
Adult Human, but Not Rodent, Spermatogonial Stem Cells Retain States with a Foetal-like Signature

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Integrated scRNA-Seq Atlases of the Post-Pubescent Human Testes and Spermatogonial Stem Cells
2.2. scRNA-Seq Analysis of Non-Adult Human and Non-Human Samples
2.3. RNA Velocity Analysis
2.4. Enrichment and Impact Analyses
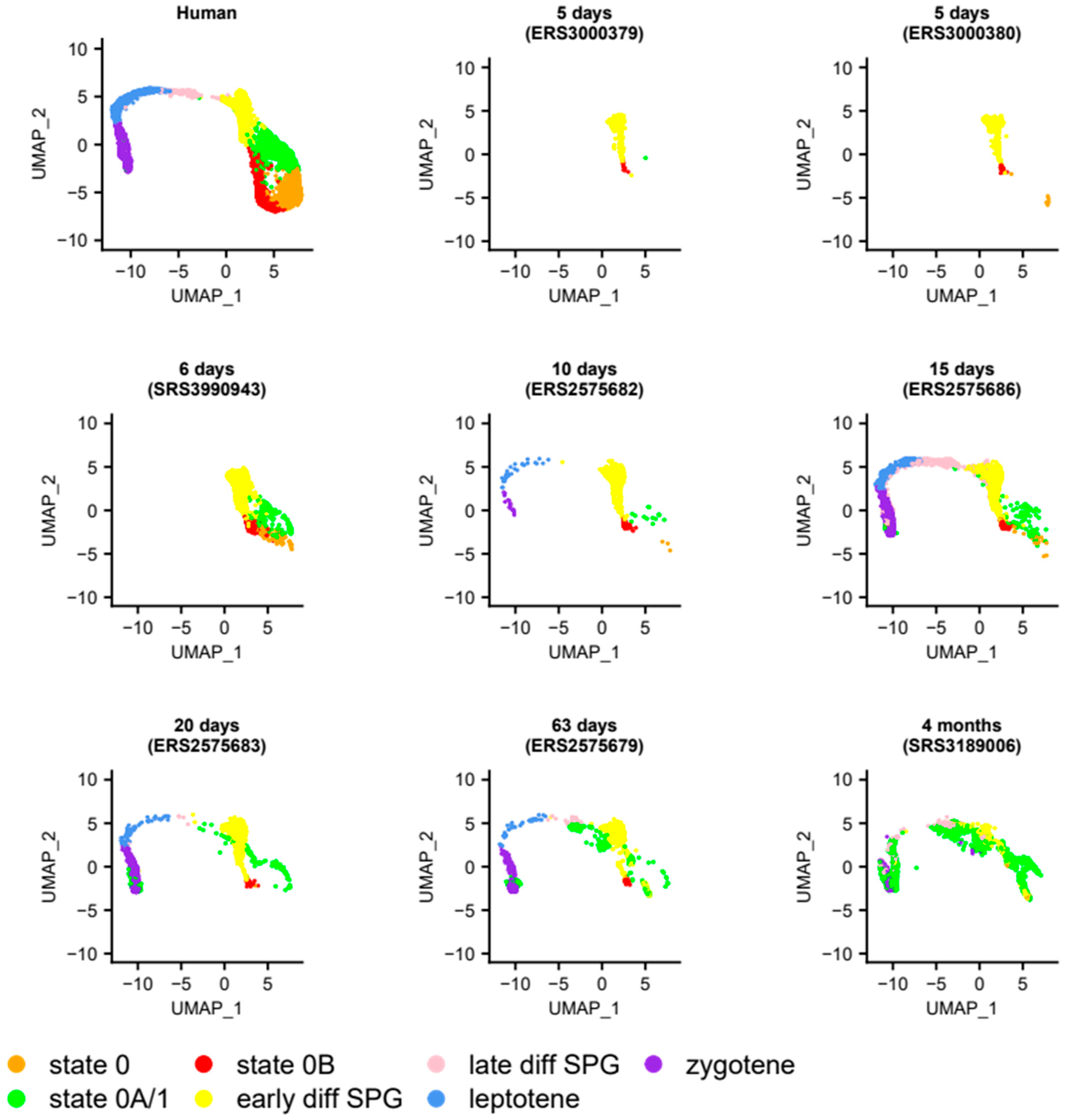
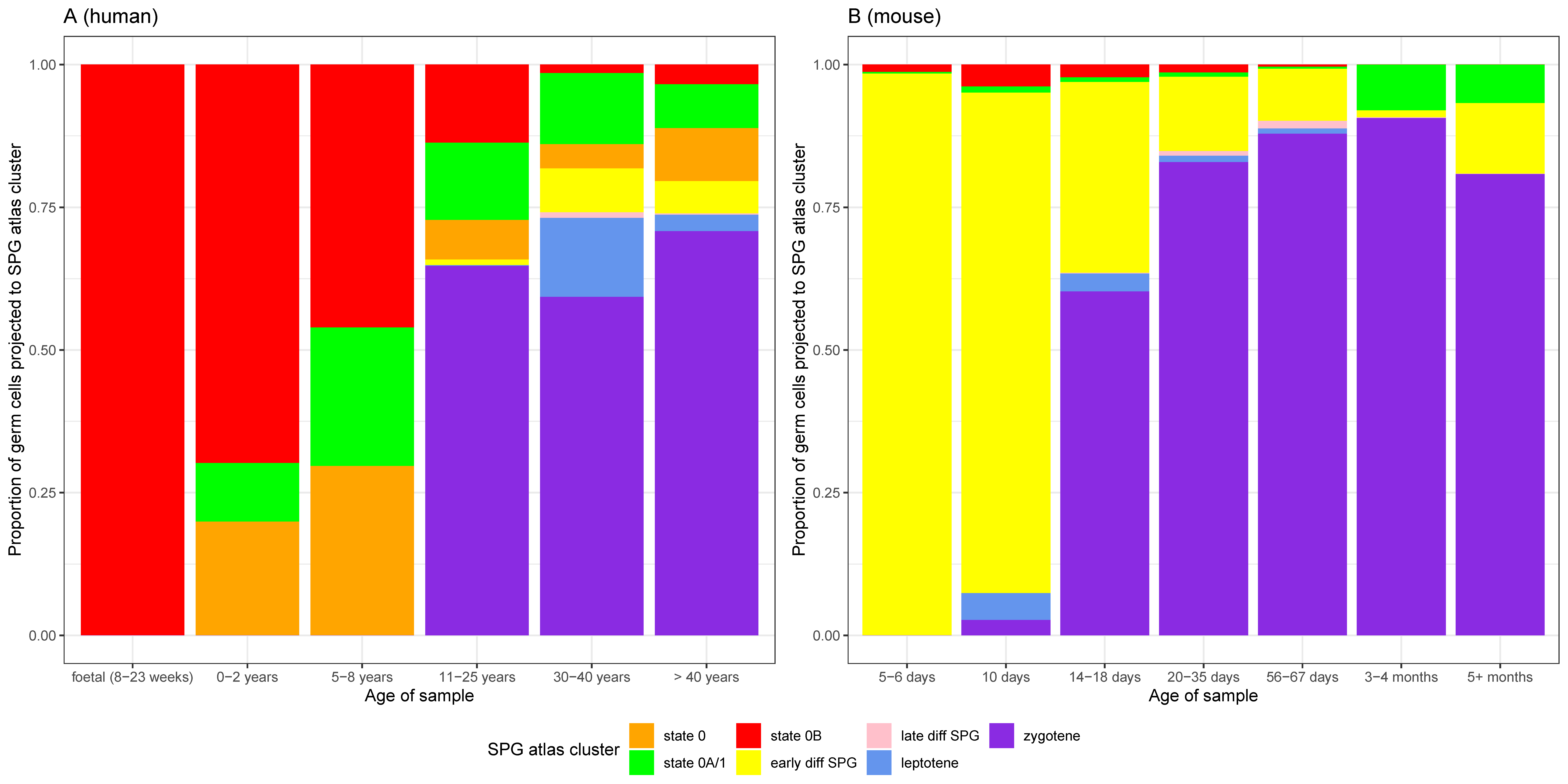
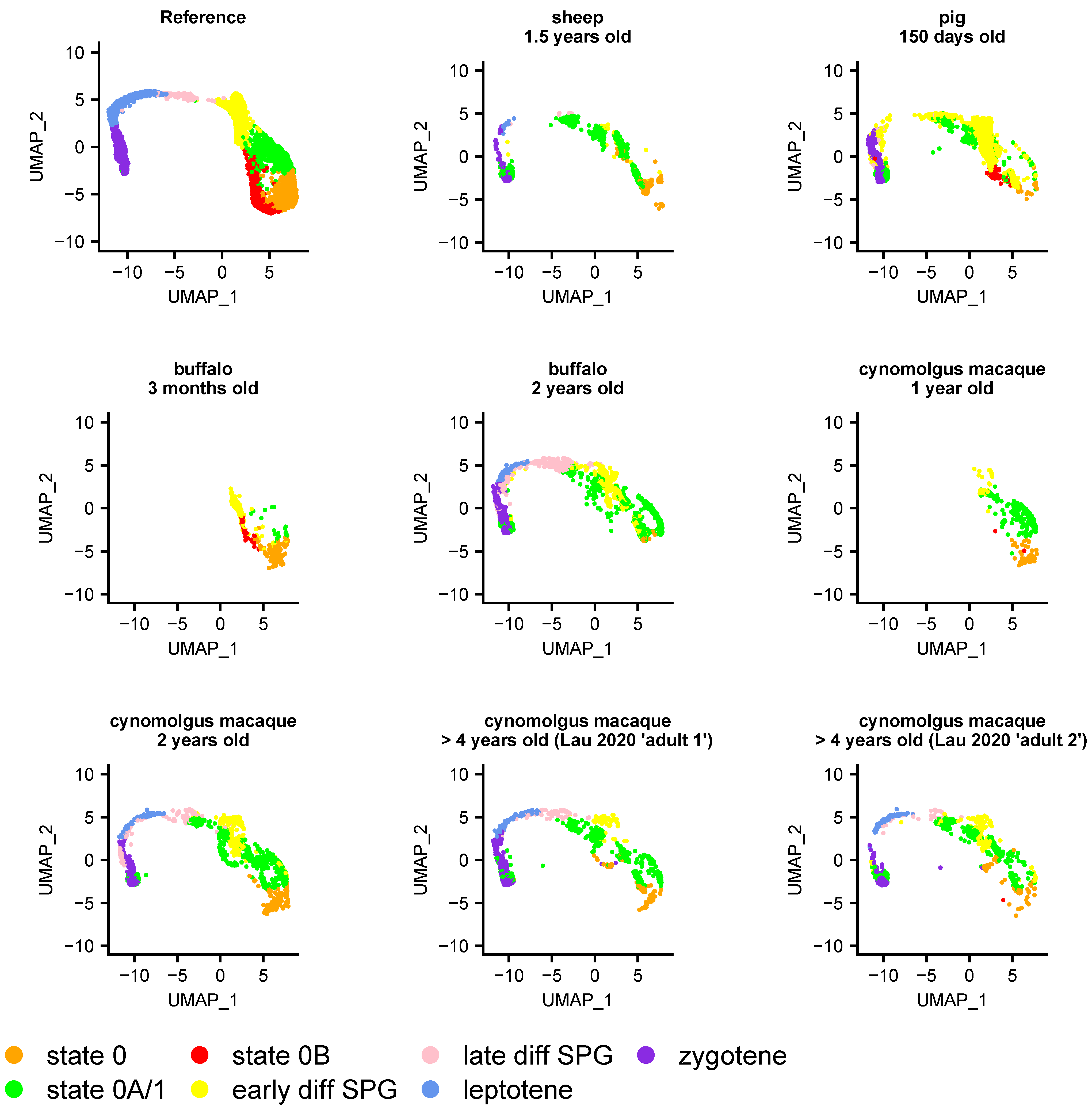
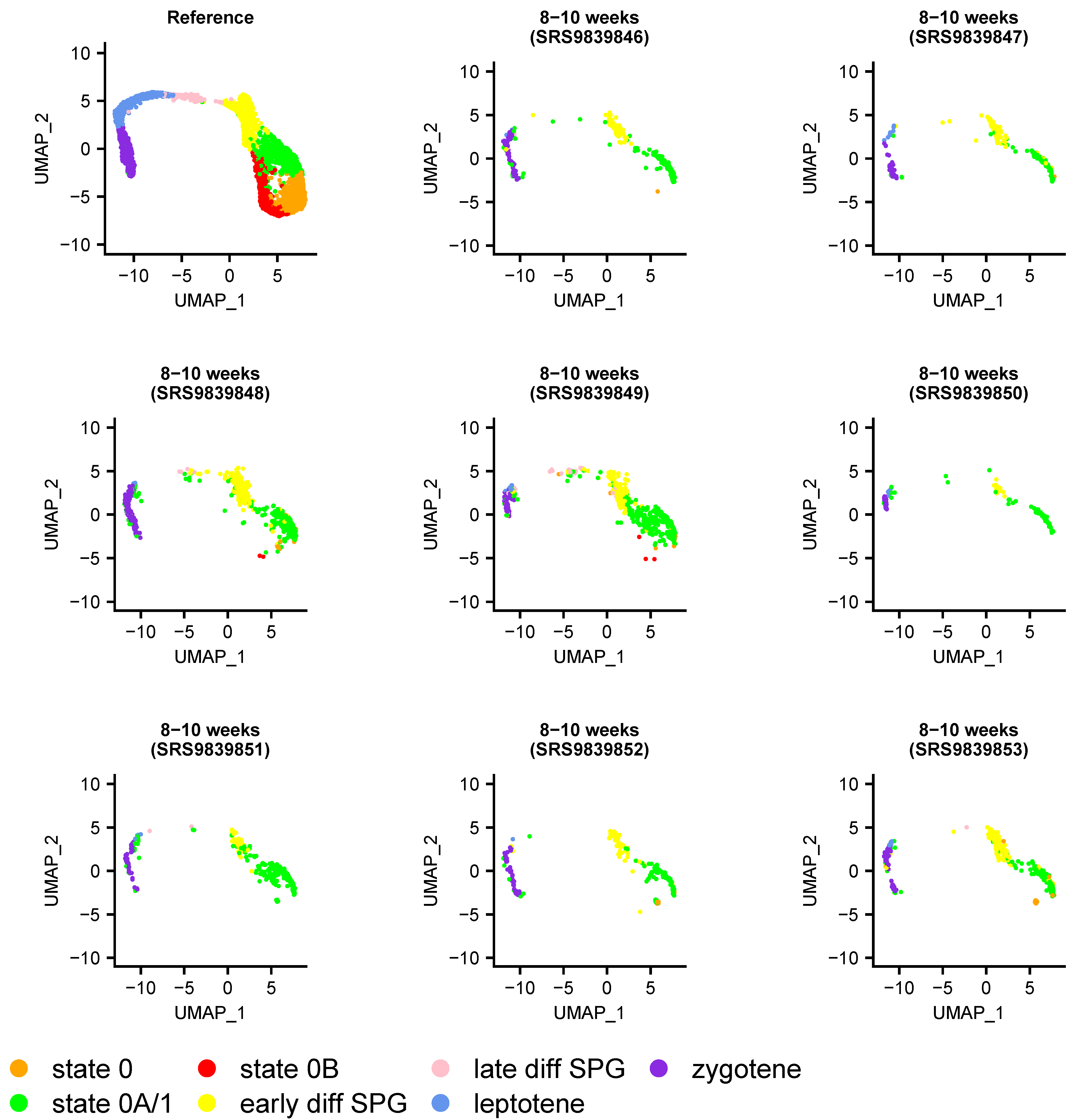
2.5. Projection of Individual Samples onto the SPG Atlas
3. Results and Discussion
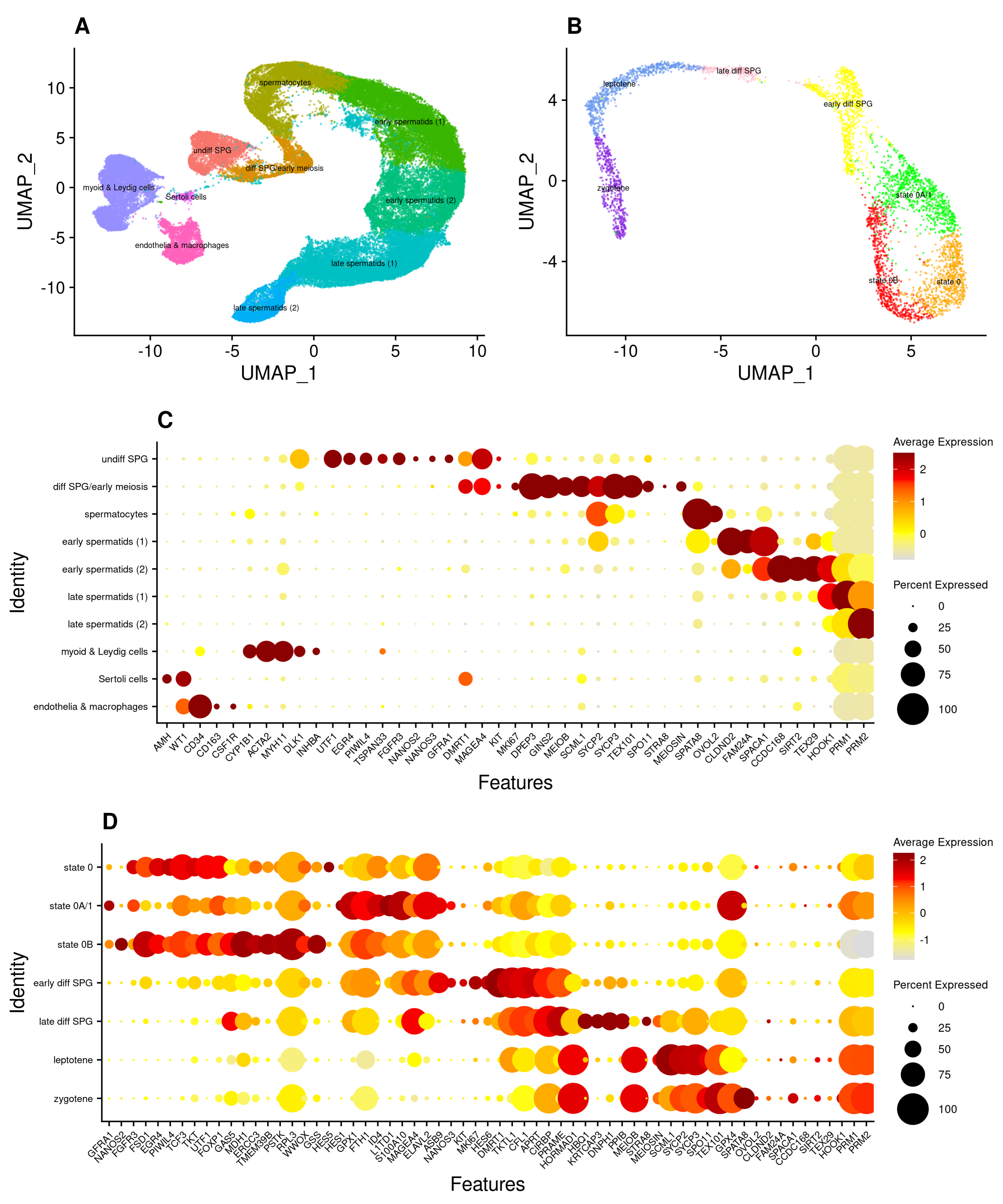
3.1. An scRNA-Seq Atlas of Adult Human Spermatogonial Stem Cells
3.2. Subpopulations of Undifferentiated Spermatogonia with Differing Metabolic Profiles
3.3. The ‘State 0’ or ‘SSC Reserve’ Cluster Has Little Transcriptomic Evidence of G0 Quiescence
3.4. The ‘State 0B’ Transcriptional Programme Is Characteristic of Foetal Germ Cells, Likely Suppresses Cell Proliferation, and Is Retained into Adulthood in Humans
3.5. Heterogeneity of Undifferentiated Spermatogonial States across Species and Development
3.6. General Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bergeron, L.A.; Besenbacher, S.; Zheng, J.; Li, P.; Bertelsen, M.F.; Quintard, B.; Hoffman, J.I.; Li, Z.; Leger, J.S.; Shao, C.; et al. Evolution of the germline mutation rate across vertebrates. Nature 2023, 615, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Seplyarskiy, V.B.; Sunyaev, S. The origin of human mutation in light of genomic data. Nat. Rev. Genet. 2021, 22, 672–686. [Google Scholar] [CrossRef] [PubMed]
- Bush, S.J.; Goriely, A. Fine-tuning germline mutation rates across evolution. Trends Genet. 2023, 39, 598–599. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Jörg, D.J.; Watanabe, H.; Mizuno, S.; Han, S.; Ikeda, T.; Omatsu, Y.; Nishimura, K.; Fujita, M.; Takahashi, S.; et al. A multistate stem cell dynamics maintains homeostasis in mouse spermatogenesis. Cell Rep. 2021, 37, 109875. [Google Scholar] [CrossRef]
- Singh, A.; Hermann, B.P. Conserved Transcriptome Features Define Prepubertal Primate Spermatogonial Stem Cells as Adark Spermatogonia and Identify Unique Regulators. Int. J. Mol. Sci. 2023, 24, 4755. [Google Scholar] [CrossRef]
- Fayomi, A.P.; Orwig, K.E. Spermatogonial stem cells and spermatogenesis in mice, monkeys and men. Stem Cell Res. 2018, 29, 207–214. [Google Scholar] [CrossRef]
- Clermont, Y. The cycle of the seminiferous epithelium in man. Am. J. Anat. 1963, 112, 35–51. [Google Scholar] [CrossRef]
- Heller, C.G.; Clermont, Y. Spermatogenesis in man: An estimate of its duration. Science 1963, 140, 184–186. [Google Scholar] [CrossRef]
- Clermont, Y. Spermatogenesis in man. A study of the spermatogonial population. Fertil. Steril. 1966, 17, 705–721. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Grow, E.J.; Mlcochova, H.; Maher, G.J.; Lindskog, C.; Nie, X.; Guo, Y.; Takei, Y.; Yun, J.; Cai, L.; et al. The adult human testis transcriptional cell atlas. Cell Res. 2018, 28, 1141–1157. [Google Scholar] [CrossRef]
- Guo, J.; Grow, E.J.; Yi, C.; Mlcochova, H.; Maher, G.J.; Lindskog, C.; Murphy, P.J.; Wike, C.L.; Carrell, D.T.; Goriely, A.; et al. Chromatin and Single-Cell RNA-Seq Profiling Reveal Dynamic Signaling and Metabolic Transitions during Human Spermatogonial Stem Cell Development. Cell Stem Cell 2017, 21, 533–546.e6. [Google Scholar] [CrossRef] [PubMed]
