

Valproic Acid Treatment after Traumatic Brain Injury in Mice Alleviates Neuronal Death and Inflammation in Association with Increased Plasma Lysophosphatidylcholines
 , , , and
, , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Drug Administration, Anesthesia, and Trauma Induction
2.3. Lipidomic and Metabolomic Analyses of Plasma
2.4. Immunofluoresence Analyses and Histology
2.5. Gene Expression Analyses
2.6. Identification of REST/NRSF Target Genes
2.7. Immunoblotting
2.8. Statistical Analyses
3. Results
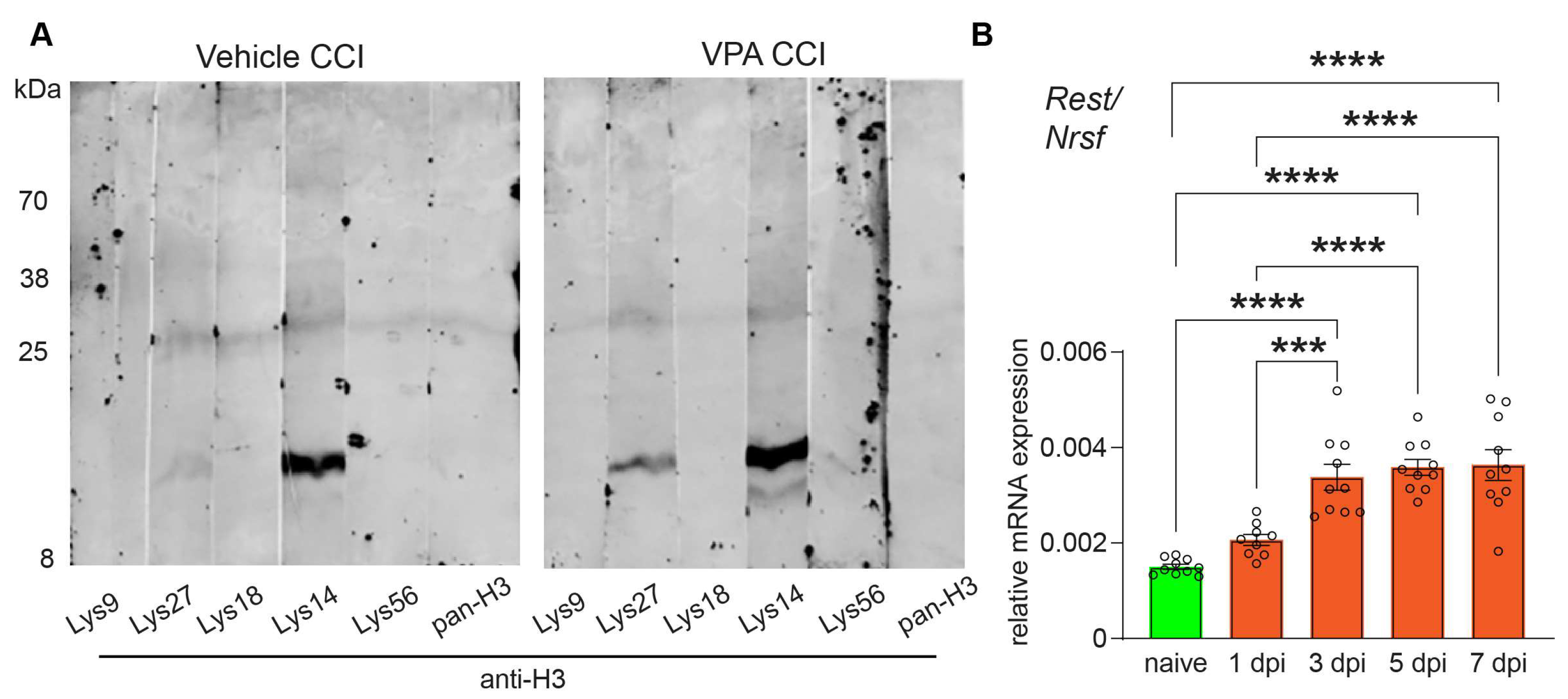
3.1. VPA Functions as HDACi and REST/NRSF Is Up-Regulated after CCI
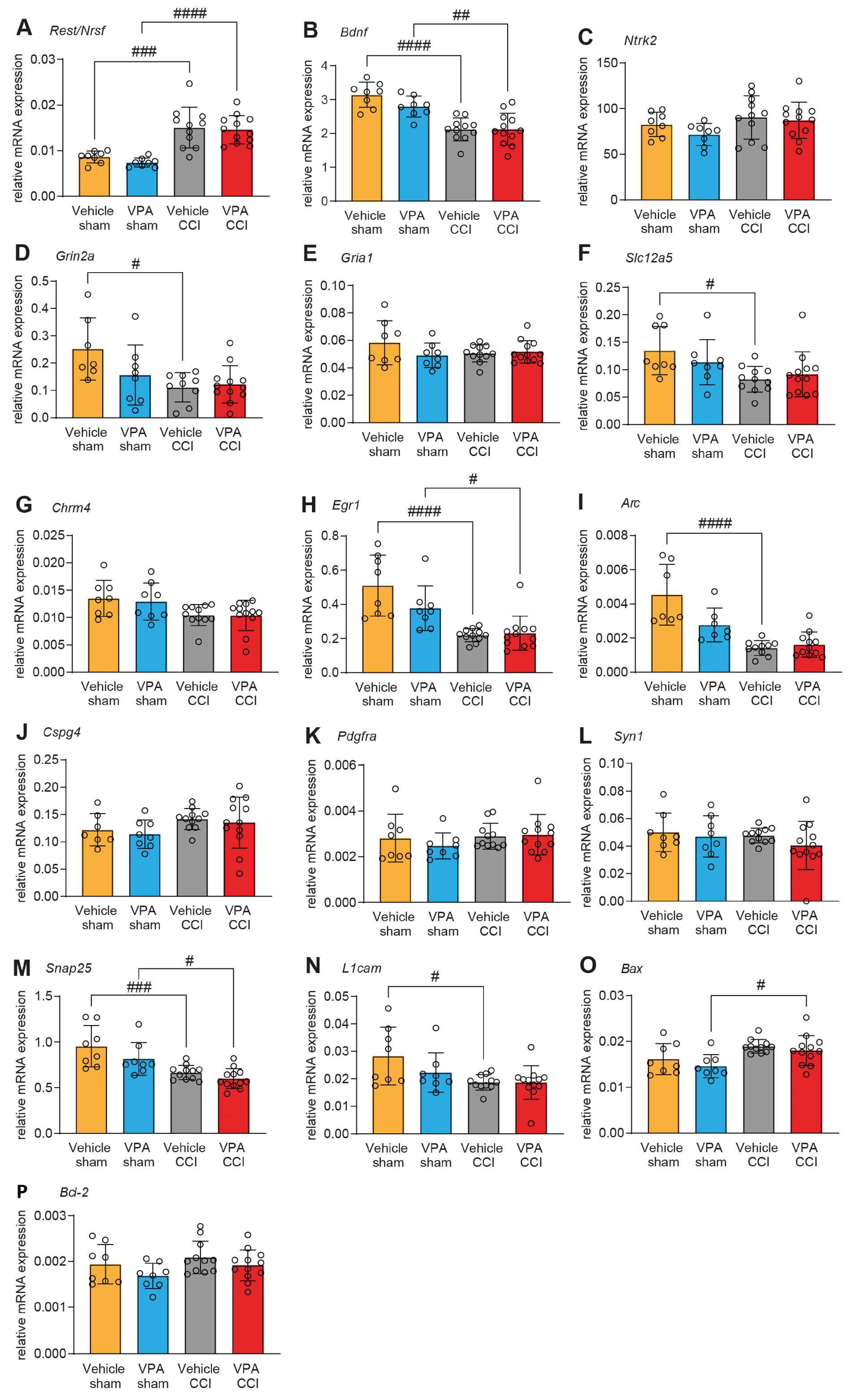
3.2. VPA Treatment Does Not Affect mRNA Expression of REST/NRSF Target Genes in the Injured Brain
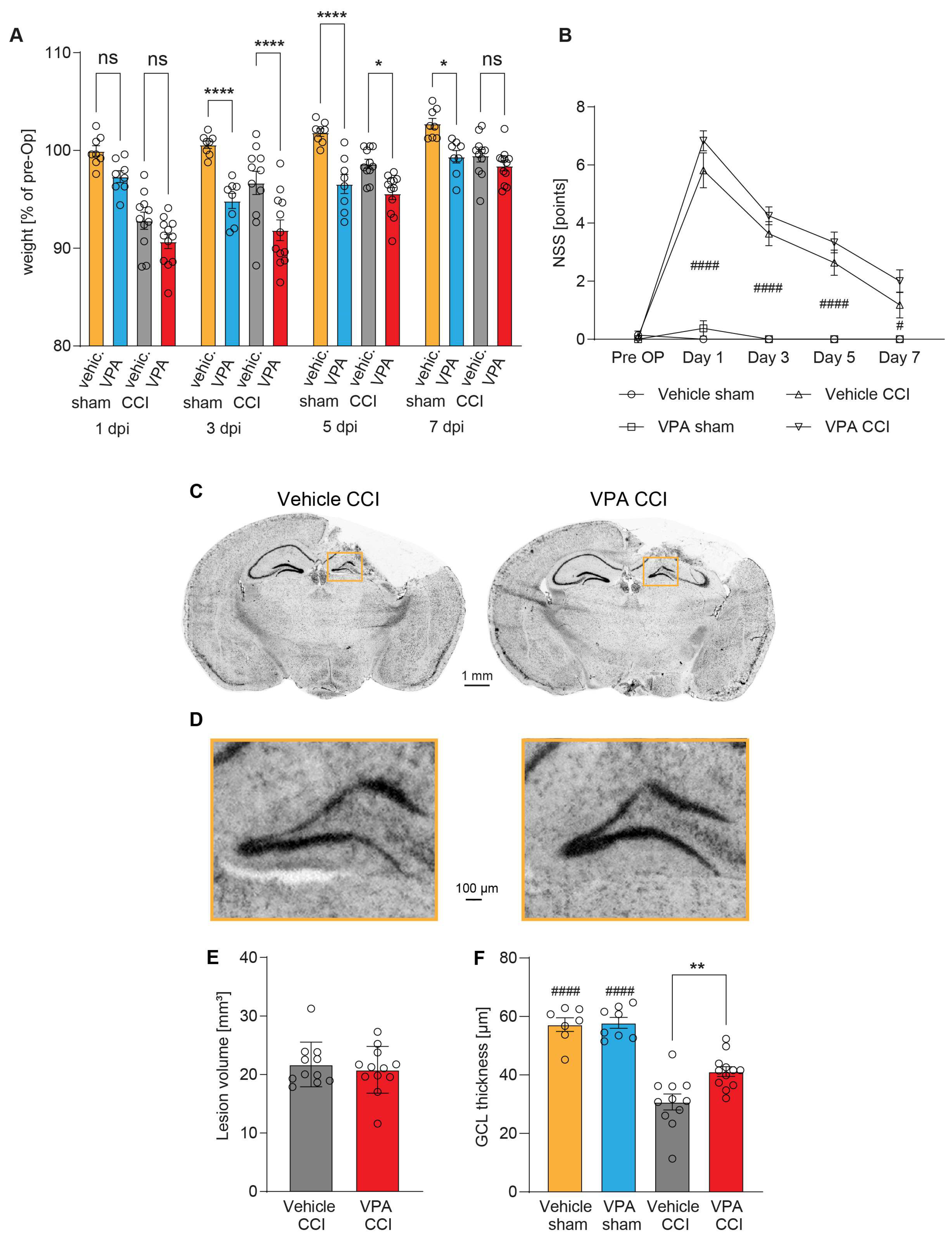
3.3. Administration of VPA Does Not Influence Acute Neurological Deficits or Brain Lesion Size 7 dpi but Attenuates Structural Damage in the Hippocampus
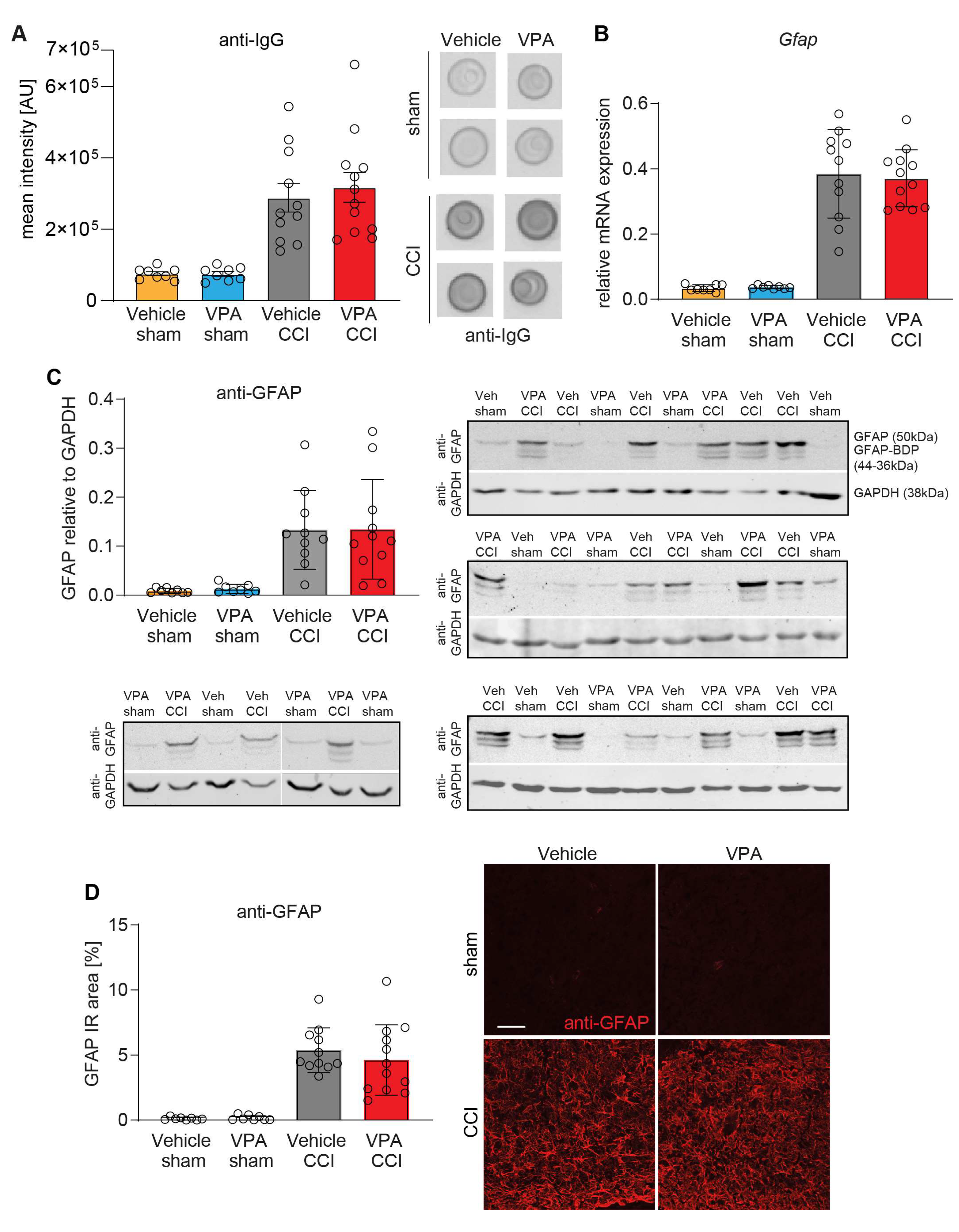
3.4. VPA Treatment Does Not Affect BBB Disruption and Astrocyte Activation at 7 dpi
3.5. VPA Attenuates Microglia Activation and Pro-Inflammatory Gene Expression at 7 dpi
3.6. VPA Treatment Is Associated with High Levels of Lysophosphatidylcholines in Plasma
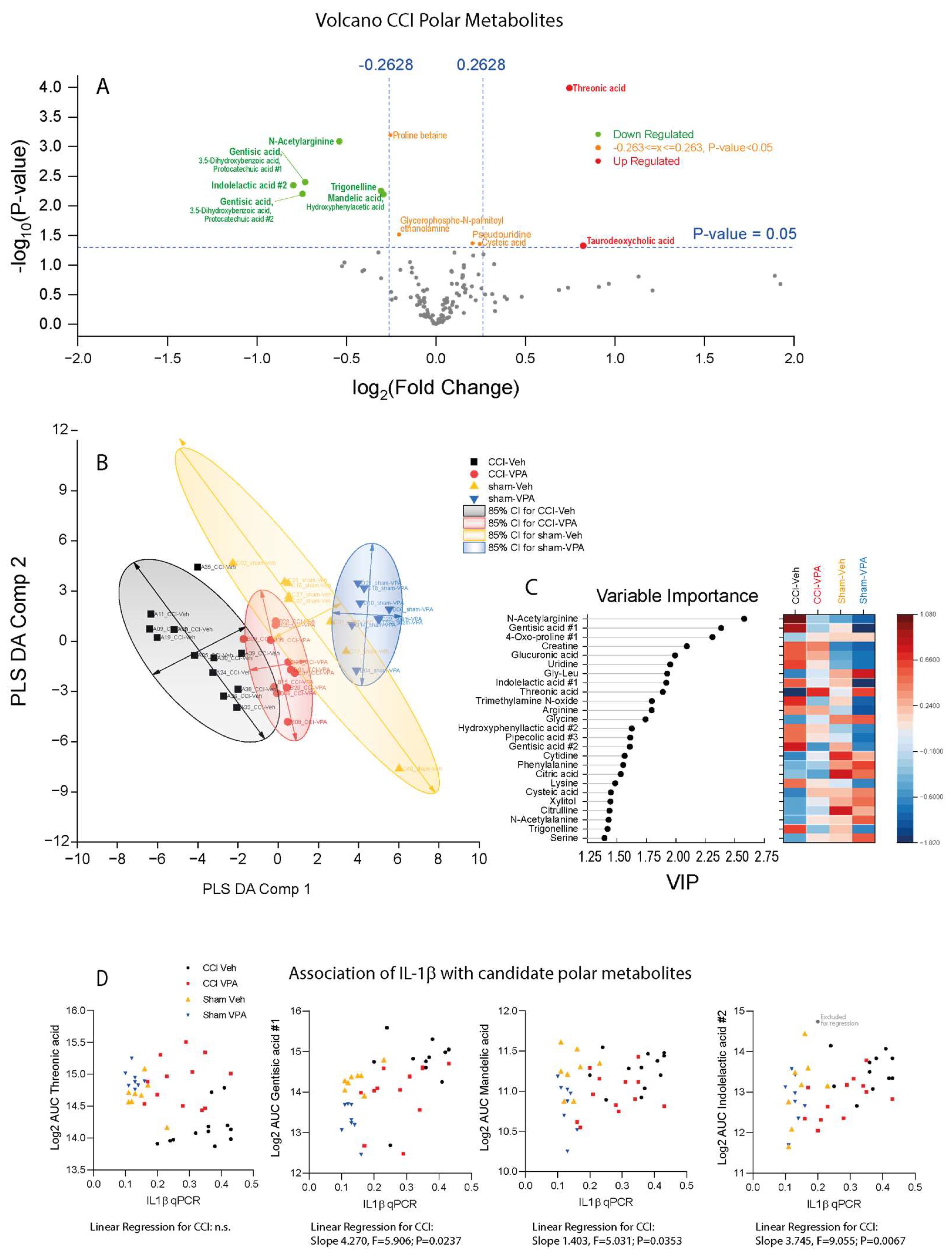
3.7. VPA Treatment Is Associated with High Threonic Acid and Low Gentisic Acid
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bramlett, H.M.; Dietrich, W.D. Long-Term Consequences of Traumatic Brain Injury: Current Status of Potential Mechanisms of Injury and Neurological Outcomes. J. Neurotrauma 2015, 32, 1834–1848. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, M.K.; Pfeiffer, A.; Jaeckel, M.; Pouya, A.; Dolga, A.M.; Methner, A. Regulators of mitochondrial Ca2+ homeostasis in cerebral ischemia. Cell Tissue Res. 2014, 357, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Cash, A.; Theus, M.H. Mechanisms of Blood-Brain Barrier Dysfunction in Traumatic Brain Injury. Int. J. Mol. Sci. 2020, 21, 3344. [Google Scholar] [CrossRef] [PubMed]
- Dinet, V.; Petry, K.G.; Badaut, J. Brain-Immune Interactions and Neuroinflammation After Traumatic Brain Injury. Front. Neurosci. 2019, 13, 1178. [Google Scholar] [CrossRef] [PubMed]
- Risbrough, V.B.; Vaughn, M.N.; Friend, S.F. Role of Inflammation in Traumatic Brain Injury-Associated Risk for Neuropsychiatric Disorders: State of the Evidence and Where Do We Go From Here. Biol. Psychiatry 2022, 91, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Eastman, C.L.; D’Ambrosio, R.; Ganesh, T. Modulating neuroinflammation and oxidative stress to prevent epilepsy and improve outcomes after traumatic brain injury. Neuropharmacology 2020, 172, 107907. [Google Scholar] [CrossRef] [PubMed]
- Begemann, M.; Leon, M.; van der Horn, H.J.; van der Naalt, J.; Sommer, I. Drugs with anti-inflammatory effects to improve outcome of traumatic brain injury: A meta-analysis. Sci. Rep. 2020, 10, 16179. [Google Scholar] [CrossRef] [PubMed]
