
Patient-Derived Organoids Recapitulate Pathological Intrinsic and Phenotypic Features of Fibrous Dysplasia
 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
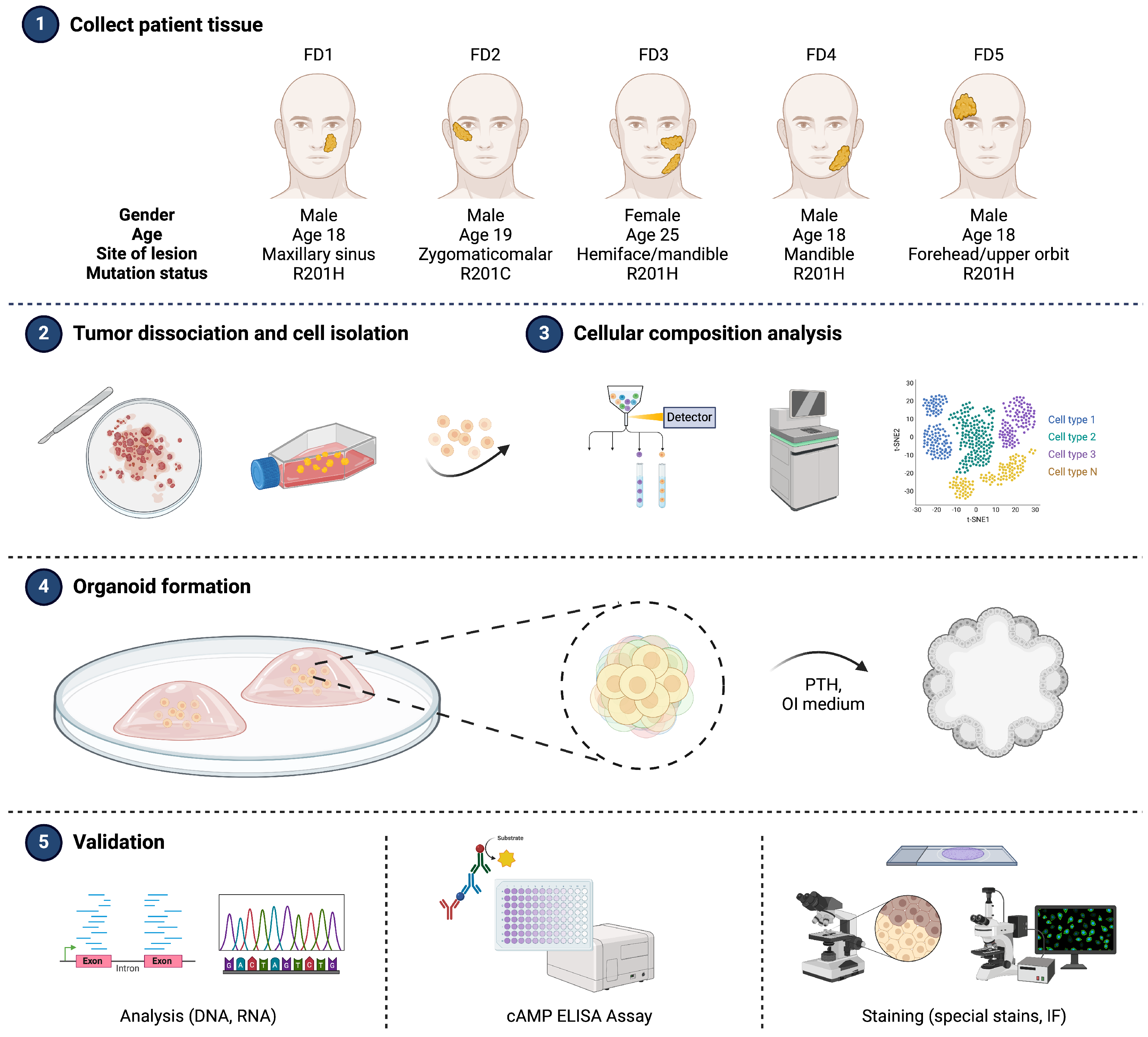
2.1. Human Primary Specimens
2.2. Genomic DNA Extraction and Single Nucleotide Polymorphism (SNP) Genotyping
2.3. Specimen Dissociation and Cell Isolation
2.4. Single-Cell RNA Sequencing (scRNA-seq) and Data Pre-Processing
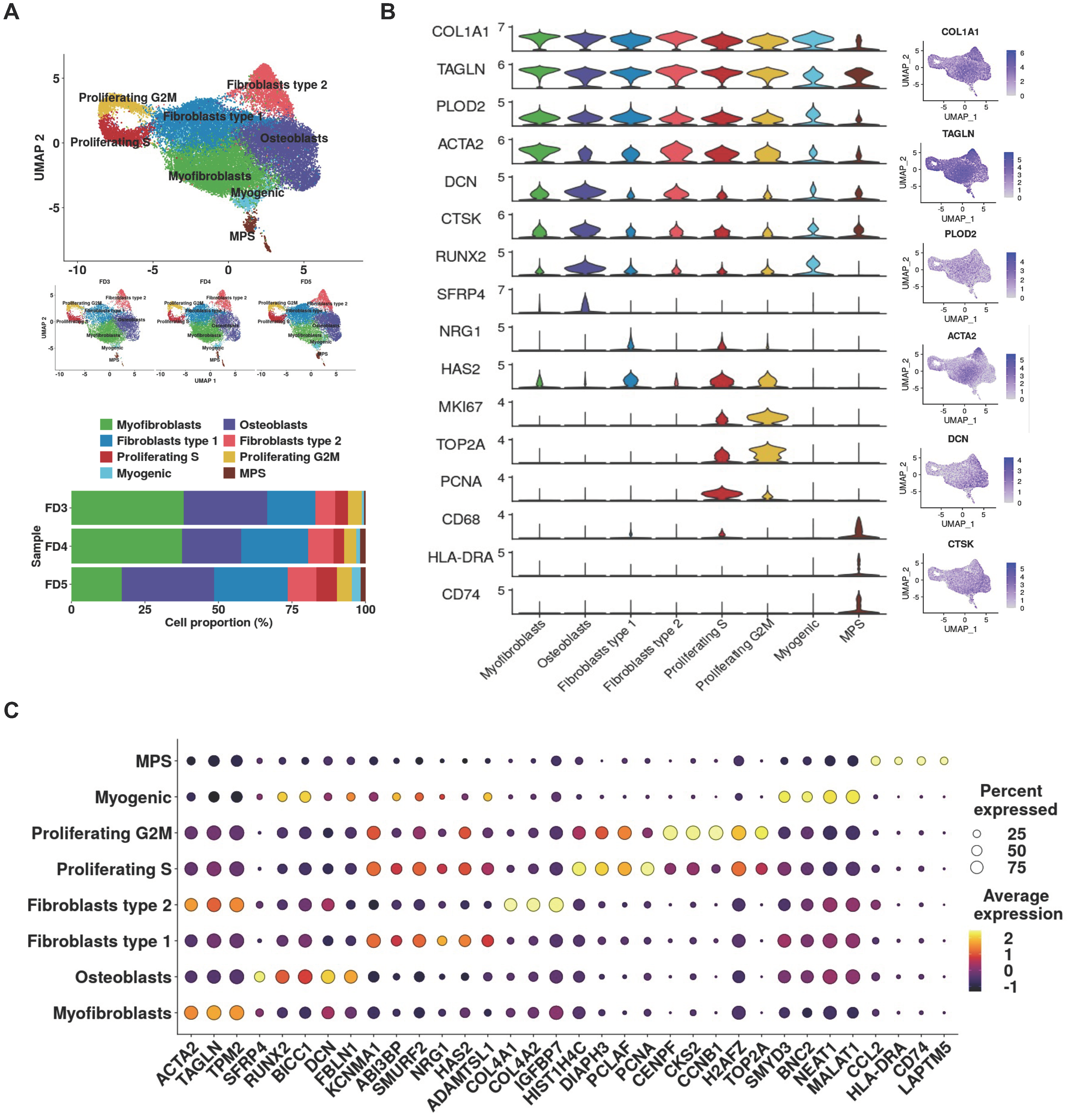
2.5. Cell-Clustering and Cluster Annotation
2.6. Organoid Development and Culture
2.7. Organoid Imaging and Quantitative Analysis
2.8. RNA Extraction and RT-qPCR
2.9. cAMP Assay
2.10. Histology
2.11. Immunofluorescence
2.12. Statistical Analysis
3. Results
3.1. Integrating Patient’s Biology into Organoid Model System
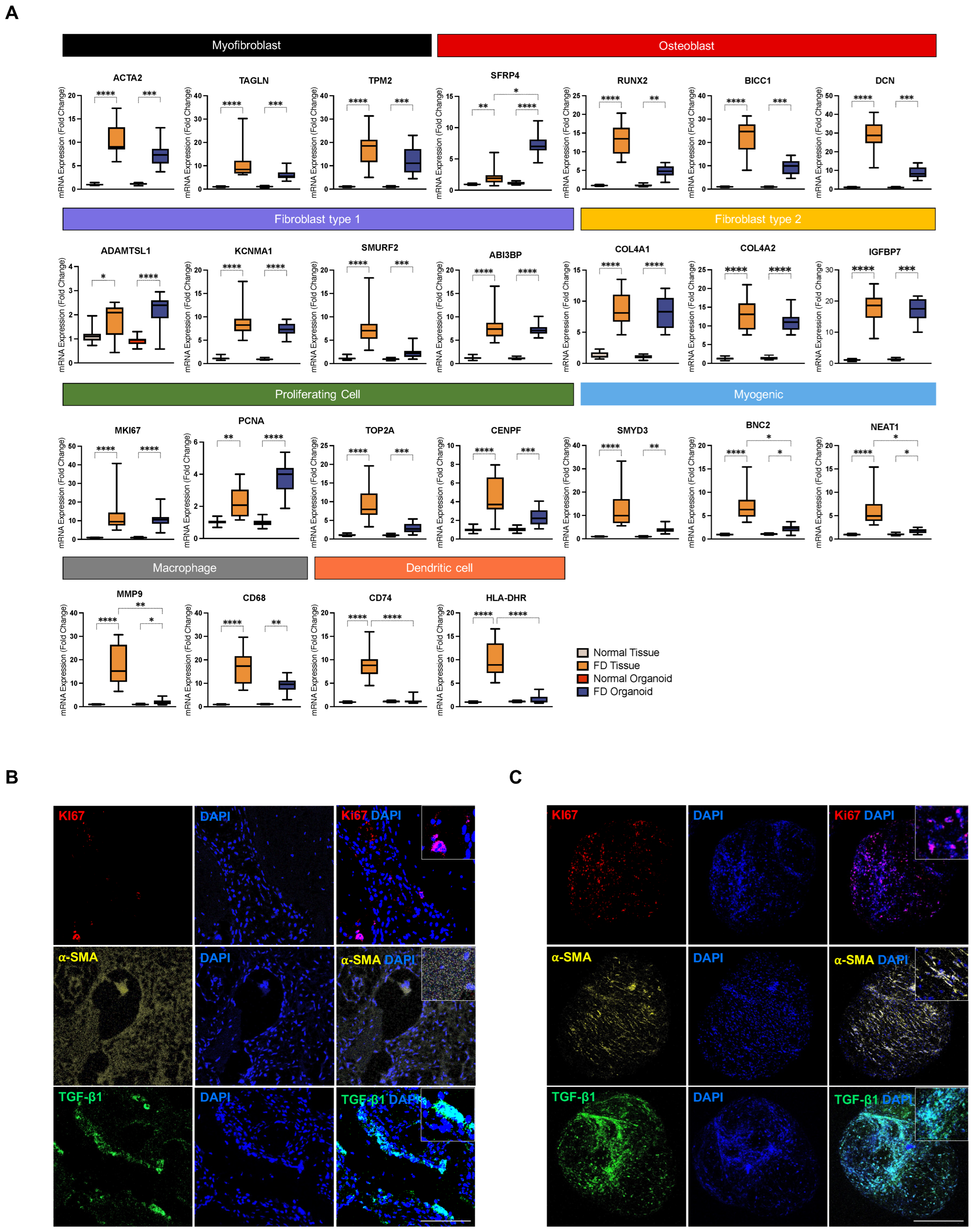
3.2. Single-Cell Analysis Unveils Pro-Fibrotic Transcriptional Signatures and Fibroblastic Cellular Heterogeneity in FD Lesions
3.3. Organoid Generation and Morphological Assessment
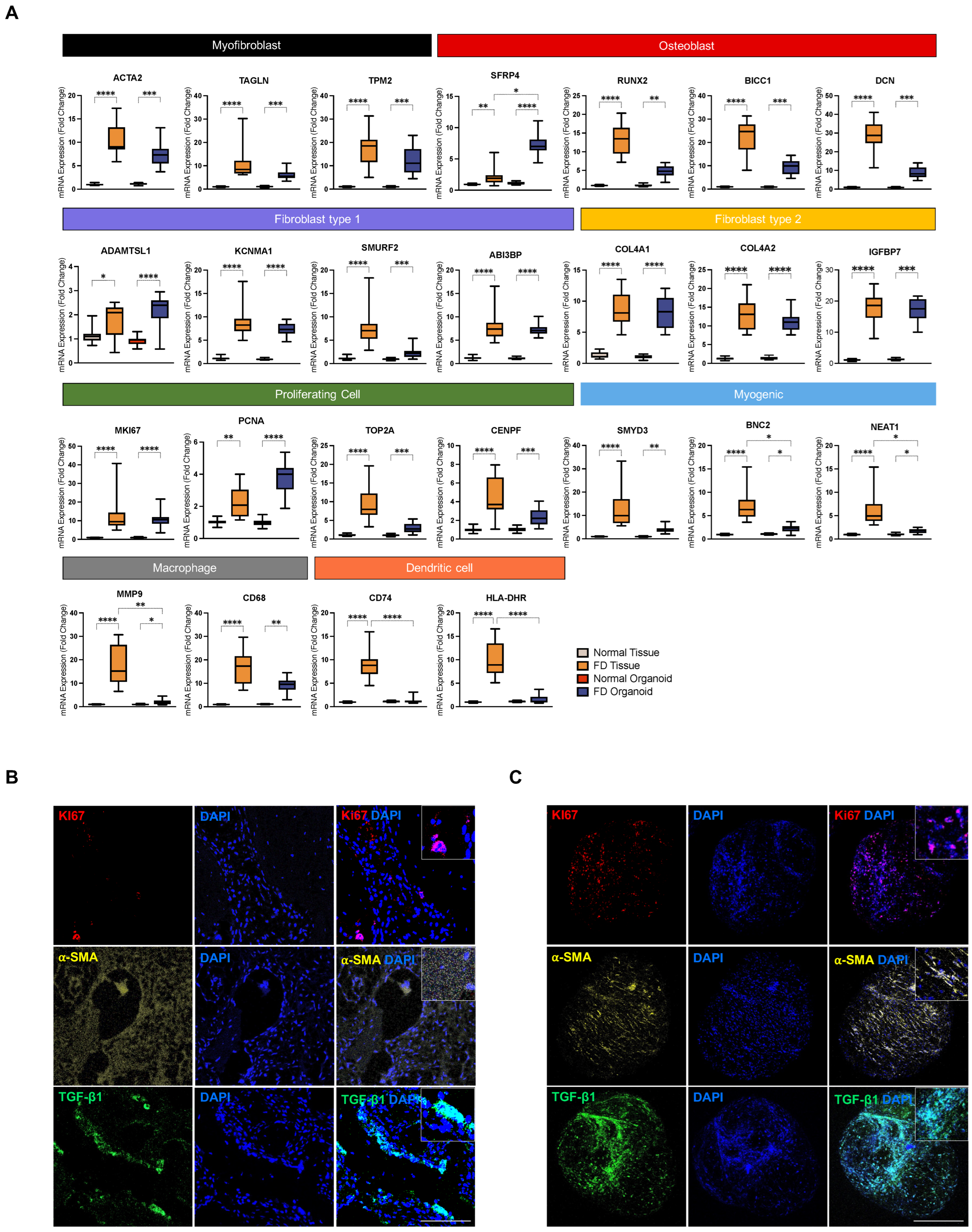
3.4. PDOs Exhibit Enrichment of FD-Relevant Gene Signatures and Cellular Microenvironment
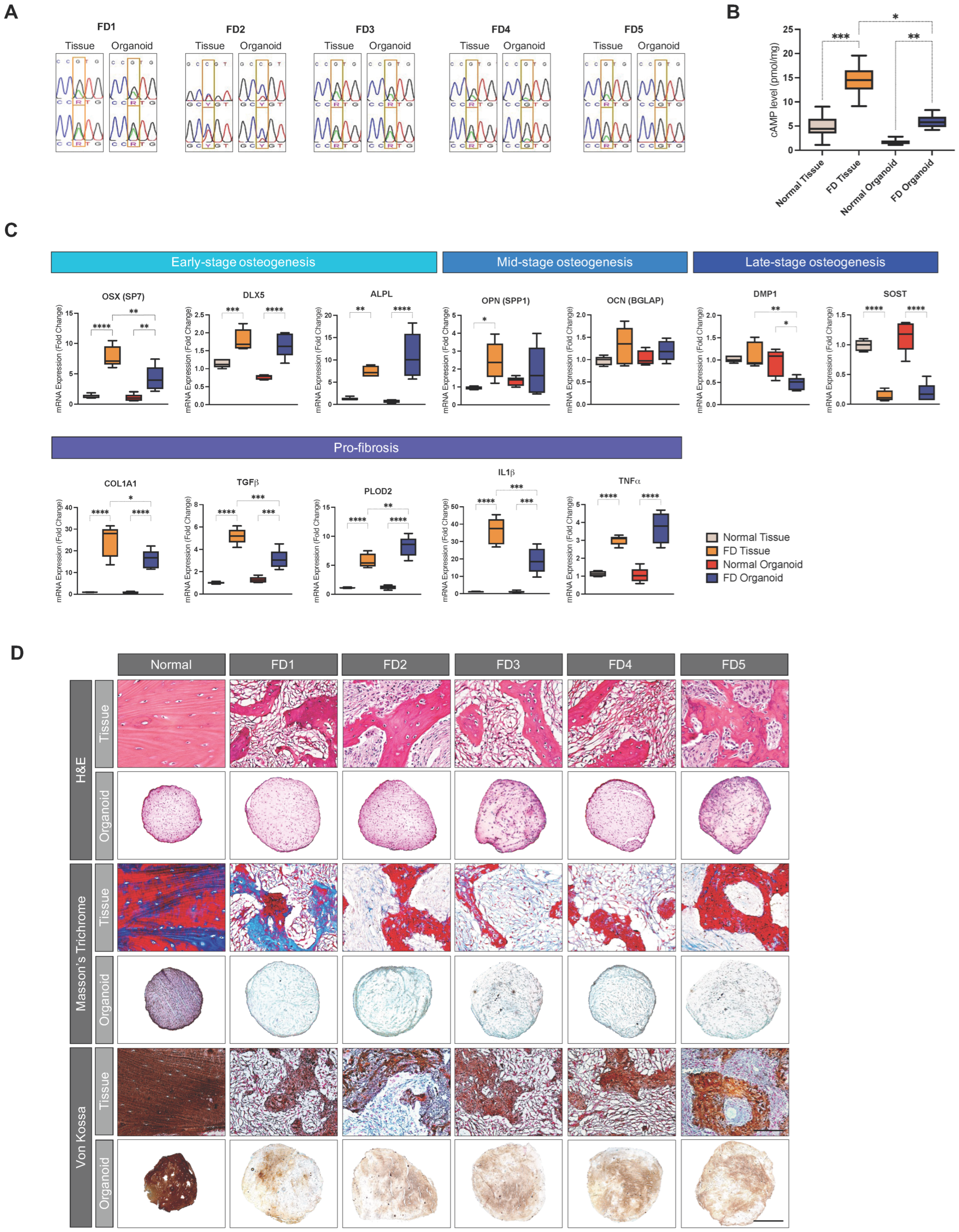
3.5. PDOs Recapitulate the Genetic Abnormality, Metabolic Alteration, and Functional Defects of Corresponding FD Tissues
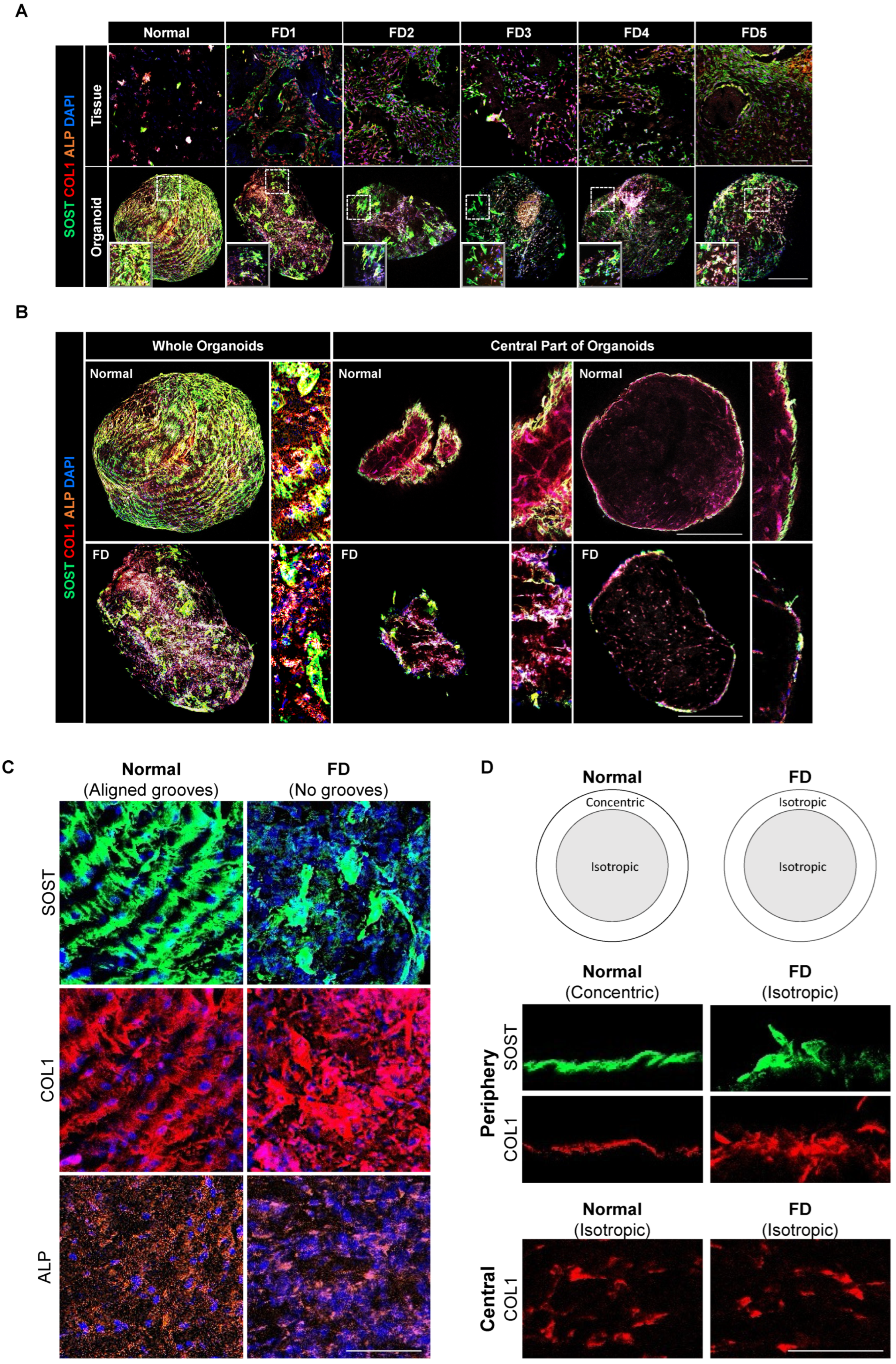
3.6. PDOs Display Non-Aligned, Isotropic Distribution Patterns of Extracellular and Cellular Components
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rosen, C.J. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Chapurlat, R.D.; Orcel, P. Fibrous Dysplasia of Bone and McCune–Albright Syndrome. Best Pract. Res. Clin. Rheumatol. 2008, 22, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Eiraku, M.; Watanabe, K.; Matsuo-Takasaki, M.; Kawada, M.; Yonemura, S.; Matsumura, M.; Wataya, T.; Nishiyama, A.; Muguruma, K.; Sasai, Y. Self-Organized Formation of Polarized Cortical Tissues from ESCs and Its Active Manipulation by Extrinsic Signals. Cell Stem Cell 2008, 3, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Unbekandt, M.; Davies, J.A. Dissociation of Embryonic Kidneys Followed by Reaggregation Allows the Formation of Renal Tissues. Kidney Int. 2010, 77, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Renner, M.; Martin, C.-A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral Organoids Model Human Brain Development and Microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Bhang, D.H.; Beede, A.; Huang, T.L.; Stripp, B.R.; Bloch, K.D.; Wagers, A.J.; Tseng, Y.-H.; Ryeom, S.; Kim, C.F. Lung Stem Cell Differentiation in Mice Directed by Endothelial Cells via a BMP4-NFATc1-Thrombospondin-1 Axis. Cell 2014, 156, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Dorrell, C.; Boj, S.F.; Van Es, J.H.; Li, V.S.W.; Van De Wetering, M.; Sato, T.; Hamer, K.; Sasaki, N.; Finegold, M.J.; et al. In Vitro Expansion of Single Lgr5+ Liver Stem Cells Induced by Wnt-Driven Regeneration. Nature 2013, 494, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Greggio, C.; De Franceschi, F.; Figueiredo-Larsen, M.; Gobaa, S.; Ranga, A.; Semb, H.; Lutolf, M.; Grapin-Botton, A. Artificial Three-Dimensional Niches Deconstruct Pancreas Development in Vitro. Dev. Camb. Engl. 2013, 140, 4452–4462. [Google Scholar]