- von Kopylow, K.; Spiess, A.-N. Human spermatogonial markers. Stem Cell Res. 2017, 25, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Di Persio, S.; Saracino, R.; Fera, S.; Muciaccia, B.; Esposito, V.; Boitani, C.; Berloco, B.P.; Nudo, F.; Spadetta, G.; Stefanini, M.; et al. Spermatogonial kinetics in humans. Development 2017, 144, 3430–3439. [Google Scholar] [CrossRef] [PubMed]
- Di Persio, S.; Neuhaus, N. Human spermatogonial stem cells and their niche in male (in)fertility: Novel concepts from single-cell RNA-sequencing. Hum. Reprod. 2023, 38, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Di Persio, S.; Tekath, T.; Siebert-Kuss, L.M.; Cremers, J.F.; Wistuba, J.; Li, X.; Zu Hörste, G.M.; Drexler, H.C.; Wyrwoll, M.J.; Tüttelmann, F.; et al. Single-cell RNA-seq unravels alterations of the human spermatogonial stem cell compartment in patients with impaired spermatogenesis. Cell Rep. Med. 2021, 2, 100395. [Google Scholar] [CrossRef]
- Sohni, A.; Tan, K.; Song, H.-W.; Burow, D.; de Rooij, D.G.; Laurent, L.; Hsieh, T.-C.; Rabah, R.; Hammoud, S.S.; Vicini, E.; et al. The Neonatal and Adult Human Testis Defined at the Single-Cell Level. Cell Rep. 2019, 26, 1501.e4–1517.e4. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Nakagawa, T.; Enomoto, H.; Suzuki, M.; Yamamoto, M.; Simons, B.D.; Yoshida, S. Mouse spermatogenic stem cells continually interconvert between equipotent singly isolated and syncytial states. Cell Stem Cell 2014, 14, 658–672. [Google Scholar] [CrossRef] [PubMed]
- Goriely, A.; Wilkie, A.O. Paternal Age Effect mutations and selfish spermatogonial selection: Causes and consequences for human disease. Am. J. Hum. Genet. 2012, 90, 175–200. [Google Scholar] [CrossRef]
- Wood, K.A.; Goriely, A. The impact of paternal age on new mutations and disease in the next generation. Fertil. Steril. 2022, 118, 1001–1012. [Google Scholar] [CrossRef]
- Shami, A.N.; Zheng, X.; Munyoki, S.K.; Ma, Q.; Manske, G.L.; Green, C.D.; Sukhwani, M.; Orwig, K.E.; Li, J.Z.; Hammoud, S.S. Single-Cell RNA Sequencing of Human, Macaque, and Mouse Testes Uncovers Conserved and Divergent Features of Mammalian Spermatogenesis. Dev. Cell 2020, 54, 529.e12–547.e12. [Google Scholar] [CrossRef]
- Murat, F.; Mbengue, N.; Winge, S.B.; Trefzer, T.; Leushkin, E.; Sepp, M.; Cardoso-Moreira, M.; Schmidt, J.; Schneider, C.; Mößinger, K.; et al. The molecular evolution of spermatogenesis across mammals. Nature 2022, 613, 308–316. [Google Scholar] [CrossRef]
- Chen, S.; An, G.; Wang, H.; Wu, X.; Ping, P.; Hu, L.; Chen, Y.; Fan, J.; Cheng, C.Y.; Sun, F. Human obstructive (postvasectomy) and nonobstructive azoospermia—Insights from scRNA-Seq and transcriptome analysis. Genes Dis. 2022, 9, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Yao, C.; Xing, X.; Jing, T.; Li, P.; Zhu, Z.; Yang, C.; Zhai, J.; Tian, R.; Chen, H.; et al. Single-cell analysis of developing and azoospermia human testicles reveals central role of Sertoli cells. Nat. Commun. 2020, 11, 5683. [Google Scholar] [CrossRef] [PubMed]
- Hermann, B.P.; Cheng, K.; Singh, A.; Roa-De La Cruz, L.; Mutoji, K.N.; Chen, I.-C.; Gildersleeve, H.; Lehle, J.D.; Mayo, M.; Westernströer, B.; et al. The Mammalian Spermatogenesis Single-Cell Transcriptome, from Spermatogonial Stem Cells to Spermatids. Cell Rep. 2018, 25, 1650.e8–1667.e8. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Nie, X.; Giebler, M.; Mlcochova, H.; Wang, Y.; Grow, E.J.; DonorConnect; Kim, R.; Tharmalingam, M.; Matilionyte, G.; et al. The Dynamic Transcriptional Cell Atlas of Testis Development during Human Puberty. Cell Stem Cell 2020, 26, 262.e4–276.e4. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Munyoki, S.K.; Sukhwani, M.; Schmid, N.; Missel, A.; Emery, B.R.; Connect, D.; Stukenborg, J.