- Helmy, A.; Guilfoyle, M.R.; Carpenter, K.L.; Pickard, J.D.; Menon, D.K.; Hutchinson, P.J. Recombinant human interleukin-1 receptor antagonist in severe traumatic brain injury: A phase II randomized control trial. J. Cereb. Blood Flow Metab. 2014, 34, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Boghdadi, A.G.; Teo, L.; Bourne, J.A. The Neuroprotective Role of Reactive Astrocytes after Central Nervous System Injury. J. Neurotrauma 2020, 37, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Krukowski, K.; Nolan, A.; Becker, M.; Picard, K.; Vernoux, N.; Frias, E.S.; Feng, X.; Tremblay, M.E.; Rosi, S. Novel microglia-mediated mechanisms underlying synaptic loss and cognitive impairment after traumatic brain injury. Brain Behav. Immun. 2021, 98, 122–135. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wernersbach, I.; Strehle, J.; Li, S.; Appel, D.; Klein, M.; Ritter, K.; Hummel, R.; Tegeder, I.; Schäfer, M.K.E. Early posttraumatic CSF1R inhibition via PLX3397 leads to time- and sex-dependent effects on inflammation and neuronal maintenance after traumatic brain injury in mice. Brain Behav. Immun. 2022, 106, 49–66. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.W.; McGeachy, M.J.; Bayır, H.; Clark, R.S.B.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 572. [Google Scholar] [CrossRef] [PubMed]
- Ghiam, M.K.; Patel, S.D.; Hoffer, A.; Selman, W.R.; Hoffer, B.J.; Hoffer, M.E. Drug Repurposing in the Treatment of Traumatic Brain Injury. Front. Neurosci. 2021, 15, 635483. [Google Scholar] [CrossRef] [PubMed]
- Stein, D.G.; Sayeed, I. Repurposing and repositioning neurosteroids in the treatment of traumatic brain injury: A report from the trenches. Neuropharmacology 2019, 147, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Romoli, M.; Mazzocchetti, P.; D’Alonzo, R.; Siliquini, S.; Rinaldi, V.E.; Verrotti, A.; Calabresi, P.; Costa, C. Valproic Acid and Epilepsy: From Molecular Mechanisms to Clinical Evidences. Curr. Neuropharmacol. 2019, 17, 926–946. [Google Scholar] [CrossRef] [PubMed]
- Pathak, S.M.; Ziechmann, R.; Menzer, J.; Hoeft, A.; Villanueva, P. Discontinuation of Levetiracetam and Valproic Acid Due to Adverse Effects in Early Post-traumatic Seizure Prophylaxis. Cureus 2023, 15, e47742. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, U.F.; Karnovsky, A.; Dennahy, I.S.; Kachman, M.; Williams, A.M.; Nikolian, V.C.; Biesterveld, B.E.; Siddiqui, A.; O’Connell, R.L.; Liu, B.; et al. Pharmacologic modulation of brain metabolism by valproic acid can induce a neuroprotective environment. J. Trauma Acute Care Surg. 2021, 90, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Wakam, G.K.; Biesterveld, B.E.; Pai, M.P.; Kemp, M.T.; O’Connell, R.L.; Rajanayake, K.K.; Chtraklin, K.; Vercruysse, C.A.; Alam, H.B. A single dose of valproic acid improves neurologic recovery and decreases brain lesion size in swine subjected to an isolated traumatic brain injury. J. Trauma Acute Care Surg. 2021, 91, 867–871. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Wu, Y.; Li, Z.; Ye, L.; Lu, Q.; Zhou, Y.; Yuan, Y.; Jiang, T.; Xie, L.; Liu, Y.; et al. Valproic acid affects neuronal fate and microglial function via enhancing autophagic flux in mice after traumatic brain injury. J. Neurochem. 2020, 154, 284–300. [Google Scholar] [CrossRef] [PubMed]