- Park, I.-H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-Specific Induced Pluripotent Stem Cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Lee, E.Y.; You, Y.-H.; Yang, H.K.; Yoon, K.-H.; Kim, J.-W. Generation of iPSC-Derived Insulin-Producing Cells from Patients with Type 1 and Type 2 Diabetes Compared with Healthy Control. Stem Cell Res. 2020, 48, 101958. [Google Scholar] [CrossRef] [PubMed]
- Mou, X.; Wu, Y.; Cao, H.; Meng, Q.; Wang, Q.; Sun, C.; Hu, S.; Ma, Y.; Zhang, H. Generation of Disease-Specific Induced Pluripotent Stem Cells from Patients with Different Karyotypes of Down Syndrome. Stem Cell Res. Ther. 2012, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Fleischer, A.; Lorenzo, I.M.; Palomino, E.; Aasen, T.; Gómez, F.; Servera, M.; Asensio, V.J.; Gálvez, V.; Izpisúa-Belmonte, J.C.; Bachiller, D. Generation of Two Induced Pluripotent Stem Cell (iPSC) Lines from p.F508del Cystic Fibrosis Patients. Stem Cell Res. 2018, 29, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Vries, R.G.; Snippert, H.J.; Van De Wetering, M.; Barker, N.; Stange, D.E.; Van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 Stem Cells Build Crypt-Villus Structures in Vitro without a Mesenchymal Niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Koo, B.-K.; Knoblich, J.A. Human Organoids: Model Systems for Human Biology and Medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef]
- Smits, L.M.; Reinhardt, L.; Reinhardt, P.; Glatza, M.; Monzel, A.S.; Stanslowsky, N.; Rosato-Siri, M.D.; Zanon, A.; Antony, P.M.; Bellmann, J.; et al. Modeling Parkinson’s Disease in Midbrain-like Organoids. NPJ Park. Dis. 2019, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, C.; Sachs, N.; Chiu, M.C.; Wong, B.H.-Y.; Chu, H.; Poon, V.K.-M.; Wang, D.; Zhao, X.; Wen, L.; et al. Differentiated Human Airway Organoids to Assess Infectivity of Emerging Influenza Virus. Proc. Natl. Acad. Sci. USA 2018, 115, 6822–6827. [Google Scholar] [CrossRef]
- Dekkers, J.F.; Wiegerinck, C.L.; De Jonge, H.R.; Bronsveld, I.; Janssens, H.M.; De Winter-de Groot, K.M.; Brandsma, A.M.; De Jong, N.W.M.; Bijvelds, M.J.C.; Scholte, B.J.; et al. A Functional CFTR Assay Using Primary Cystic Fibrosis Intestinal Organoids. Nat. Med. 2013, 19, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Sachs, N.; Papaspyropoulos, A.; Zomer-van Ommen, D.D.; Heo, I.; Böttinger, L.; Klay, D.; Weeber, F.; Huelsz-Prince, G.; Iakobachvili, N.; Amatngalim, G.D.; et al. Long-Term Expanding Human Airway Organoids for Disease Modeling. EMBO J. 2019, 38, e100300. [Google Scholar] [CrossRef] [PubMed]
- Sampaziotis, F.; de Brito, M.C.; Madrigal, P.; Bertero, A.; Saeb-Parsy, K.; Soares, F.A.C.; Schrumpf, E.; Melum, E.; Karlsen, T.H.; Bradley, J.A.; et al. Cholangiocytes Derived from Human Induced Pluripotent Stem Cells for Disease Modeling and Drug Validation. Nat. Biotechnol. 2015, 33, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Bertaux-Skeirik, N.; Centeno, J.; Gao, J.; Gabre, J.; Zavros, Y. Oncogenic Transformation of Human-Derived Gastric Organoids. In Organoids; Turksen, K., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; Volume 1576, pp. 205–213. [Google Scholar]
- Mahe, M.M.; Aihara, E.; Schumacher, M.A.; Zavros, Y.; Montrose, M.H.; Helmrath, M.A.; Sato, T.; Shroyer, N.F. Establishment of Gastrointestinal Epithelial Organoids. Curr. Protoc. Mouse Biol. 2013, 3, 217–240. [Google Scholar] [CrossRef] [PubMed]