-B.; Mayerhofer, A.; Orwig, K.E.; et al. Single-cell analysis of human testis aging and correlation with elevated body mass index. Dev. Cell 2022, 57, 1160.e5–1176.e5. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Sosa, E.; Chitiashvili, T.; Nie, X.; Rojas, E.J.; Oliver, E.; Plath, K.; Hotaling, J.M.; Stukenborg, J.-B.; Clark, A.T.; et al. Single-cell analysis of the developing human testis reveals somatic niche cell specification and fetal germline stem cell establishment. Cell Stem Cell 2021, 28, 764.e4–778.e4. [Google Scholar] [CrossRef]
- Voigt, A.L.; Dardari, R.; Su, L.; Lara, N.L.M.; Sinha, S.; Jaffer, A.; Munyoki, S.K.; Alpaugh, W.; Dufour, A.; Biernaskie, J.; et al. Metabolic transitions define spermatogonial stem cell maturation. Hum. Reprod. 2022, 37, 2095–2112. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Liu, X.; Li, L.; Yang, M.; Yong, J.; Zhai, F.; Wen, L.; Yan, L.; Qiao, J.; Tang, F. Dissecting Human Gonadal Cell Lineage Specification and Sex Determination Using a Single-cell RNA-seq Approach. Genom. Proteom. Bioinform. 2022, 20, 223–245. [Google Scholar] [CrossRef] [PubMed]
- Lau, X.; Munusamy, P.; Ng, M.J.; Sangrithi, M. Single-Cell RNA Sequencing of the Cynomolgus Macaque Testis Reveals Conserved Transcriptional Profiles during Mammalian Spermatogenesis. Dev. Cell 2020, 54, 548–566.e7. [Google Scholar] [CrossRef]
- Ernst, C.; Eling, N.; Martinez-Jimenez, C.P.; Marioni, J.C.; Odom, D.T. Staged developmental mapping and X chromo-some transcriptional dynamics during mouse spermatogenesis. Nat. Commun. 2019, 10, 1251. [Google Scholar] [CrossRef] [PubMed]
- Grive, K.J.; Hu, Y.; Shu, E.; Grimson, A.; Elemento, O.; Grenier, J.K.; Cohen, P.E. Dynamic transcriptome profiles within spermatogonial and spermatocyte populations during postnatal testis maturation revealed by single-cell sequencing. PLoS Genet. 2019, 15, e1007810. [Google Scholar] [CrossRef] [PubMed]
- Green, C.D.; Ma, Q.; Manske, G.L.; Shami, A.N.; Zheng, X.; Marini, S.; Moritz, L.; Sultan, C.; Gurczynski, S.J.; Moore, B.B.; et al. A Comprehensive Roadmap of Murine Spermatogenesis Defined by Single-Cell RNA-Seq. Dev. Cell 2018, 46, 651–667.e10. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Wells, D.; Rusch, J.; Ahmad, S.; Marchini, J.; Myers, S.R.; Conrad, D.F. Unified single-cell analysis of testis gene regulation and pathology in five mouse strains. eLife 2019, 8, e43966. [Google Scholar] [CrossRef] [PubMed]
- Whelan, E.C.; Yang, F.; Avarbock, M.R.; Sullivan, M.C.; Beiting, D.P.; Brinster, R.L. Reestablishment of spermatogenesis after more than 20 years of cryopreservation of rat spermatogonial stem cells reveals an important impact in differentiation capacity. PLoS Biol. 2022, 20, e3001618. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, F.; Lei, P.; Guo, M.; Liu, R.; Wang, L.; Yu, T.; Lv, Y.; Zhang, T.; Zeng, W.; et al. Single-cell RNA-sequencing reveals the dynamic process and novel markers in porcine spermatogenesis. J. Anim. Sci. Biotechnol. 2021, 12, 122. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ma, J.; Wan, Z.; Wang, Q.; Wang, Z.; Zhao, J.; Wang, F.; Zhang, Y. Characterization of sheep spermatogenesis through single-cell RNA sequencing. FASEB J. 2020, 35, e21187. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Zhang, J.; Zhang, P.; Huang, X.; Yang, W.; Liu, R.; Sun, Q.; Lu, Y.; Zhang, M.; Fu, Q. Single-cell RNA sequencing uncovers dynamic roadmap and cell-cell communication during buffalo spermatogenesis. iScience 2023, 26, 105733. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, N.; Yoon, J.; Terwort, N.; Kliesch, S.; Seggewiss, J.; Huge, A.; Voss, R.; Schlatt, S.; Grindberg, R.; Schöler, H. Single-cell gene expression analysis reveals diversity among human spermatogonia. Mol. Hum. Reprod. 2017, 23, 79–90. [Google Scholar] [CrossRef]
