Clinical Phenotypes, Serological Biomarkers, and Synovial Features Defining Seropositive and Seronegative Rheumatoid Arthritis: A Literature Review

 , and
, and
Abstract
:1. Introduction
2. Epidemiology, Genetics, and Clinical Phenotypes: Similarities and Differences between Seropositive and Seronegative RA
2.1. Epidemiology of Rheumatoid Arthritis: What Is Changing?
2.2. Genetic and Environmental Factors Differences
2.3. Immunopathogenesis of Synovial Inflammation in Seropositive Versus Seronegative RA
2.4. Clinical Features and Long-Term Outcomes: Is Seronegative RA a Different Disease?
2.5. Radiological Differences and Erosive Burden
2.5.1. X-ray
2.5.2. Ultrasound
2.5.3. Magnetic Resonance Imaging (MRI)
3. The Role of Peripheral Biomarkers in Rheumatoid Arthritis: From Autoantibodies to microRNA
3.1. Rheumatoid Factor and Anti-CCP Antibodies
3.1.1. Rheumatoid Factor (RF)
3.1.2. Anti-Cyclic Citrullinated Peptide Antibodies
3.2. Novel Biomarkers in “Seronegative” Arthritis
3.2.1. Anti-Carbamylated Protein Antibodies
3.2.2. Other Autoantibody Candidates
3.2.3. Long Pentraxin 3
3.2.4. Peripheral Blood MicroRNA

{kind=link}
{kind=link}
| Novel Biomarkers in Rheumatoid Arthritis | Available Statistics | Target | Features and Phase of Disease |
|---|---|---|---|
| Anti-carbamylated protein antibodies (anti-CarP) [71,108,110,113,114,115,116] |
| Carbamylated proteins |
|
| Anti-Peptidyl-Arginine Deaminase Antibodies (anti-PAD) [117,118,119,120,121,122,123,124] |
| Peptidyl-arginine deaminase enzymes (5 different isoforms) |
|
| Anti-mitochondrial antibodies (AMAs) [125,126] | n/a | Extracellular mitochondria |
|
| Anti-RA33 [135] |
| RA33—a nuclear protein involved in splicing of mRNA |
|