- Teodorescu, P.; Pasca, S.; Jurj, A.; Gafencu, G.; Joelsson, J.; Selicean, S.; Moldovan, C.; Munteanu, R.; Onaciu, A.; Tigu, A.; et al. Transforming Growth Factor Β-mediated Micromechanics Modulates Disease Progression in Primary Myelofibrosis. J. Cell. Mol. Med. 2020, 24, 11100–11110. [Google Scholar] [CrossRef]
- Elbadawy, M.; Yamanaka, M.; Goto, Y.; Hayashi, K.; Tsunedomi, R.; Hazama, S.; Nagano, H.; Yoshida, T.; Shibutani, M.; Ichikawa, R.; et al. Efficacy of Primary Liver Organoid Culture from Different Stages of Non-Alcoholic Steatohepatitis (NASH) Mouse Model. Biomaterials 2020, 237, 119823. [Google Scholar] [CrossRef] [PubMed]
- Ramón Y Cajal, S.; Sesé, M.; Capdevila, C.; Aasen, T.; De Mattos-Arruda, L.; Diaz-Cano, S.J.; Hernández-Losa, J.; Castellví, J. Clinical Implications of Intratumor Heterogeneity: Challenges and Opportunities. J. Mol. Med. 2020, 98, 161–177. [Google Scholar] [CrossRef]
- Persichetti, A.; Milanetti, E.; Palmisano, B.; Di Filippo, A.; Spica, E.; Donsante, S.; Coletta, I.; Venti, M.D.S.; Ippolito, E.; Corsi, A.; et al. Nanostring Technology on Fibrous Dysplasia Bone Biopsies. A Pilot Study Suggesting Different Histology-Related Molecular Profiles. Bone Rep. 2022, 16, 101156. [Google Scholar] [CrossRef]
- Latham, P.D.; Athanasouz, N.A.; Woods, C.G. Fibrous Dysplasia with Locally Aggressive Malignant Change. Arch. Orthop. Trauma Surg. 1992, 111, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Glickman, I. Fibrous Dysplasia in Alveolar Bone. Oral Surg. Oral Med. Oral Pathol. 1948, 1, 895–916. [Google Scholar] [CrossRef]
- Weinstein, L.S. Other Skeletal Diseases Resulting from G Protein Defects. In Principles of Bone Biology; Elsevier: Amsterdam, The Netherlands, 2002; pp. 1165–1176. [Google Scholar]
- Saggio, I.; Remoli, C.; Spica, E.; Cersosimo, S.; Sacchetti, B.; Robey, P.G.; Holmbeck, K.; Cumano, A.; Boyde, A.; Bianco, P.; et al. Constitutive Expression of Gsα R201C in Mice Produces a Heritable, Direct Replica of Human Fibrous Dysplasia Bone Pathology and Demonstrates Its Natural History: Mouse Model of Human Fibrous Dysplasia. J. Bone Miner. Res. 2014, 29, 2357–2368. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Deng, P.; Iglesias-Bartolome, R.; Amornphimoltham, P.; Steffen, D.J.; Jin, Y.; Molinolo, A.A.; De Castro, L.F.; Ovejero, D.; Yuan, Q.; et al. Expression of an Active Gα s Mutant in Skeletal Stem Cells Is Sufficient and Necessary for Fibrous Dysplasia Initiation and Maintenance. Proc. Natl. Acad. Sci. USA 2018, 115, E428–E437. [Google Scholar] [CrossRef]
- Bianco, P.; Kuznetsov, S.A.; Riminucci, M.; Fisher, L.W.; Spiegel, A.M.; Robey, P.G. Reproduction of Human Fibrous Dysplasia of Bone in Immunocompromised Mice by Transplanted Mosaics of Normal and Gsalpha-Mutated Skeletal Progenitor Cells. J. Clin. Investig. 1998, 101, 1737–1744. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Fu, Y.; Zhu, W.; Xu, R.; Xu, L.; Zhang, P.; Du, Y.; Cheng, J.; Jiang, H. HDAC8, A Potential Therapeutic Target, Regulates Proliferation and Differentiation of Bone Marrow Stromal Cells in Fibrous Dysplasia. Stem Cells Transl. Med. 2019, 8, 148–161. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Liu, J.; Zhang, L.; Li, X.; Sun, Z.; Dai, Z.; Ma, J.; Jiao, G.; Chen, Y. WTAP-Mediated m6A Modification Modulates Bone Marrow Mesenchymal Stem Cells Differentiation Potential and Osteoporosis. Cell Death Dis. 2023, 14, 33. [Google Scholar] [CrossRef] [PubMed]