- Li, L.; Dong, J.; Yan, L.; Yong, J.; Liu, X.; Hu, Y.; Fan, X.; Wu, X.; Guo, H.; Wang, X.; et al. Single-Cell RNA-Seq Analysis Maps Development of Human Germline Cells and Gonadal Niche Interactions. Cell Stem Cell 2017, 20, 858.e4–873.e4. [Google Scholar] [CrossRef]
- Wang, M.; Liu, X.; Chang, G.; Chen, Y.; An, G.; Yan, L.; Gao, S.; Xu, Y.; Cui, Y.; Dong, J.; et al. Single-Cell RNA Sequencing Analysis Reveals Sequential Cell Fate Transition during Human Spermatogenesis. Cell Stem Cell 2018, 23, 599.e4–614.e4. [Google Scholar] [CrossRef] [PubMed]
- Picelli, S.; Bjorklund, A.K.; Faridani, O.R.; Sagasser, S.; Winberg, G.; Sandberg, R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 2013, 10, 1096–1098. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.; Song, H.-W.; Thompson, M.; Munyoki, S.; Sukhwani, M.; Hsieh, T.-C.; Orwig, K.E.; Wilkinson, M.F. Transcriptome profiling reveals signaling conditions dictating human spermatogonia fate in vitro. Proc. Natl. Acad. Sci. USA 2020, 117, 17832–17841. [Google Scholar] [CrossRef] [PubMed]
- Laurentino, S.; Heckmann, L.; Di Persio, S.; Li, X.; Meyer zu Hörste, G.; Wistuba, J.; Cremers, J.F.; Gromoll, J.; Kliesch, S.; Schlatt, S.; et al. High-resolution analysis of germ cells from men with sex chromosomal aneuploidies reveals normal transcriptome but impaired imprinting. Clin. Epigenet. 2019, 11, 127. [Google Scholar] [CrossRef]
- Melsted, P.; Booeshaghi, A.S.; Liu, L.; Gao, F.; Lu, L.; Min, K.H.; Beltrame, E.d.V.; Hjörleifsson, K.E.; Gehring, J.; Pachter, L. Modular, efficient and constant-memory single-cell RNA-seq preprocessing. Nat. Biotechnol. 2021, 39, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, R.J.; Kähäri, A.; Haider, S.; Zamora, J.; Proctor, G.; Spudich, G.; Almeida-King, J.; Staines, D.; Derwent, P.; Kerhornou, A.; et al. Ensembl BioMarts: A hub for data retrieval across taxonomic space. Database 2011, 2011, bar030. [Google Scholar] [CrossRef]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef]
- Hafemeister, C.; Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019, 20, 296. [Google Scholar] [CrossRef]
- Ahlmann-Eltze, C.; Huber, W. glmGamPoi: Fitting Gamma-Poisson generalized linear models on single cell count data. Bioinformatics 2020, 36, 5701–5702. [Google Scholar] [CrossRef]
- Dominguez, D.; Tsai, Y.-H.; Gomez, N.; Jha, D.K.; Davis, I.; Wang, Z. A high-resolution transcriptome map of cell cycle reveals novel connections between periodic genes and cancer. Cell Res. 2016, 26, 946–962. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Lu, P.; Wan, C.; Huang, Y.; Cui, M.; Yang, X.; Hu, Y.; Zheng, Y.; Dong, J.; Wang, M.; et al. Cell-fate transition and determination analysis of mouse male germ cells throughout development. Nat. Commun. 2021, 12, 6839. [Google Scholar] [CrossRef] [PubMed]
- McInnes, L.; Healy, J.; Melville, J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Waltman, L.; van Eck, N.J. A smart local moving algorithm for large-scale modularity-based community detection. Eur. Phys. J. B 2013, 86, 471. [Google Scholar] [CrossRef]
- Zappia, L.; Oshlack, A. Clustering trees: A visualization for evaluating clusterings at multiple resolutions. GigaScience 2018, 7, giy083. [Google Scholar] [CrossRef] [PubMed]
- Bergen, V.; Lange, M.; Peidli, S.; Wolf, F.A.; Theis, F.J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 2020, 38, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A.; Rahnenführer, J.; Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 2006, 22, 1600–1607. [Google Scholar] [CrossRef] [PubMed]