| Antibodies against advanced glycation end-products and malondialdehyde–acetaldehyde adducts (Anti-AGE and MAAs) [128,129,130] | n/a |
|
|
| Peripheral Blood MicroRNA [152] | n/a | n/a |
|
| Long pentraxin 3 [136,144] | n/a | n/a |
|
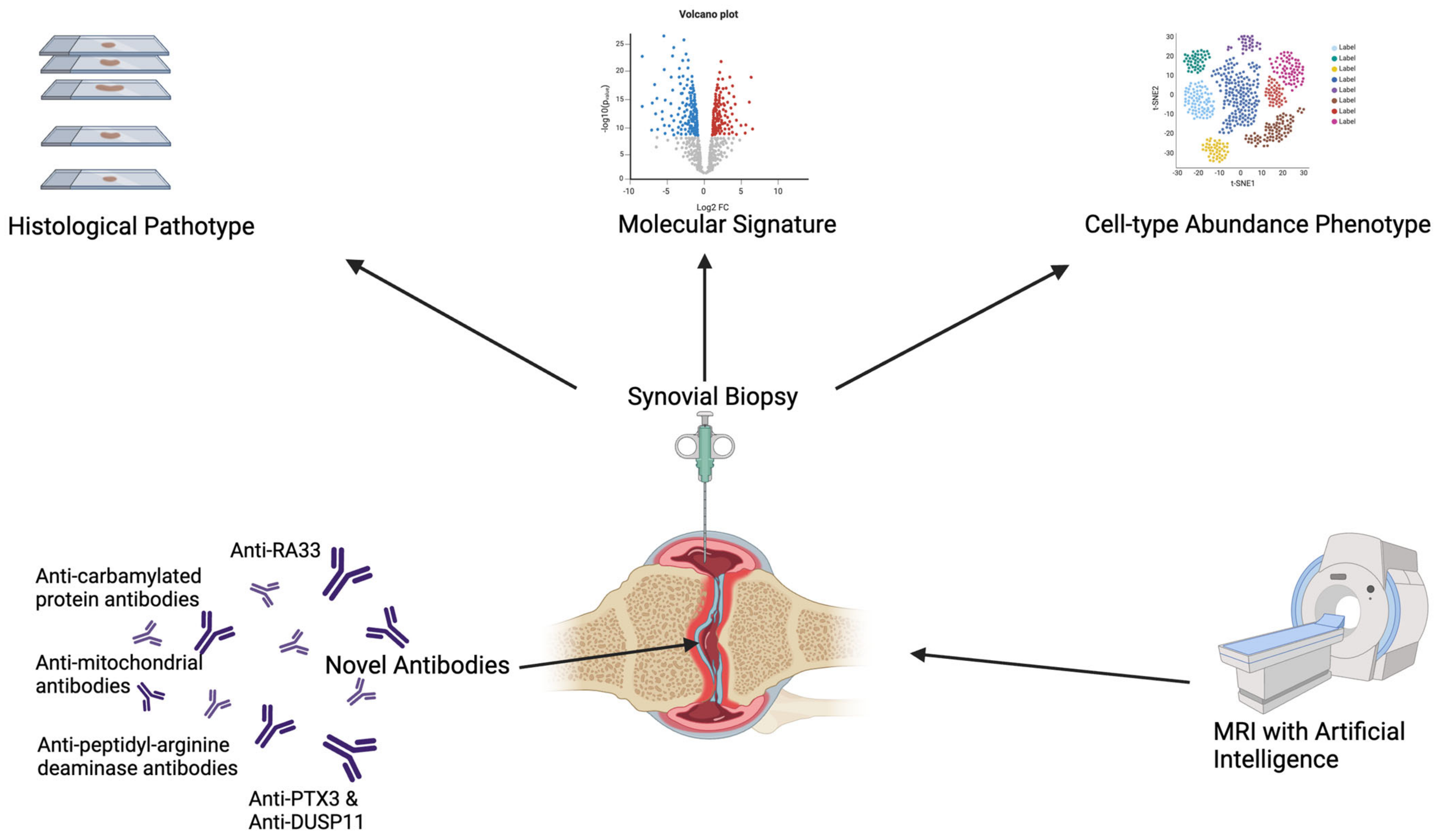
4. Synovial Tissue Histopathology and Relationship with Autoantibody Status
4.1. Synovial Tissue in RA
4.1.1. Histological Pathotypes and Immuno-Serological Status
4.1.2. Focus on Ectopic Lymphoid Structures
4.1.3. Transcriptomic Phenotypes
4.2. Synovial Tissue in Other Seronegative Diseases
4.3. Other Models to Study Seropositive and Seronegative RA
5. Conclusions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gravallese, E.M.; Firestein, G.S. Rheumatoid Arthritis—Common Origins, Divergent Mechanisms. N. Engl. J. Med. 2023, 388, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Prevalence and Incidence|Background Information|Rheumatoid Arthritis|CKS|NICE. Available online: https://cks.nice.org.uk/topics/rheumatoid-arthritis/background-information/prevalence-incidence/ (accessed on 30 July 2023).
- Services for People with Rheumatoid Arthritis—National Audit Office (NAO) Report. Available online: https://www.nao.org.uk/reports/services-for-people-with-rheumatoid-arthritis/ (accessed on 28 October 2023).
- Rheumatoid Arthritis in Adults: Management|Guidance|NICE. Available online: https://www.nice.org.uk/guidance/cg79 (accessed on 30 October 2023).
- York Health Economics. The Cost of Arthritis: Calculation Conducted on Behalf of Arthritis Research UK; York Health Economics: York, UK, 2017. [Google Scholar]
- Health Matters: Health and Work. Available online: https://www.gov.uk/government/publications/health-matters-health-and-work/health-matters-health-and-work (accessed on 28 January 2024).