- Riminucci, M.; Collins, M.T.; Corsi, A.; Boyde, A.; Murphey, M.D.; Wientroub, S.; Kuznetsov, S.A.; Cherman, N.; Robey, P.G.; Bianco, P. Gnathodiaphyseal Dysplasia: A Syndrome of Fibro-Osseous Lesions of Jawbones, Bone Fragility, and Long Bone Bowing. J. Bone Miner. Res. 2001, 16, 1710–1718. [Google Scholar] [CrossRef] [PubMed]
- Piersanti, S.; Remoli, C.; Saggio, I.; Funari, A.; Michienzi, S.; Sacchetti, B.; Robey, P.G.; Riminucci, M.; Bianco, P. Transfer, Analysis and Reversion of the Fibrous Dysplasia Cellular Phenotype in Human Skeletal Progenitors. J. Bone Miner. Res. 2009, 25, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Corsi, A.; Palmisano, B.; Spica, E.; Di Filippo, A.; Coletta, I.; Dello Spedale Venti, M.; Labella, R.; Fabretti, F.; Donsante, S.; Remoli, C.; et al. Zoledronic Acid in a Mouse Model of Human Fibrous Dysplasia: Ineffectiveness on Tissue Pathology, Formation of “Giant Osteoclasts” and Pathogenetic Implications. Calcif. Tissue Int. 2020, 107, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Bianco, P.; Riminucci, M.; Majolagbe, A.; Kuznetsov, S.A.; Collins, M.T.; Mankani, M.H.; Corsi, A.; Bone, H.G.; Wientroub, S.; Spiegel, A.M.; et al. Mutations of the GNAS1 Gene, Stromal Cell Dysfunction, and Osteomalacic Changes in Non–McCune–Albright Fibrous Dysplasia of Bone. J. Bone Miner. Res. 2000, 15, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, S.A.; Cherman, N.; Riminucci, M.; Collins, M.T.; Robey, P.G.; Bianco, P. Age-Dependent Demise of GNAS -Mutated Skeletal Stem Cells and “Normalization” of Fibrous Dysplasia of Bone. J. Bone Miner. Res. 2008, 23, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- De Castro, L.F.; Burke, A.B.; Wang, H.D.; Tsai, J.; Florenzano, P.; Pan, K.S.; Bhattacharyya, N.; Boyce, A.M.; Gafni, R.I.; Molinolo, A.A.; et al. Activation of RANK/RANKL/OPG Pathway Is Involved in the Pathophysiology of Fibrous Dysplasia and Associated With Disease Burden. J. Bone Miner. Res. 2019, 34, 290–294. [Google Scholar] [CrossRef]
- Peyser, R.; MacDonnell, S.; Gao, Y.; Cheng, L.; Kim, Y.; Kaplan, T.; Ruan, Q.; Wei, Y.; Ni, M.; Adler, C.; et al. Defining the Activated Fibroblast Population in Lung Fibrosis Using Single-Cell Sequencing. Am. J. Respir. Cell Mol. Biol. 2019, 61, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. Regulation of Proliferation, Differentiation and Functions of Osteoblasts by Runx2. Int. J. Mol. Sci. 2019, 20, 1694. [Google Scholar] [CrossRef] [PubMed]
- Silvent, J.; Nassif, N.; Helary, C.; Azaïs, T.; Sire, J.-Y.; Guille, M.M.G. Collagen Osteoid-Like Model Allows Kinetic Gene Expression Studies of Non-Collagenous Proteins in Relation with Mineral Development to Understand Bone Biomineralization. PLoS ONE 2013, 8, e57344. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Guo, Q.; Hei, H.; Tao, J.; Zhou, Y.; Dong, J.; Xin, H.; Cai, H.; Gao, J.; Yu, K.; et al. BK Ablation Attenuates Osteoblast Bone Formation via Integrin Pathway. Cell Death Dis. 2019, 10, 738. [Google Scholar] [CrossRef] [PubMed]