- Tarca, A.L.; Draghici, S.; Khatri, P.; Hassan, S.S.; Mittal, P.; Kim, J.-S.; Kim, C.J.; Kusanovic, J.P.; Romero, R. A novel signaling pathway impact analysis. Bioinformatics 2008, 25, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Mitrea, C.; Taghavi, Z.; Bokanizad, B.; Hanoudi, S.; Tagett, R.; Donato, M.; Voichiţa, C.; Drăghici, S. Methods and approaches in the topology-based analysis of biological pathways. Front. Physiol. 2013, 4, 278. [Google Scholar] [CrossRef]
- Suzuki, S.; Diaz, V.D.; Hermann, B.P. What has single-cell RNA-seq taught us about mammalian spermatogenesis? Biol. Reprod. 2019, 101, 617–634. [Google Scholar] [CrossRef] [PubMed]
- Patterson-Cross, R.B.; Levine, A.J.; Menon, V. Selecting single cell clustering parameter values using subsampling-based robustness metrics. BMC Bioinform. 2021, 22, 39. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, F.R.; Lepelley, A.; Seeley, J.J.; Hayden, M.S.; Ghosh, S. An Essential Role for ECSIT in Mitochondrial Complex I Assembly and Mitophagy in Macrophages. Cell Rep. 2018, 22, 2654–2666. [Google Scholar] [CrossRef] [PubMed]
- Kathagen-Buhmann, A.; Schulte, A.; Weller, J.; Holz, M.; Herold-Mende, C.; Glass, R.; Lamszus, K. Glycolysis and the pentose phosphate pathway are differentially associated with the dichotomous regulation of glioblastoma cell migration versus proliferation. Neuro-Oncology 2016, 18, 1219–1229. [Google Scholar] [CrossRef] [PubMed]
- van Velthoven, C.T.J.; Rando, T.A. Stem Cell Quiescence: Dynamism, Restraint, and Cellular Idling. Cell Stem Cell 2019, 24, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Llorens-Bobadilla, E.; Zhao, S.; Baser, A.; Saiz-Castro, G.; Zwadlo, K.; Martin-Villalba, A. Single-Cell Transcriptomics Reveals a Population of Dormant Neural Stem Cells that Become Acti-vated upon Brain Injury. Cell Stem Cell 2015, 17, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Simsek, T.; Kocabas, F.; Zheng, J.; DeBerardinis, R.J.; Mahmoud, A.I.; Olson, E.N.; Schneider, J.W.; Zhang, C.C.; Sadek, H.A. The Distinct Metabolic Profile of Hematopoietic Stem Cells Reflects Their Location in a Hypoxic Niche. Cell Stem Cell 2010, 7, 380–390. [Google Scholar] [CrossRef]
- Cheung, T.H.; Rando, T.A. Molecular regulation of stem cell quiescence. Nat. Rev. Mol. Cell Biol. 2013, 14, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Li, J. Quiescence regulators for hematopoietic stem cell. Exp. Hematol. 2011, 39, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Nakamura-Ishizu, A.; Takizawa, H.; Suda, T. The analysis, roles and regulation of quiescence in hematopoietic stem cells. Development 2014, 141, 4656–4666. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Mess, J.; Aizarani, N.; Mishra, P.; Johnson, C.; Romero-Mulero, M.C.; Rettkowski, J.; Schönberger, K.; Obier, N.; Jäcklein, K.; et al. Hyaluronic acid–GPRC5C signalling promotes dormancy in haematopoietic stem cells. Nature 2022, 24, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.-F.; Chen, W.S.C.; Chen, Y.-H.; Kao, T.-H.; Tseng, Y.-T.; Lee, C.-Y.; Chiu, Y.-C.; Lee, P.-L.; Lin, Q.-J.; Ching, Y.-H.; et al. DNMT3L promotes quiescence in postnatal spermatogonial progenitor cells. Development 2014, 141, 2402–2413. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.Y.; Cutler, S.; Lin, J.-J.; Tsai, C.-H.; Tsai, H.-K.; Biggins, S.; Tsukiyama, T.; Lo, Y.-C.; Kao, C.-F. H3K4 methylation at active genes mitigates transcription-replication conflicts during replication stress. Nat. Commun. 2020, 11, 809. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jin, C.; Li, P.; Li, Y.; Tang, J.; Yu, Z.; Jiao, T.; Ou, J.; Wang, H.; Zou, D.; et al. Identification of quiescent FOXC2(+) spermatogonial stem cells in adult mammals. eLife 2023, 12, RP85380. [Google Scholar] [CrossRef] [PubMed]