- Dash, P.K.; Orsi, S.A.; Zhang, M.; Grill, R.J.; Pati, S.; Zhao, J.; Moore, A.N. Valproate administered after traumatic brain injury provides neuroprotection and improves cognitive function in rats. PLoS ONE 2010, 5, e11383. [Google Scholar] [CrossRef] [PubMed]
- Jepsen, C.H.; deMoya, M.A.; Perner, A.; Sillesen, M.; Ostrowski, S.R.; Alam, H.B.; Johansson, P.I. Effect of valproic acid and injury on lesion size and endothelial glycocalyx shedding in a rodent model of isolated traumatic brain injury. J. Trauma Acute Care Surg. 2014, 77, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Bertogliat, M.J.; Morris-Blanco, K.C.; Vemuganti, R. Epigenetic mechanisms of neurodegenerative diseases and acute brain injury. Neurochem. Int. 2020, 133, 104642. [Google Scholar] [CrossRef] [PubMed]
- Phiel, C.J.; Zhang, F.; Huang, E.Y.; Guenther, M.G.; Lazar, M.A.; Klein, P.S. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J. Biol. Chem. 2001, 276, 36734–36741. [Google Scholar] [CrossRef] [PubMed]
- Higgins, G.A.; Georgoff, P.; Nikolian, V.; Allyn-Feuer, A.; Pauls, B.; Higgins, R.; Athey, B.D.; Alam, H.E. Network Reconstruction Reveals that Valproic Acid Activates Neurogenic Transcriptional Programs in Adult Brain Following Traumatic Injury. Pharm. Res. 2017, 34, 1658–1672. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, H.; Zhou, M.; Li, X.; Fang, Z.; Gao, H.; Li, Y.; Hu, W. Valproic Acid Attenuates Traumatic Brain Injury-Induced Inflammation in Vivo: Involvement of Autophagy and the Nrf2/ARE Signaling Pathway. Front. Mol. Neurosci. 2018, 11, 117. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhu, M.; Yu, Y.; Qiu, L.; Zhang, Y.; He, L.; Zhang, J. Brain REST/NRSF Is Not Only a Silent Repressor but Also an Active Protector. Mol. Neurobiol. 2017, 54, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Baldelli, P.; Meldolesi, J. The Transcription Repressor REST in Adult Neurons: Physiology, Pathology, and Diseases. Eneuro 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Wartman, C.; VandenBerg, A. Valproate: Not All Boxed Warnings Are Created Equal. Ann. Pharmacother. 2022, 56, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Chen, Y.; Ma, Y.; Liu, T.; Zhao, M.; Wang, Z.; Zhao, L. Lipidomic Profiling Reveals Disruption of Lipid Metabolism in Valproic Acid-Induced Hepatotoxicity. Front. Pharmacol. 2019, 10, 819. [Google Scholar] [CrossRef] [PubMed]
- Huo, T.; Chen, X.; Lu, X.; Qu, L.; Liu, Y.; Cai, S. An effective assessment of valproate sodium-induced hepatotoxicity with UPLC-MS and (1)HNMR-based metabonomics approach. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 969, 109–116. [Google Scholar] [CrossRef]
- Hahnefeld, L.; Vogel, A.; Gurke, R.; Geisslinger, G.; Schäfer, M.K.E.; Tegeder, I. Phosphatidylethanolamine Deficiency and Triglyceride Overload in Perilesional Cortex Contribute to Non-Goal-Directed Hyperactivity after Traumatic Brain Injury in Mice. Biomedicines 2022, 10, 914. [Google Scholar] [CrossRef] [PubMed]
- Pulliam, A.N.; Gier, E.C.; Gaul, D.A.; Moore, S.G.; Fernández, F.M.; LaPlaca, M.C. Comparing Brain and Blood Lipidome Changes following Single and Repetitive Mild Traumatic Brain Injury in Rats. ACS Chem. Neurosci. 2024, 15, 300–314. [Google Scholar] [CrossRef] [PubMed]