- Hodgkinson, C.P.; Naidoo, V.; Patti, K.G.; Gomez, J.A.; Schmeckpeper, J.; Zhang, Z.; Davis, B.; Pratt, R.E.; Mirotsou, M.; Dzau, V.J. Abi3bp Is a Multifunctional Autocrine/Paracrine Factor That Regulates Mesenchymal Stem Cell Biology. Stem Cells 2013, 31, 1669–1682. [Google Scholar] [CrossRef] [PubMed]
- Manikoth Ayyathan, D.; Koganti, P.; Marcu-Malina, V.; Litmanovitch, T.; Trakhtenbrot, L.; Emanuelli, A.; Apel-Sarid, L.; Blank, M. SMURF2 Prevents Detrimental Changes to Chromatin, Protecting Human Dermal Fibroblasts from Chromosomal Instability and Tumorigenesis. Oncogene 2020, 39, 3396–3410. [Google Scholar] [CrossRef] [PubMed]
- Rypdal, K.B.; Erusappan, P.M.; Melleby, A.O.; Seifert, D.E.; Palmero, S.; Strand, M.E.; Tønnessen, T.; Dahl, C.P.; Almaas, V.; Hubmacher, D.; et al. The Extracellular Matrix Glycoprotein ADAMTSL2 Is Increased in Heart Failure and Inhibits TGFβ Signalling in Cardiac Fibroblasts. Sci. Rep. 2021, 11, 19757. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-S.; Choi, I.-G.; Lee, B.-C.; Park, J.-B.; Kim, J.-H.; Jeong, J.H.; Jeong, J.H.; Seo, C.H. Neuregulin Induces CTGF Expression in Hypertrophic Scarring Fibroblasts. Mol. Cell. Biochem. 2012, 365, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jiang, D.; Liang, J.; Meltzer, E.B.; Gray, A.; Miura, R.; Wogensen, L.; Yamaguchi, Y.; Noble, P.W. Severe Lung Fibrosis Requires an Invasive Fibroblast Phenotype Regulated by Hyaluronan and CD44. J. Exp. Med. 2011, 208, 1459–1471. [Google Scholar] [CrossRef] [PubMed]
- Kreus, M.; Lehtonen, S.; Skarp, S.; Kaarteenaho, R. Extracellular Matrix Proteins Produced by Stromal Cells in Idiopathic Pulmonary Fibrosis and Lung Adenocarcinoma. PLoS ONE 2021, 16, e0250109. [Google Scholar] [CrossRef] [PubMed]
- Lendahl, U.; Muhl, L.; Betsholtz, C. Identification, Discrimination and Heterogeneity of Fibroblasts. Nat. Commun. 2022, 13, 3409. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wang, T.; Shelp-Peck, E.; Wu, S.-P.; DeMayo, F.J. The Single-Cell Atlas of Cultured Human Endometrial Stromal Cells. FS Sci. 2022, 3, 349–366. [Google Scholar] [CrossRef] [PubMed]
- Codato, R.; Perichon, M.; Divol, A.; Fung, E.; Sotiropoulos, A.; Bigot, A.; Weitzman, J.B.; Medjkane, S. The SMYD3 Methyltransferase Promotes Myogenesis by Activating the Myogenin Regulatory Network. Sci. Rep. 2019, 9, 17298. [Google Scholar] [CrossRef] [PubMed]
- Bobowski-Gerard, M.; Boulet, C.; Zummo, F.P.; Dubois-Chevalier, J.; Gheeraert, C.; Bou Saleh, M.; Strub, J.-M.; Farce, A.; Ploton, M.; Guille, L.; et al. Functional Genomics Uncovers the Transcription Factor BNC2 as Required for Myofibroblastic Activation in Fibrosis. Nat. Commun. 2022, 13, 5324. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Gao, X.; Li, Y.; Sun, W.; Xu, Y.; Tan, Y.; Du, R.; Zhong, G.; Zhao, D.; Liu, Z.; et al. The Mechanosensitive lncRNA Neat1 Promotes Osteoblast Function through Paraspeckle-Dependent Smurf1 mRNA Retention. Bone Res. 2022, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; He, L.; Zhao, Y.; Li, Y.; Zhang, S.; Sun, K.; So, K.; Chen, F.; Zhou, L.; Lu, L.; et al. Malat1 Regulates Myogenic Differentiation and Muscle Regeneration through Modulating MyoD Transcriptional Activity. Cell Discov. 2017, 3, 17002. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jeong, J.; Song, W. Molecular Characteristics and Distribution of Adult Human Corneal Immune Cell Types. Front. Immunol. 2022, 13, 798346. [Google Scholar] [CrossRef] [PubMed]