- Caldeira-Brant, A.L.; Martinelli, L.M.; Marques, M.M.; Reis, A.B.; Martello, R.; Almeida, F.R.C.L.; Chiarini-Garcia, H. A subpopulation of human Adark spermatogonia behaves as the reserve stem cell. Reproduction 2020, 159, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Dovey, S.L.; Valli, H.; Hermann, B.P.; Sukhwani, M.; Donohue, J.; Castro, C.A.; Chu, T.; Sanfilippo, J.S.; Orwig, K.E. Eliminating malignant contamination from therapeutic human spermatogonial stem cells. J. Clin. Investig. 2013, 123, 1833–1843. [Google Scholar] [CrossRef] [PubMed]
- Sada, A.; Hasegawa, K.; Pin, P.H.; Saga, Y. NANOS2 acts downstream of glial cell line-derived neurotrophic factor signal-ing to suppress differentiation of spermatogonial stem cells. Stem Cells 2012, 30, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Sada, A.; Suzuki, A.; Suzuki, H.; Saga, Y. The RNA-binding protein NANOS2 is required to maintain murine spermatogonial stem cells. Science 2009, 325, 1394–1398. [Google Scholar] [CrossRef]
- Pandey, V.; Tripathi, A.; Dubey, P.K. Expression and intracellular localization of NANOS2-homologue protein in primordial germ cells and spermatogonial stem cells. Zygote 2019, 27, 82–88. [Google Scholar] [CrossRef]
- Bowles, J.; Knight, D.; Smith, C.; Wilhelm, D.; Richman, J.; Mamiya, S.; Yashiro, K.; Chawengsaksophak, K.; Wilson, M.J.; Rossant, J.; et al. Retinoid Signaling Determines Germ Cell Fate in Mice. Science 2006, 312, 596–600. [Google Scholar] [CrossRef]
- Koubova, J.; Menke, D.B.; Zhou, Q.; Capel, B.; Griswold, M.D.; Page, D.C. Retinoic acid regulates sex-specific timing of meiotic initiation in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 2474–2479. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Katsuki, T.; Kokubo, H.; Masuda, A.; Saga, Y. Dazl is a target RNA suppressed by mammalian NANOS2 in sexually differentiating male germ cells. Nat. Commun. 2016, 7, 11272. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Igarashi, K.; Aisaki, K.-I.; Kanno, J.; Saga, Y. NANOS2 interacts with the CCR4-NOT deadenylation complex and leads to suppression of specific RNAs. Proc. Natl. Acad. Sci. USA 2010, 107, 3594–3599. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Saba, R.; Miyoshi, K.; Morita, Y.; Saga, Y. Interaction between NANOS2 and the CCR4-NOT deadenylation complex is essential for male germ cell development in mouse. PLoS ONE 2012, 7, e33558. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Niimi, Y.; Saga, Y. Interaction of NANOS2 and NANOS3 with different components of the CNOT complex may contribute to the functional differences in mouse male germ cells. Biol. Open 2014, 3, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Shimada, R.; Koike, H.; Hirano, T.; Kato, Y.; Saga, Y. NANOS2 suppresses the cell cycle by repressing mTORC1 activators in embryonic male germ cells. iScience 2021, 24, 102890. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Shirakawa, T.; Ohbo, K.; Sada, A.; Wu, Q.; Hasegawa, K.; Saba, R.; Saga, Y. RNA Binding Protein Nanos2 Organizes Post-transcriptional Buffering System to Retain Primitive State of Mouse Spermatogonial Stem Cells. Dev. Cell 2015, 34, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Lu, J.; Lou, N.; Lu, W.; Xu, J.; Jiang, H.; Ye, G. Long noncoding RNA GAS5 inhibits proliferation and metastasis in papillary thyroid carcinoma through the IFN/STAT1 signaling pathway. Pathol. Res. Pr. 2022, 233, 153856. [Google Scholar] [CrossRef]