- Pulliam, A.N.; Pybus, A.F.; Gaul, D.A.; Moore, S.G.; Wood, L.B.; Fernández, F.M.; LaPlaca, M.C. Integrative Analysis of Cytokine and Lipidomics Datasets Following Mild Traumatic Brain Injury in Rats. Metabolites 2024, 14, 133. [Google Scholar] [CrossRef] [PubMed]
- Pasvogel, A.E.; Miketova, P.; Moore, I.M. Differences in CSF phospholipid concentration by traumatic brain injury outcome. Biol. Res. Nurs. 2010, 11, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Thomas, I.; Dickens, A.M.; Posti, J.P.; Czeiter, E.; Duberg, D.; Sinioja, T.; Kråkström, M.; Retel Helmrich, I.R.A.; Wang, K.K.W.; Maas, A.I.R.; et al. Serum metabolome associated with severity of acute traumatic brain injury. Nat. Commun. 2022, 13, 2545. [Google Scholar] [CrossRef] [PubMed]
- Palafox-Sánchez, V.; Ying, Z.; Royes, L.F.F.; Gomez-Pinilla, F. The interaction between brain and liver regulates lipid metabolism in the TBI pathology. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166078. [Google Scholar] [CrossRef] [PubMed]
- Gölz, C.; Kirchhoff, F.P.; Westerhorstmann, J.; Schmidt, M.; Hirnet, T.; Rune, G.M.; Bender, R.A.; Schäfer, M.K.E. Sex hormones modulate pathogenic processes in experimental traumatic brain injury. J. Neurochem. 2019, 150, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Ohdo, S.; Nakano, S.; Ogawa, N. Chronotoxicity of sodium valproate and its mechanisms in mice: Dose-concentration-response relationship. Chronobiol. Int. 1989, 6, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Hummel, R.; Ulbrich, S.; Appel, D.; Li, S.; Hirnet, T.; Zander, S.; Bobkiewicz, W.; Gölz, C.; Schäfer, M.K.E. Administration of all-trans retinoic acid after experimental traumatic brain injury is brain protective. Br. J. Pharmacol. 2020, 177, 5208–5223. [Google Scholar] [CrossRef] [PubMed]
- Krämer, T.; Grob, T.; Menzel, L.; Hirnet, T.; Griemert, E.; Radyushkin, K.; Thal, S.C.; Methner, A.; Schäfer, M.K.E. Dimethyl fumarate treatment after traumatic brain injury prevents depletion of antioxidative brain glutathione and confers neuroprotection. J. Neurochem. 2017, 143, 523–533. [Google Scholar] [CrossRef]
- Sens, A.; Rischke, S.; Hahnefeld, L.; Dorochow, E.; Schäfer, S.M.G.; Thomas, D.; Köhm, M.; Geisslinger, G.; Behrens, F.; Gurke, R. Pre-analytical sample handling standardization for reliable measurement of metabolites and lipids in LC-MS-based clinical research. J. Mass. Spectrom. Adv. Clin. Lab. 2023, 28, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Dieterle, F.; Ross, A.; Schlotterbeck, G.; Senn, H. Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in 1H NMR metabonomics. Anal. Chem. 2006, 78, 4281–4290. [Google Scholar] [CrossRef] [PubMed]
- Schaible, E.V.; Windschugl, J.; Bobkiewicz, W.; Kaburov, Y.; Dangel, L.; Kramer, T.; Huang, C.; Sebastiani, A.; Luh, C.; Werner, C.; et al. 2-Methoxyestradiol confers neuroprotection and inhibits a maladaptive HIF-1alpha response after traumatic brain injury in mice. J. Neurochem. 2014, 129, 940–954. [Google Scholar] [CrossRef] [PubMed]