- Bisgaard, L.S.; Mogensen, C.K.; Rosendahl, A.; Cucak, H.; Nielsen, L.B.; Rasmussen, S.E.; Pedersen, T.X. Bone Marrow-Derived and Peritoneal Macrophages Have Different Inflammatory Response to oxLDL and M1/M2 Marker Expression—Implications for Atherosclerosis Research. Sci. Rep. 2016, 6, 35234. [Google Scholar] [CrossRef]
- Haraguchi, R.; Kitazawa, R.; Mori, K.; Tachibana, R.; Kiyonari, H.; Imai, Y.; Abe, T.; Kitazawa, S. sFRP4-Dependent Wnt Signal Modulation Is Critical for Bone Remodeling during Postnatal Development and Age-Related Bone Loss. Sci. Rep. 2016, 6, 25198. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, Y.; Li, X.-H.; Cao, C.; Zhang, H.-X.; Zhou, C.; Chen, Y.; Gong, Y.; Yang, J.-X.; Cheng, L.; et al. Dissection of Cellular Communication between Human Primary Osteoblasts and Bone Marrow Mesenchymal Stem Cells in Osteoarthritis at Single-Cell Resolution. Int. J. Stem Cells 2023, 16, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Keshav, R.; Narayanappa, U. Expression of Proliferating Cell Nuclear Antigen (PCNA) in Oral Submucous Fibrosis: An Immunohistochemical Study. J. Clin. Diagn. Res. JCDR 2015, 9, ZC20–ZC23. [Google Scholar] [PubMed]
- Stewart, C.A.; Dell’orco, R.T. Age Related Decline in the Expression of Proliferating Cell Nuclear Antigen in Human Diploid Fibroblasts. Mech. Ageing Dev. 1992, 66, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Mariot, V.; Wu, J.Y.; Aydin, C.; Mantovani, G.; Mahon, M.J.; Linglart, A.; Bastepe, M. Potent Constitutive Cyclic AMP-Generating Activity of XLαs Implicates This Imprinted GNAS Product in the Pathogenesis of McCune–Albright Syndrome and Fibrous Dysplasia of Bone. Bone 2011, 48, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Kaplan, F.; Shore, E. Different Roles of GNAS and cAMP Signaling During Early and Late Stages of Osteogenic Differentiation. Horm. Metab. Res. 2012, 44, 724–731. [Google Scholar] [CrossRef] [PubMed]
- De Castro, L.F.; Whitlock, J.M.; Michel, Z.; Pan, K.; Taylor, J.; Szymczuk, V.; Boyce, B.; Martin, D.; Kram, V.; Galisteo, R.; et al. RANKL Inhibition Reduces Lesional Cellularity and Gαs Variant Expression and Enables Osteogenic Maturation in Fibrous Dysplasia. Bone Res. 2024, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Sorrell, J.M.; Caplan, A.I. Chapter 4 Fibroblasts—A Diverse Population at the Center of It All. In International Review of Cell and Molecular Biology; Elsevier: Amsterdam, The Netherlands, 2009; Volume 276, pp. 161–214. [Google Scholar]
- Guerrero-Juarez, C.F.; Dedhia, P.H.; Jin, S.; Ruiz-Vega, R.; Ma, D.; Liu, Y.; Yamaga, K.; Shestova, O.; Gay, D.L.; Yang, Z.; et al. Single-Cell Analysis Reveals Fibroblast Heterogeneity and Myeloid-Derived Adipocyte Progenitors in Murine Skin Wounds. Nat. Commun. 2019, 10, 650. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.; Friedenstein, A.J. Stromal Stem Cells: Marrow-Derived Osteogenic Precursors. Ciba Found. Symp. 1988, 136, 42–60. [Google Scholar] [PubMed]