- Lee, S.M.; Kim, J.H.; Cho, E.J.; Youn, H.D. A nucleocytoplasmic malate dehydrogenase regulates p53 transcriptional ac-tivity in response to metabolic stress. Cell Death Differ. 2009, 16, 738–748. [Google Scholar] [CrossRef]
- Qadri, I.; Iwahashi, M.; Simon, F. Hepatitis C virus NS5A protein binds TBP and p53, inhibiting their DNA binding and p53 interactions with TBP and ERCC3. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2002, 1592, 193–204. [Google Scholar] [CrossRef]
- Liu, R.; Long, Y.; Liu, R.; Song, G.; Li, Q.; Yan, H.; Cui, Z. Understanding the Function and Mechanism of Zebrafish Tmem39b in Regulating Cold Resistance. Int. J. Mol. Sci. 2022, 23, 11442. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Li, C.; Wang, S.; Cang, Y.; Song, Y.; Liu, X.; Li, X.; Mohan, C.; Wu, T.; Hu, D.; et al. PSTK is a novel gene associated with early lung injury in Paraquat Poisoning. Life Sci. 2015, 123, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Esposito, D.; Crescenzi, E.; Sagar, V.; Loreni, F.; Russo, A.; Russo, G. Human rpL3 plays a crucial role in cell response to nucleolar stress induced by 5-FU and L-OHP. Oncotarget 2014, 5, 11737–11751. [Google Scholar] [CrossRef]
- Abu-Odeh, M.; Salah, Z.; Herbel, C.; Hofmann, T.G.; Aqeilan, R.I. WWOX, the common fragile site FRA16D gene product, regulates ATM activation and the DNA damage response. Proc. Natl. Acad. Sci. USA 2014, 111, E4716–E4725. [Google Scholar] [CrossRef] [PubMed]
- Abu-Odeh, M.; Hereema, N.A.; Aqeilan, R.I. WWOX modulates the ATR-mediated DNA damage checkpoint response. Oncotarget 2015, 7, 4344–4355. [Google Scholar] [CrossRef] [PubMed]
- Geyer, C.B. Setting the Stage: The First Round of Spermatogenesis. In The Biology of Mammalian Spermatogonia; Oatley, J.M., Griswold, M.D., Eds.; Springer: New York, NY, USA, 2017; pp. 39–63. [Google Scholar]
- La, H.M.; Mäkelä, J.-A.; Chan, A.-L.; Rossello, F.J.; Nefzger, C.M.; Legrand, J.M.D.; De Seram, M.; Polo, J.M.; Hobbs, R.M. Identification of dynamic undifferentiated cell states within the male germline. Nat. Commun. 2018, 9, 2819. [Google Scholar] [CrossRef] [PubMed]
- Law, N.C.; Oatley, M.J.; Oatley, J.M. Developmental kinetics and transcriptome dynamics of stem cell specification in the spermatogenic lineage. Nat. Commun. 2019, 10, 2787. [Google Scholar] [CrossRef] [PubMed]
- McNamara, K.J. Heterochrony: The Evolution of Development. Evol. Educ. Outreach 2012, 5, 203–218. [Google Scholar] [CrossRef]
- Ramm, S.A.; Schärer, L.; Ehmcke, J.; Wistuba, J. Sperm competition and the evolution of spermatogenesis. Mol. Hum. Reprod. 2014, 20, 1169–1179. [Google Scholar] [CrossRef]
- Trumpp, A.; Essers, M.; Wilson, A. Awakening dormant haematopoietic stem cells. Nat. Rev. Immunol. 2010, 10, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Lillie, S.H.; Pringle, J.R. Reserve carbohydrate metabolism in Saccharomyces cerevisiae: Responses to nutrient limitation. J. Bacteriol. 1980, 143, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Otsuki, L.; Brand, A.H. Cell cycle heterogeneity directs the timing of neural stem cell activation from quiescence. Science 2018, 360, 99–102. [Google Scholar] [CrossRef]
- Otsuki, L.; Brand, A.H. Quiescent Neural Stem Cells for Brain Repair and Regeneration: Lessons from Model Systems. Trends Neurosci. 2020, 43, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.; Kadekar, P.; Jurczak, E.; Roy, R. Germline stem cell integrity and quiescence are controlled by an AMPK-dependent neuronal trafficking pathway. PLoS Genet. 2023, 19, e1010716. [Google Scholar] [CrossRef] [PubMed]