- Menzel, L.; Kleber, L.; Friedrich, C.; Hummel, R.; Dangel, L.; Winter, J.; Schmitz, K.; Tegeder, I.; Schafer, M.K. Progranulin protects against exaggerated axonal injury and astrogliosis following traumatic brain injury. Glia 2017, 65, 278–292. [Google Scholar] [CrossRef] [PubMed]
- Thal, S.C.; Wyschkon, S.; Pieter, D.; Engelhard, K.; Werner, C. Selection of endogenous control genes for normalization of gene expression analysis after experimental brain trauma in mice. J. Neurotrauma 2008, 25, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Zhou, G.; Ewald, J.; Chang, L.; Hacariz, O.; Basu, N.; Xia, J. Using MetaboAnalyst 5.0 for LC-HRMS spectra processing, multi-omics integration and covariate adjustment of global metabolomics data. Nat. Protoc. 2022, 17, 1735–1761. [Google Scholar] [CrossRef]
- Kim, S.J.; Lee, B.H.; Lee, Y.S.; Kang, K.S. Defective cholesterol traffic and neuronal differentiation in neural stem cells of Niemann-Pick type C disease improved by valproic acid, a histone deacetylase inhibitor. Biochem. Biophys. Res. Commun. 2007, 360, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Schoenherr, C.J.; Paquette, A.J.; Anderson, D.J. Identification of potential target genes for the neuron-restrictive silencer factor. Proc. Natl. Acad. Sci. USA 1996, 93, 9881–9886. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zhao, L.; Guo, Z.; Hou, Y.; Jiang, J.; Song, Y. Valproate Sodium Protects Blood Brain Barrier Integrity in Intracerebral Hemorrhage Mice. Oxidative Med. Cell. Longev. 2020, 2020, 8884320. [Google Scholar] [CrossRef]
- Vogel, A.; Wilken-Schmitz, A.; Hummel, R.; Lang, M.; Gurke, R.; Schreiber, Y.; Schäfer, M.K.E.; Tegeder, I. Low brain endocannabinoids associated with persistent non-goal directed nighttime hyperactivity after traumatic brain injury in mice. Sci. Rep. 2020, 10, 14929. [Google Scholar] [CrossRef]
- Huang, C.; Sakry, D.; Menzel, L.; Dangel, L.; Sebastiani, A.; Kramer, T.; Karram, K.; Engelhard, K.; Trotter, J.; Schafer, M.K. Lack of NG2 exacerbates neurological outcome and modulates glial responses after traumatic brain injury. Glia 2016, 64, 507–523. [Google Scholar] [CrossRef] [PubMed]
- Timaru-Kast, R.; Luh, C.; Gotthardt, P.; Huang, C.; Schafer, M.K.; Engelhard, K.; Thal, S.C. Influence of age on brain edema formation, secondary brain damage and inflammatory response after brain trauma in mice. PLoS ONE 2012, 7, e43829. [Google Scholar] [CrossRef] [PubMed]
- Colicos, M.A.; Dixon, C.E.; Dash, P.K. Delayed, selective neuronal death following experimental cortical impact injury in rats: Possible role in memory deficits. Brain Res. 1996, 739, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Soria-Castro, R.; Schcolnik-Cabrera, A.; Rodríguez-López, G.; Campillo-Navarro, M.; Puebla-Osorio, N.; Estrada-Parra, S.; Estrada-García, I.; Chacón-Salinas, R.; Chávez-Blanco, A.D. Exploring the Drug Repurposing Versatility of Valproic Acid as a Multifunctional Regulator of Innate and Adaptive Immune Cells. J. Immunol. Res. 2019, 2019, 9678098. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Cobos, S.N.; Bennett, S.A.; Torrente, M.P. The impact of histone post-translational modifications in neurodegenerative diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1982–1991. [Google Scholar] [CrossRef] [PubMed]