- Salliot, C.; Finckh, A.; Katchamart, W.; Lu, Y.; Sun, Y.; Bombardier, C.; Keystone, E. Indirect Comparisons of the Efficacy of Biological Antirheumatic Agents in Rheumatoid Arthritis in Patients with an Inadequate Response to Conventional Disease-Modifying Antirheumatic Drugs or to an Anti-Tumour Necrosis Factor Agent: A Meta-Analysis. Ann. Rheum. Dis. 2011, 70, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Nagy, G.; Roodenrijs, N.M.; Welsing, P.M.; Kedves, M.; Hamar, A.; van der Goes, M.C.; Kent, A.; Bakkers, M.; Blaas, E.; Senolt, L.; et al. EULAR Definition of Difficult-to-Treat Rheumatoid Arthritis. Ann. Rheum. Dis. 2021, 80, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Overview|Rheumatoid Arthritis in Adults: Management|Guidance|NICE. Available online: https://www.nice.org.uk/guidance/ng100 (accessed on 21 January 2024).
- Aletaha, D. Precision Medicine and Management of Rheumatoid Arthritis. J. Autoimmun. 2020, 110, 102405. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O.; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 Rheumatoid Arthritis Classification Criteria: An American College of Rheumatology/European League Against Rheumatism Collaborative Initiative. Arthritis Rheum. 2010, 62, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- Arnett, F.C.; Edworthy, S.M.; Bloch, D.A.; McShane, D.J.; Fries, J.F.; Cooper, N.S.; Healey, L.A.; Kaplan, S.R.; Liang, M.H.; Luthra, H.S. The American Rheumatism Association 1987 Revised Criteria for the Classification of Rheumatoid Arthritis. Arthritis Rheum. 1988, 31, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Kuwana, M.; Kameda, H.; Takeuchi, T. Sensitivity and Specificity of 2010 Rheumatoid Arthritis Classification Criteria. Rheumatology 2011, 50, 1268–1274. [Google Scholar] [CrossRef] [PubMed]
- Perniola, S.; Chimenti, M.S.; Spinelli, F.R.; Frediani, B.; Foti, R.; Ferrigno, S.; Garufi, C.; Cassone, G.; Venerito, V.; Atzeni, F.; et al. Rheumatoid Arthritis from Easy to Complex Disease: From the “2022 GISEA International Symposium”. J. Clin. Med. 2023, 12, 2781. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, L.; Bugatti, S.; Mazzucchelli, I.; Rossi, S.; Xoxi, B.; Bozzalla Cassione, E.; Luvaro, T.; Montecucco, C.; Manzo, A. Synovial and Serum B-Cell Signature of Autoantibody-Negative Rheumatoid Arthritis versus Autoantibody-Positive Rheumatoid Arthritis and Psoriatic Arthritis. Rheumatology 2023, kead378. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, L.; D’Onofrio, B.; Gandolfo, S.; Bozzalla Cassione, E.; Mauro, D.; Manzo, A.; Ciccia, F.; Bugatti, S. Seronegative Rheumatoid Arthritis: One Year in Review 2023. Clin. Exp. Rheumatol. 2023, 41, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Myasoedova, E.; Davis, J.; Matteson, E.L.; Crowson, C.S. Is the Epidemiology of Rheumatoid Arthritis Changing? Results from a Population-Based Incidence Study, 1985–2014. Ann. Rheum. Dis. 2020, 79, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Muilu, P.; Rantalaiho, V.; Kautiainen, H.; Virta, L.J.; Eriksson, J.G.; Puolakka, K. Increasing Incidence and Shifting Profile of Idiopathic Inflammatory Rheumatic Diseases in Adults during This Millennium. Clin. Rheumatol. 2019, 38, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Matthijssen, X.M.E.; Huizinga, T.W.J.; van der Helm-van Mil, A.H.M. Increasing Incidence of Autoantibody-Negative RA Is Replicated and Is Partly Explained by an Aging Population. Ann. Rheum. Dis. 2022, 81, e69. [Google Scholar] [CrossRef] [PubMed]