- Kadekar, P.; Chaouni, R.; Clark, E.; Kazanets, A.; Roy, R. Genome-wide surveys reveal polarity and cytoskeletal regulators mediate LKB1-associated germline stem cell quiescence. BMC Genom. 2018, 19, 462. [Google Scholar] [CrossRef]
- Nguyen, P.D.; Gurevich, D.B.; Sonntag, C.; Hersey, L.; Alaei, S.; Nim, H.T.; Siegel, A.; Hall, T.E.; Rossello, F.J.; Boyd, S.E.; et al. Muscle Stem Cells Undergo Extensive Clonal Drift during Tissue Growth via Meox1-Mediated Induction of G2 Cell-Cycle Arrest. Cell Stem Cell 2017, 21, 107.e6–119.e6. [Google Scholar] [CrossRef]
- Sutcu, H.H.; Ricchetti, M. Loss of heterogeneity, quiescence, and differentiation in muscle stem cells. Stem Cell Investig. 2018, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Montazid, S.; Bandyopadhyay, S.; Hart, D.W.; Gao, N.; Johnson, B.; Thrumurthy, S.G.; Penn, D.J.; Wernisch, B.; Bansal, M.; Altrock, P.M.; et al. Adult stem cell activity in naked mole rats for long-term tissue maintenance. Nat. Commun. 2023, 14, 8484. [Google Scholar] [CrossRef]
- Buzgariu, W.; Crescenzi, M.; Galliot, B. Robust G2 pausing of adult stem cells in Hydra. Differentiation 2014, 87, 83–99. [Google Scholar] [CrossRef]
- Baudat, F.; Buard, J.; Grey, C.; Fledel-Alon, A.; Ober, C.; Przeworski, M.; Coop, G.; de Massy, B. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 2010, 327, 836–840. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Niimi, Y.; Shinmyozu, K.; Zhou, Z.; Kiso, M.; Saga, Y. Dead end1 is an essential partner of NANOS2 for selective binding of target RNAs in male germ cell devel-opment. EMBO Rep. 2016, 17, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S. Open niche regulation of mouse spermatogenic stem cells. Dev. Growth Differ. 2018, 60, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Irie, N.; Weinberger, L.; Tang, W.W.; Kobayashi, T.; Viukov, S.; Manor, Y.S.; Dietmann, S.; Hanna, J.H.; Surani, M.A. SOX17 Is a Critical Specifier of Human Primordial Germ Cell Fate. Cell 2014, 160, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Mitsunaga, S.; Shioda, T. Evolutionarily diverse mechanisms of germline specification among mammals: What about us? Stem Cell Investig. 2018, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Tacutu, R.; Thornton, D.; Johnson, E.; Budovsky, A.; Barardo, D.; Craig, T.; Diana, E.; Lehmann, G.; Toren, D.; Wang, J.; et al. Human Ageing Genomic Resources: New and updated databases. Nucleic Acids Res. 2017, 46, D1083–D1090. [Google Scholar] [CrossRef]
- Ouyang, J.F.; Kamaraj, U.S.; Cao, E.Y.; Rackham, O.J.L. ShinyCell: Simple and sharable visualization of single-cell gene expression data. Bioinformatics 2021, 37, 3374–3376. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bush, S.J.; Nikola, R.; Han, S.; Suzuki, S.; Yoshida, S.; Simons, B.D.; Goriely, A. Adult Human, but Not Rodent, Spermatogonial Stem Cells Retain States with a Foetal-like Signature. Cells 2024, 13, 742. https://doi.org/10.3390/cells13090742
Bush SJ, Nikola R, Han S, Suzuki S, Yoshida S, Simons BD, Goriely A. Adult Human, but Not Rodent, Spermatogonial Stem Cells Retain States with a Foetal-like Signature. Cells. 2024; 13(9):742. https://doi.org/10.3390/cells13090742
Chicago/Turabian StyleBush, Stephen J., Rafail Nikola, Seungmin Han, Shinnosuke Suzuki, Shosei Yoshida, Benjamin D. Simons, and Anne Goriely. 2024. "Adult Human, but Not Rodent, Spermatogonial Stem Cells Retain States with a Foetal-like Signature" Cells 13, no. 9: 742. https://doi.org/10.3390/cells13090742