- Giallongo, S.; Longhitano, L.; Denaro, S.; D’Aprile, S.; Torrisi, F.; La Spina, E.; Giallongo, C.; Mannino, G.; Lo Furno, D.; Zappalà, A.; et al. The Role of Epigenetics in Neuroinflammatory-Driven Diseases. Int. J. Mol. Sci. 2022, 23, 15218. [Google Scholar] [CrossRef]
- Shein, N.A.; Shohami, E. Histone deacetylase inhibitors as therapeutic agents for acute central nervous system injuries. Mol. Med. 2011, 17, 448–456. [Google Scholar] [CrossRef]
- Warburton, A.; Savage, A.L.; Myers, P.; Peeney, D.; Bubb, V.J.; Quinn, J.P. Molecular signatures of mood stabilisers highlight the role of the transcription factor REST/NRSF. J. Affect. Disord. 2015, 172, 63–73. [Google Scholar] [CrossRef]
- Singh, D.; Gupta, S.; Verma, I.; Morsy, M.A.; Nair, A.B.; Ahmed, A.-S.F. Hidden pharmacological activities of valproic acid: A new insight. Biomed. Pharmacother. 2021, 142, 112021. [Google Scholar] [CrossRef]
- Lotfy, D.M.; Safar, M.M.; Mohamed, S.H.; Kenawy, S.A. Effect of valproic acid alone or combined with low dose gamma irradiation in modulating PTZ-induced convulsions in rats involving AKT/m-TOR pathway. Life Sci. 2018, 212, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.; Walker, M.C.; Williams, R.S. Seizure-induced reduction in PIP3 levels contributes to seizure-activity and is rescued by valproic acid. Neurobiol. Dis. 2014, 62, 296–306. [Google Scholar] [CrossRef]
- Kelly, E.; Sharma, D.; Wilkinson, C.J.; Williams, R.S.B. Diacylglycerol kinase (DGKA) regulates the effect of the epilepsy and bipolar disorder treatment valproic acid in Dictyostelium discoideum. Dis. Models Mech. 2018, 11, dmm035600. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, S.; Russo, S.; Cowart, L.A.; Greenberg, M.L. Inositol Depletion Induced by Acute Treatment of the Bipolar Disorder Drug Valproate Increases Levels of Phytosphingosine. J. Biol. Chem. 2017, 292, 4953–4959. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Qin, X.; Liang, X.; Liu, M.; Zhang, X. Lipidomic characteristics and clinical findings of epileptic patients treated with valproic acid. J. Cell Mol. Med. 2019, 23, 6017–6023. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.L.; Wang, W.J.; Dong, N.; Zhao, Y.T.; Dai, H.R.; Hu, Y.H.; Zhang, Y.Y.; Wang, J.; Qiu, J.C.; Lu, X.P.; et al. Integrating metabolomics and lipidomics revealed a decrease in plasma fatty acids but an increase in triglycerides in children with drug-refractory epilepsy. Epilepsia Open 2023, 8, 466–478. [Google Scholar] [CrossRef]
- Brunkhorst-Kanaan, N.; Klatt-Schreiner, K.; Hackel, J.; Schroter, K.; Trautmann, S.; Hahnefeld, L.; Wicker, S.; Reif, A.; Thomas, D.; Geisslinger, G.; et al. Targeted lipidomics reveal derangement of ceramides in major depression and bipolar disorder. Metabolism 2019, 95, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Nessel, I.; Michael-Titus, A.T. Lipid profiling of brain tissue and blood after traumatic brain injury: A review of human and experimental studies. Semin. Cell Dev. Biol. 2021, 112, 145–156. [Google Scholar] [CrossRef]
- Esposito, E.; Cordaro, M.; Cuzzocrea, S. Roles of fatty acid ethanolamides (FAE) in traumatic and ischemic brain injury. Pharmacol. Res. 2014, 86, 26–31. [Google Scholar] [CrossRef]