- Carbonell-Bobadilla, N.; Soto-Fajardo, C.; Amezcua-Guerra, L.M.; Batres-Marroquín, A.B.; Vargas, T.; Hernández-Diazcouder, A.; Jiménez-Rojas, V.; Medina-García, A.C.; Pineda, C.; Silveira, L.H. Patients with Seronegative Rheumatoid Arthritis Have a Different Phenotype than Seropositive Patients: A Clinical and Ultrasound Study. Front. Med. 2022, 9, 978351. [Google Scholar] [CrossRef] [PubMed]
- Paroli, M.; Sirinian, M.I. When Autoantibodies Are Missing: The Challenge of Seronegative Rheumatoid Arthritis. Antibodies 2023, 12, 69. [Google Scholar] [CrossRef] [PubMed]
- Deane, K.D.; Demoruelle, M.K.; Kelmenson, L.B.; Kuhn, K.A.; Norris, J.M.; Holers, V.M. Genetic and Environmental Risk Factors for Rheumatoid Arthritis. Best Pract. Res. Clin. Rheumatol. 2017, 31, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Hadwen, B.; Yu, R.; Cairns, E.; Barra, L. Presence of Autoantibodies in Males and Females with Rheumatoid Arthritis: A Systematic Review and Metaanalysis. J. Rheumatol. 2022, 49, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Guekos, A.; Saxer, J.; Salinas Gallegos, D.; Schweinhardt, P. Healthy Women Show More Experimentally Induced Central Sensitization Compared with Men. Pain 2024. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, P.K.; Silver, J.; Winchester, R.J. The Shared Epitope Hypothesis. an Approach to Understanding the Molecular Genetics of Susceptibility to Rheumatoid Arthritis. Arthritis Rheum. 1987, 30, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Padyukov, L. Genetics of Rheumatoid Arthritis. Semin. Immunopathol. 2022, 44, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Verpoort, K.N.; van Gaalen, F.A.; van der Helm-van Mil, A.H.M.; Schreuder, G.M.T.; Breedveld, F.C.; Huizinga, T.W.J.; de Vries, R.R.P.; Toes, R.E.M. Association of HLA-DR3 with Anti-Cyclic Citrullinated Peptide Antibody-Negative Rheumatoid Arthritis. Arthritis Rheum. 2005, 52, 3058–3062. [Google Scholar] [CrossRef] [PubMed]
- Bossini-Castillo, L.; de Kovel, C.; Kallberg, H.; van ’t Slot, R.; Italiaander, A.; Coenen, M.; Tak, P.P.; Posthumus, M.D.; Wijmenga, C.; Huizinga, T.; et al. A Genome-Wide Association Study of Rheumatoid Arthritis without Antibodies against Citrullinated Peptides. Ann. Rheum. Dis. 2015, 74, e15. [Google Scholar] [CrossRef] [PubMed]
- Eyre, S.; Bowes, J.; Diogo, D.; Lee, A.; Barton, A.; Martin, P.; Zhernakova, A.; Stahl, E.; Viatte, S.; McAllister, K.; et al. High-Density Genetic Mapping Identifies New Susceptibility Loci for Rheumatoid Arthritis. Nat. Genet. 2012, 44, 1336–1340. [Google Scholar] [CrossRef] [PubMed]