- Denu, R.A.; Nemcek, S.; Bloom, D.D.; Goodrich, A.D.; Kim, J.; Mosher, D.F.; Hematti, P. Fibroblasts and Mesenchymal Stromal/Stem Cells Are Phenotypically Indistinguishable. Acta Haematol. 2016, 136, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, D.; Šumová, B.; Mallano, T.; Chen, C.-W.; Distler, A.; Bergmann, C.; Ludolph, I.; Horch, R.E.; Gelse, K.; Ramming, A.; et al. Activation of STAT3 Integrates Common Profibrotic Pathways to Promote Fibroblast Activation and Tissue Fibrosis. Nat. Commun. 2017, 8, 1130. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.A.R.; Evans, M.F.; Ghule, P.N.; Lee, K.; Vacek, P.; Sprague, B.L.; Weaver, D.L.; Stein, G.S.; Stein, J.L. Identification of Molecularly Unique Tumor-Associated Mesenchymal Stromal Cells in Breast Cancer Patients. PLoS ONE 2023, 18, e0282473. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.; Benhammou, J.N.; Darci-Maher, N.; French, S.W.; Han, S.B.; Sinsheimer, J.S.; Agopian, V.G.; Pisegna, J.R.; Pajukanta, P. Human Liver Single Nucleus and Single Cell RNA Sequencing Identify a Hepatocellular Carcinoma-Associated Cell-Type Affecting Survival. Genome Med. 2022, 14, 50. [Google Scholar] [CrossRef] [PubMed]
- Fallon, M.; Sopata, M.; Dragon, E.; Brown, M.T.; Viktrup, L.; West, C.R.; Bao, W.; Agyemang, A. A Randomized Placebo-Controlled Trial of the Anti-Nerve Growth Factor Antibody Tanezumab in Subjects With Cancer Pain Due to Bone Metastasis. Oncologist 2023, 28, e1268–e1278. [Google Scholar] [CrossRef]
- Cao, B.; Sun, H.; Fan, Z.; Khawar, M.B.; Cai, L.; Yu, S.; Liang, Z.; Lv, D.; Wang, N.; Bi, C.; et al. Integrative Analyses of Bulk Microarray Data to Discover Genes, Pathways, and Immune Infiltration Characteristics Associated with Targeting of Ewing Sarcoma. J. Cancer Res. Clin. Oncol. 2023, 149, 6967–6977. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Chen, L.; Zhang, W.; Li, Y.; Zhang, Y.; Gao, Y.; Teng, X.; Zou, L.; Wang, Q.; Jia, H.; et al. TOP2A and CENPF Are Synergistic Master Regulators Activated in Cervical Cancer. BMC Med. Genomics 2020, 13, 145. [Google Scholar] [CrossRef]
- Toscano-Marquez, F.; Romero, Y.; Espina-Ordoñez, M.; Cisneros, J. Absence of HDAC3 by Matrix Stiffness Promotes Chromatin Remodeling and Fibroblast Activation in Idiopathic Pulmonary Fibrosis. Cells 2023, 12, 1020. [Google Scholar] [CrossRef] [PubMed]
- Nevzorova, Y.A.; Bangen, J.-M.; Hu, W.; Haas, U.; Weiskirchen, R.; Gassler, N.; Huss, S.; Tacke, F.; Sicinski, P.; Trautwein, C.; et al. Cyclin E1 Controls Proliferation of Hepatic Stellate Cells and Is Essential for Liver Fibrogenesis in Mice. Hepatology 2012, 56, 1140–1149. [Google Scholar] [CrossRef] [PubMed]
- Demarchi, L.M.M.F.; Reis, M.M.; Palomino, S.A.P.; Farhat, C.; Takagaki, T.Y.; Beyruti, R.; Saldiva, P.H.N.; Capelozzi, V.L. Prognostic Values of Stromal Proportion and PCNA, Ki-67, and P53 Proteins in Patients with Resected Adenocarcinoma of the Lung. Mod. Pathol. 2000, 13, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Wei, M.; Hodge, L.; Fanburg-Smith, J.C.; Nelson, A.; Miettinen, M.; Foss, R.D.; Wang, G. Quantitative Analysis of Activating Alpha Subunit of the G Protein (Gsα) Mutation by Pyrosequencing in Fibrous Dysplasia and Other Bone Lesions. J. Mol. Diagn. 2011, 13, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.; Collins, M.T.; Boyce, A.M. Fibrous Dysplasia/McCune-Albright Syndrome: Clinical and Translational Perspectives. Curr. Osteoporos. Rep. 2016, 14, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Leupin, O.; Kramer, I.; Collette, N.M.; Loots, G.G.; Natt, F.; Kneissel, M.; Keller, H. Control of the SOST Bone Enhancer by PTH Using MEF2 Transcription Factors. J. Bone Miner. Res. 2007, 22, 1957–1967. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Takahashi, H.; Tabata-Okubo, K.; Nagaoka, N.; Tokunaga, K.; Matsumori, H.; Ishihara, Y.; Kaku, M.; Iimura, T.; Hara, T.; et al. Bundling of Collagen Fibrils Influences Osteocyte Network Formation during Bone Modeling. Sci. Rep. 2023, 13, 22028. [Google Scholar] [CrossRef] [PubMed]