- Plemel, J.R.; Michaels, N.J.; Weishaupt, N.; Caprariello, A.V.; Keough, M.B.; Rogers, J.A.; Yukseloglu, A.; Lim, J.; Patel, V.V.; Rawji, K.S.; et al. Mechanisms of lysophosphatidylcholine-induced demyelination: A primary lipid disrupting myelinopathy. Glia 2018, 66, 327–347. [Google Scholar] [CrossRef] [PubMed]
- Scholz, H.; Eder, C. Lysophosphatidylcholine activates caspase-1 in microglia via a novel pathway involving two inflammasomes. J. Neuroimmunol. 2017, 310, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Adibhatla, R.M.; Hatcher, J.F. Altered lipid metabolism in brain injury and disorders. Subcell. Biochem. 2008, 49, 241–268. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Ko, H.J.; Song, D.K.; Jung, Y.J. Lysophosphatidylcholine Promotes Phagosome Maturation and Regulates Inflammatory Mediator Production Through the Protein Kinase A-Phosphatidylinositol 3 Kinase-p38 Mitogen-Activated Protein Kinase Signaling Pathway During Mycobacterium tuberculosis Infection in Mouse Macrophages. Front. Immunol. 2018, 9, 920. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, C.E.; Bergmann, A. The Sound of Silence: Signaling by Apoptotic Cells. Curr. Top. Dev. Biol. 2015, 114, 241–265. [Google Scholar] [CrossRef] [PubMed]
- Cunha, M.I.; Su, M.; Cantuti-Castelvetri, L.; Müller, S.A.; Schifferer, M.; Djannatian, M.; Alexopoulos, I.; van der Meer, F.; Winkler, A.; van Ham, T.J.; et al. Pro-inflammatory activation following demyelination is required for myelin clearance and oligodendrogenesis. J. Exp. Med. 2020, 217, e20191390. [Google Scholar] [CrossRef] [PubMed]
- Lo Van, A.; Bernoud-Hubac, N.; Lagarde, M. Esterification of Docosahexaenoic Acid Enhances Its Transport to the Brain and Its Potential Therapeutic Use in Brain Diseases. Nutrients 2022, 14, 4550. [Google Scholar] [CrossRef] [PubMed]
- Georgoff, P.E.; Nikolian, V.C.; Bonham, T.; Pai, M.P.; Tafatia, C.; Halaweish, I.; To, K.; Watcharotone, K.; Parameswaran, A.; Luo, R.; et al. Safety and Tolerability of Intravenous Valproic Acid in Healthy Subjects: A Phase I Dose-Escalation Trial. Clin. Pharmacokinet. 2018, 57, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Alam, H.B.; Shuja, F.; Butt, M.U.; Duggan, M.; Li, Y.; Zacharias, N.; Fukudome, E.Y.; Liu, B.; Demoya, M.; Velmahos, G.C. Surviving blood loss without blood transfusion in a swine poly-trauma model. Surgery 2009, 146, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W. Serum protein binding and pharmacokinetics of valproate in man, dog, rat and mouse. J. Pharmacol. Exp. Ther. 1978, 204, 255–261. [Google Scholar] [PubMed]
- Methaneethorn, J. A systematic review of population pharmacokinetics of valproic acid. Br. J. Clin. Pharmacol. 2018, 84, 816–834. [Google Scholar] [CrossRef] [PubMed]
- Cornford, E.M.; Diep, C.P.; Pardridge, W.M. Blood–Brain Barrier Transport of Valproic Acid. J. Neurochem. 1985, 44, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Kühne, M.; Kretzer, C.; Lindemann, H.; Godmann, M.; Heinze, T.; Werz, O.; Heinzel, T. Biocompatible valproic acid-coupled nanoparticles attenuate lipopolysaccharide-induced inflammation. Int. J. Pharm. 2021, 601, 120567. [Google Scholar] [CrossRef] [PubMed]