- Saevarsdottir, S.; Stefansdottir, L.; Sulem, P.; Thorleifsson, G.; Ferkingstad, E.; Rutsdottir, G.; Glintborg, B.; Westerlind, H.; Grondal, G.; Loft, I.C.; et al. Multiomics Analysis of Rheumatoid Arthritis Yields Sequence Variants That Have Large Effects on Risk of the Seropositive Subset. Ann. Rheum. Dis. 2022, 81, 1085–1095. [Google Scholar] [CrossRef]
- Pombo-Suarez, M.; Sanchez-Piedra, C.; Gómez-Reino, J.; Lauper, K.; Mongin, D.; Iannone, F.; Pavelka, K.; Nordström, D.C.; Inanc, N.; Codreanu, C.; et al. After JAK Inhibitor Failure: To Cycle or to Switch, That Is the Question—Data from the JAK-Pot Collaboration of Registries. Ann. Rheum. Dis. 2023, 82, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Molina, C.; Gich, I.; Diaz-Torné, C.; Park, H.S.; Feliu, A.; Vidal, S.; Corominas, H. Patient-Related Factors Influencing the Effectiveness and Safety of Janus Kinase Inhibitors in Rheumatoid Arthritis: A Real-World Study. Sci. Rep. 2024, 14, 172. [Google Scholar] [CrossRef] [PubMed]
- Frisell, T.; Holmqvist, M.; Källberg, H.; Klareskog, L.; Alfredsson, L.; Askling, J. Familial Risks and Heritability of Rheumatoid Arthritis: Role of Rheumatoid Factor/Anti-Citrullinated Protein Antibody Status, Number and Type of Affected Relatives, Sex, and Age. Arthritis Rheum. 2013, 65, 2773–2782. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Mazmanian, S.K. Has the Microbiota Played a Critical Role in the Evolution of the Adaptive Immune System? Science 2010, 330, 1768–1773. [Google Scholar] [CrossRef] [PubMed]
- Koboziev, I.; Reinoso Webb, C.; Furr, K.L.; Grisham, M.B. Role of the Enteric Microbiota in Intestinal Homeostasis and Inflammation. Free Radic. Biol. Med. 2014, 68, 122–133. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, D.; Jia, H.; Feng, Q.; Wang, D.; Liang, D.; Wu, X.; Li, J.; Tang, L.; Li, Y.; et al. The Oral and Gut Microbiomes Are Perturbed in Rheumatoid Arthritis and Partly Normalized after Treatment. Nat. Med. 2015, 21, 895–905. [Google Scholar] [CrossRef]
- Horta-Baas, G.; Romero-Figueroa, M.d.S.; Montiel-Jarquín, A.J.; Pizano-Zárate, M.L.; García-Mena, J.; Ramírez-Durán, N. Intestinal Dysbiosis and Rheumatoid Arthritis: A Link between Gut Microbiota and the Pathogenesis of Rheumatoid Arthritis. J. Immunol. Res. 2017, 2017, 4835189. [Google Scholar] [CrossRef] [PubMed]
- Chiang, H.-I.; Li, J.-R.; Liu, C.-C.; Liu, P.-Y.; Chen, H.-H.; Chen, Y.-M.; Lan, J.-L.; Chen, D.-Y. An Association of Gut Microbiota with Different Phenotypes in Chinese Patients with Rheumatoid Arthritis. J. Clin. Med. 2019, 8, 1770. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Chu, Y.; Li, J.; Meng, Q.; Liu, Y.; Jin, J.; Wang, Y.; Wang, J.; Huang, B.; Shi, L.; et al. Intestinal Butyrate-Metabolizing Species Contribute to Autoantibody Production and Bone Erosion in Rheumatoid Arthritis. Sci. Adv. 2022, 8, eabm1511. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wang, M.; Zhao, L.; Liu, Y.; Zhang, X. ACPA-Negative Rheumatoid Arthritis: From Immune Mechanisms to Clinical Translation. eBioMedicine 2022, 83, 104233. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 Paradigm of Macrophage Activation: Time for Reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, Y.; Jin, S.; Wang, M.; Jiao, Y.; Yang, B.; Lu, X.; Ji, X.; Fei, Y.; Yang, H.; et al. Single-Cell Sequencing of Immune Cells from Anticitrullinated Peptide Antibody Positive and Negative Rheumatoid Arthritis. Nat. Commun. 2021, 12, 4977. [Google Scholar] [CrossRef] [PubMed]