- Dede Eren, A.; Eren, E.D.; Wilting, T.J.S.; De Boer, J.; Gelderblom, H.; Foolen, J. Self-Agglomerated Collagen Patterns Govern Cell Behaviour. Sci. Rep. 2021, 11, 1516. [Google Scholar] [CrossRef] [PubMed]
- Rabelo, G.D.; Vom Scheidt, A.; Klebig, F.; Hemmatian, H.; Citak, M.; Amling, M.; Busse, B.; Jähn, K. Multiscale Bone Quality Analysis in Osteoarthritic Knee Joints Reveal a Role of the Mechanosensory Osteocyte Network in Osteophytes. Sci. Rep. 2020, 10, 673. [Google Scholar] [CrossRef] [PubMed]
- Burr, D.B. Bone Morphology and Organization. In Basic and Applied Bone Biology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 3–26. [Google Scholar]
- Nair, A.K.; Gautieri, A.; Chang, S.-W.; Buehler, M.J. Molecular Mechanics of Mineralized Collagen Fibrils in Bone. Nat. Commun. 2013, 4, 1724. [Google Scholar] [CrossRef]
- Toyosawa, S.; Yuki, M.; Kishino, M.; Ogawa, Y.; Ueda, T.; Murakami, S.; Konishi, E.; Iida, S.; Kogo, M.; Komori, T.; et al. Ossifying Fibroma vs Fibrous Dysplasia of the Jaw: Molecular and Immunological Characterization. Mod. Pathol. 2007, 20, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.-H.; Yang, W.-J.; Liu, S.-W.; Li, J.; Zhang, C.-Y.; Zhu, Y.; Zhang, C.-P. Gene Expression Profiling of Craniofacial Fibrous Dysplasia Reveals ADAMTS2 Overexpression as a Potential Marker. Int. J. Clin. Exp. Pathol. 2014, 7, 8532–8541. [Google Scholar] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Donor | Gender | Age | Site | Mutation |
|---|---|---|---|---|
| FD1 | M | 18 | Nasal cavity/maxillary sinus | R201H |
| FD2 | M | 19 | Zygomaticomaxillary | R201C |
| FD3 | F | 25 | Hemiface/mandible | R201H |
| FD4 | M | 12 | Mandible | R201H |
| FD5 | M | 14 | Forehead/upper orbit | R201H |
| HV1 | F | 28 | Zygomatic/mandible | None |
| HV2 | F | 23 | Hemiface/mandible | None |
| HV3 | M | 22 | Zygomatic/mandible | None |
| HV4 | M | 19 | Zygomatic/mandible | None |
| HV5 | F | 25 | Zygomatic/hemiface | None |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.-Y.; Charton, C.; Shim, J.H.; Lim, S.Y.; Kim, J.; Lee, S.; Ohn, J.H.; Kim, B.K.; Heo, C.Y. Patient-Derived Organoids Recapitulate Pathological Intrinsic and Phenotypic Features of Fibrous Dysplasia. Cells 2024, 13, 729. https://doi.org/10.3390/cells13090729
Kim H-Y, Charton C, Shim JH, Lim SY, Kim J, Lee S, Ohn JH, Kim BK, Heo CY. Patient-Derived Organoids Recapitulate Pathological Intrinsic and Phenotypic Features of Fibrous Dysplasia. Cells. 2024; 13(9):729. https://doi.org/10.3390/cells13090729
Chicago/Turabian StyleKim, Ha-Young, Clémentine Charton, Jung Hee Shim, So Young Lim, Jinho Kim, Sejoon Lee, Jung Hun Ohn, Baek Kyu Kim, and Chan Yeong Heo. 2024. "Patient-Derived Organoids Recapitulate Pathological Intrinsic and Phenotypic Features of Fibrous Dysplasia" Cells 13, no. 9: 729. https://doi.org/10.3390/cells13090729