- Ghodke-Puranik, Y.; Thorn, C.F.; Lamba, J.K.; Leeder, J.S.; Song, W.; Birnbaum, A.K.; Altman, R.B.; Klein, T.E. Valproic acid pathway: Pharmacokinetics and pharmacodynamics. Pharmacogenet. Genom. 2013, 23, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Ezhilarasan, D.; Mani, U. Valproic acid induced liver injury: An insight into molecular toxicological mechanism. Environ. Toxicol. Pharmacol. 2022, 95, 103967. [Google Scholar] [CrossRef] [PubMed]
- Mallah, K.; Quanico, J.; Raffo-Romero, A.; Cardon, T.; Aboulouard, S.; Devos, D.; Kobeissy, F.; Zibara, K.; Salzet, M.; Fournier, I. Matrix-Assisted Laser Desorption/Ionization-Mass Spectrometry Imaging of Lipids in Experimental Model of Traumatic Brain Injury Detecting Acylcarnitines as Injury Related Markers. Anal. Chem. 2019, 91, 11879–11887. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.G.W.; Ro, Y.S.; Jung, E.; Moon, S.B.; Park, G.J.; Yoon, H.; Park, J.H.; Shin, S.D. Serum Acylcarnitine and Long-Term Functional Prognosis after Traumatic Brain Injury with Intracranial Injury: A Multi-Center Prospective Study. J. Neurotrauma 2023, 40, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Sales, S.; Graessler, J.; Ciucci, S.; Al-Atrib, R.; Vihervaara, T.; Schuhmann, K.; Kauhanen, D.; Sysi-Aho, M.; Bornstein, S.R.; Bickle, M.; et al. Gender, Contraceptives and Individual Metabolic Predisposition Shape a Healthy Plasma Lipidome. Sci. Rep. 2016, 6, 27710. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biological Processes | Neurotrophic Factors | Ion Channel/ Transporter Activity | Transcription Factors | Oligodendrocyte Differentiation | Synapse Plasticity /Organization | Apoptosis |
|---|---|---|---|---|---|---|
| GO terms | GO:0031547 | GO:0015075 GO:0005216 GO:0006811 | GO:0003700 | GO:0048709 GO:0048008 | GO:0051823 GO:0050808 | (GO:0097190) GO:0006915 |
| target genes | Bdnf Ntrk2 | Glutamate: Grin2a Gria1 Chloride: Slc12a5 Acetylcholine: Chrm4 | Egr1 Arc | Cspg4 Pdgfra | Syn1 Snap25 L1cam | Bax Bcl-2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hummel, R.; Dorochow, E.; Zander, S.; Ritter, K.; Hahnefeld, L.; Gurke, R.; Tegeder, I.; Schäfer, M.K.E. Valproic Acid Treatment after Traumatic Brain Injury in Mice Alleviates Neuronal Death and Inflammation in Association with Increased Plasma Lysophosphatidylcholines. Cells 2024, 13, 734. https://doi.org/10.3390/cells13090734
Hummel R, Dorochow E, Zander S, Ritter K, Hahnefeld L, Gurke R, Tegeder I, Schäfer MKE. Valproic Acid Treatment after Traumatic Brain Injury in Mice Alleviates Neuronal Death and Inflammation in Association with Increased Plasma Lysophosphatidylcholines. Cells. 2024; 13(9):734. https://doi.org/10.3390/cells13090734
Chicago/Turabian StyleHummel, Regina, Erika Dorochow, Sonja Zander, Katharina Ritter, Lisa Hahnefeld, Robert Gurke, Irmgard Tegeder, and Michael K. E. Schäfer. 2024. "Valproic Acid Treatment after Traumatic Brain Injury in Mice Alleviates Neuronal Death and Inflammation in Association with Increased Plasma Lysophosphatidylcholines" Cells 13, no. 9: 734. https://doi.org/10.3390/cells13090734