- van Lieshout, A.W.T.; Fransen, J.; Flendrie, M.; Eijsbouts, A.M.M.; van den Hoogen, F.H.J.; van Riel, P.L.C.M.; Radstake, T.R.D.J. Circulating Levels of the Chemokine CCL18 but Not CXCL16 Are Elevated and Correlate with Disease Activity in Rheumatoid Arthritis. Ann. Rheum. Dis. 2007, 66, 1334–1338. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, T.; Okamoto, H.; Iikuni, N.; Takeuchi, M.; Toyama, Y.; Tomatsu, T.; Kamatani, N.; Momohara, S. Monocyte Chemoattractant Protein-4 (MCP-4)/CCL13 Is Highly Expressed in Cartilage from Patients with Rheumatoid Arthritis. Rheumatology 2006, 45, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, T.; Okamoto, H.; Toyama, Y.; Momohara, S. Molecular Aspects of Rheumatoid Arthritis: Chemokines in the Joints of Patients. FEBS J. 2008, 275, 4448–4455. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.; Neidhöfer, S.; Reuter, S.; Matthias, T. MMP3 Is a Reliable Marker for Disease Activity, Radiological Monitoring, Disease Outcome Predictability, and Therapeutic Response in Rheumatoid Arthritis. Best Pract. Res. Clin. Rheumatol. 2018, 32, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Louis, C.; Souza-Fonseca-Guimaraes, F.; Yang, Y.; D’Silva, D.; Kratina, T.; Dagley, L.; Hediyeh-Zadeh, S.; Rautela, J.; Masters, S.L.; Davis, M.J.; et al. NK Cell-Derived GM-CSF Potentiates Inflammatory Arthritis and Is Negatively Regulated by CIS. J. Exp. Med. 2020, 217, e20191421. [Google Scholar] [CrossRef] [PubMed]
- Pickens, S.R.; Chamberlain, N.D.; Volin, M.V.; Pope, R.M.; Mandelin, A.M.; Shahrara, S. Characterization of CCL19 and CCL21 in Rheumatoid Arthritis. Arthritis Rheum. 2011, 63, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Van Raemdonck, K.; Umar, S.; Palasiewicz, K.; Volkov, S.; Volin, M.V.; Arami, S.; Chang, H.J.; Zanotti, B.; Sweiss, N.; Shahrara, S. CCL21/CCR7 Signaling in Macrophages Promotes Joint Inflammation and Th17-Mediated Osteoclast Formation in Rheumatoid Arthritis. Cell. Mol. Life Sci. CMLS 2020, 77, 1387–1399. [Google Scholar] [CrossRef] [PubMed]
- Sigurdsson, S.; Padyukov, L.; Kurreeman, F.A.S.; Liljedahl, U.; Wiman, A.-C.; Alfredsson, L.; Toes, R.; Rönnelid, J.; Klareskog, L.; Huizinga, T.W.J.; et al. Association of a Haplotype in the Promoter Region of the Interferon Regulatory Factor 5 Gene with Rheumatoid Arthritis. Arthritis Rheum. 2007, 56, 2202–2210. [Google Scholar] [CrossRef] [PubMed]
- Pratt, A.G.; Swan, D.C.; Richardson, S.; Wilson, G.; Hilkens, C.M.U.; Young, D.A.; Isaacs, J.D. A CD4 T Cell Gene Signature for Early Rheumatoid Arthritis Implicates Interleukin 6-Mediated STAT3 Signalling, Particularly in Anti-Citrullinated Peptide Antibody-Negative Disease. Ann. Rheum. Dis. 2012, 71, 1374–1381. [Google Scholar] [CrossRef] [PubMed]
- Floudas, A.; Canavan, M.; McGarry, T.; Mullan, R.; Nagpal, S.; Veale, D.J.; Fearon, U. ACPA Status Correlates with Differential Immune Profile in Patients with Rheumatoid Arthritis. Cells 2021, 10, 647. [Google Scholar] [CrossRef] [PubMed]
