar] [CrossRef]
- Meade, J.A.; Alkhlaif, Y.; Contreras, K.M.; Obeng, S.; Toma, W.; Sim-Selley, L.J.; Selley, D.E.; Damaj, M.I. Kappa Opioid Receptors Mediate an Initial Aversive Component of Paclitaxel-Induced Neuropathy. Psychopharmacology 2020, 237, 2777–2793. [Google Scholar] [CrossRef] [PubMed]
- Cahill, C.M.; Lueptow, L.; Kim, H.; Shusharla, R.; Bishop, A.; Evans, C.J. Kappa Opioid Signaling at the Crossroads of Chronic Pain and Opioid Addiction. Handb. Exp. Pharmacol. 2022, 271, 315–350. [Google Scholar] [CrossRef]
- Shimizu, M.; Yoshimura, M.; Baba, K.; Ikeda, N.; Nonaka, Y.; Maruyama, T.; Onaka, T.; Ueta, Y. Deschloroclozapine Exhibits an Exquisite Agonistic Effect at Lower Concentration Compared to Clozapine-N-Oxide in HM3Dq Expressing Chemogenetically Modified Rats. Front. Neurosci. 2023, 17, 1301515. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Miyakawa, N.; Takuwa, H.; Hori, Y.; Oyama, K.; Ji, B.; Takahashi, M.; Huang, X.P.; Slocum, S.T.; DiBerto, J.F.; et al. Deschloroclozapine, a Potent and Selective Chemogenetic Actuator Enables Rapid Neuronal and Behavioral Modulations in Mice and Monkeys. Nat. Neurosci. 2020, 23, 1157–1167. [Google Scholar] [CrossRef]
- Urban, D.J.; Roth, B.L. DREADDs (Designer Receptors Exclusively Activated by Designer Drugs): Chemogenetic Tools with Therapeutic Utility. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 399–417. [Google Scholar] [CrossRef]
- Burnett, C.J.; Krashes, M.J. Resolving Behavioral Output via Chemogenetic Designer Receptors Exclusively Activated by Designer Drugs. J. Neurosci. 2016, 36, 9268. [Google Scholar] [CrossRef][Green Version]
- Smith, K.S.; Bucci, D.J.; Luikart, B.W.; Mahler, S.V. Dreadds: Use and Application in Behavioral Neuroscience. Behav. Neurosci. 2021, 135, 89–107. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, G.; Presto, P.; Kiritoshi, T.; Chen, Y.; Navratilova, E.; Porreca, F.; Neugebauer, V. Chemogenetic Manipulation of Amygdala Kappa Opioid Receptor Neurons Modulates Amygdala Neuronal Activity and Neuropathic Pain Behaviors. Cells 2024, 13, 705. https://doi.org/10.3390/cells13080705
Ji G, Presto P, Kiritoshi T, Chen Y, Navratilova E, Porreca F, Neugebauer V. Chemogenetic Manipulation of Amygdala Kappa Opioid Receptor Neurons Modulates Amygdala Neuronal Activity and Neuropathic Pain Behaviors. Cells. 2024; 13(8):705. https://doi.org/10.3390/cells13080705
Chicago/Turabian StyleJi, Guangchen, Peyton Presto, Takaki Kiritoshi, Yong Chen, Edita Navratilova, Frank Porreca, and Volker Neugebauer. 2024. "Chemogenetic Manipulation of Amygdala Kappa Opioid Receptor Neurons Modulates Amygdala Neuronal Activity and Neuropathic Pain Behaviors" Cells 13, no. 8: 705. https://doi.org/10.3390/cells13080705
APA StyleJi, G., Presto, P., Kiritoshi, T., Chen, Y., Navratilova, E., Porreca, F., & Neugebauer, V. (2024). Chemogenetic Manipulation of Amygdala Kappa Opioid Receptor Neurons Modulates Amygdala Neuronal Activity and Neuropathic Pain Behaviors. Cells, 13(8), 705. https://doi.org/10.3390/cells13080705