- Alivernini, S.; Bruno, D.; Tolusso, B.; Bui, L.; Petricca, L.; Gigante, M.R.; Birra, D.; Fedele, A.L.; Peluso, G.; Federico, F.; et al. Differential Synovial Tissue Biomarkers among Psoriatic Arthritis and Rheumatoid Factor/Anti-Citrulline Antibody-Negative Rheumatoid Arthritis. Arthritis Res. Ther. 2019, 21, 116. [Google Scholar] [CrossRef] [PubMed]
- van Oosterhout, M.; Bajema, I.; Levarht, E.W.N.; Toes, R.E.M.; Huizinga, T.W.J.; van Laar, J.M. Differences in Synovial Tissue Infiltrates between Anti-Cyclic Citrullinated Peptide-Positive Rheumatoid Arthritis and Anti-Cyclic Citrullinated Peptide-Negative Rheumatoid Arthritis. Arthritis Rheum. 2008, 58, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, G.; Firestein, G.S. Restoring Synovial Homeostasis in Rheumatoid Arthritis by Targeting Fibroblast-like Synoviocytes. Nat. Rev. Rheumatol. 2020, 16, 316–333. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.; Nguyen, H.N.; Brenner, M.B. Fibroblast Pathology in Inflammatory Diseases. J. Clin. Investig. 2021, 131, e149538. [Google Scholar] [CrossRef] [PubMed]
- Ronnelid, J.; Wick, M.; Lampa, J.; Lindblad, S.; Nordmark, B.; Klareskog, L.; van Vollenhoven, R.F. Longitudinal Analysis of Citrullinated Protein/Peptide Antibodies (Anti-CP) during 5 Year Follow up in Early Rheumatoid Arthritis: Anti-CP Status Predicts Worse Disease Activity and Greater Radiological Progression. Ann. Rheum. Dis. 2005, 64, 1744–1749. [Google Scholar] [CrossRef] [PubMed]
- van den Broek, M.; Dirven, L.; Klarenbeek, N.B.; Molenaar, T.H.E.; Han, K.H.; Kerstens, P.J.S.M.; Huizinga, T.W.J.; Dijkmans, B.a.C.; Allaart, C.F. The Association of Treatment Response and Joint Damage with ACPA-Status in Recent-Onset RA: A Subanalysis of the 8-Year Follow-up of the BeSt Study. Ann. Rheum. Dis. 2012, 71, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Hecht, C.; Englbrecht, M.; Rech, J.; Schmidt, S.; Araujo, E.; Engelke, K.; Finzel, S.; Schett, G. Additive Effect of Anti-Citrullinated Protein Antibodies and Rheumatoid Factor on Bone Erosions in Patients with RA. Ann. Rheum. Dis. 2015, 74, 2151–2156. [Google Scholar] [CrossRef] [PubMed]
- Farragher, T.M.; Lunt, M.; Plant, D.; Bunn, D.K.; Barton, A.; Symmons, D.P.M. Benefit of Early Treatment in Inflammatory Polyarthritis Patients with Anti–Cyclic Citrullinated Peptide Antibodies versus Those without Antibodies. Arthritis Care Res. 2010, 62, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, L.B.; Lillegraven, S.; Lie, E.; Aga, A.-B.; Olsen, I.C.; Hammer, H.B.; Uhlig, T.; Jonsson, M.K.; van der Heijde, D.; Kvien, T.K.; et al. Patients with Seronegative RA Have More Inflammatory Activity Compared with Patients with Seropositive RA in an Inception Cohort of DMARD-Naïve Patients Classified According to the 2010 ACR/EULAR Criteria. Ann. Rheum. Dis. 2017, 76, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Koslow, M.; Young, J.R.; Yi, E.S.; Baqir, M.; Decker, P.A.; Johnson, G.B.; Ryu, J.H. Rheumatoid Pulmonary Nodules: Clinical and Imaging Features Compared with Malignancy. Eur. Radiol. 2019, 29, 1684–1692. [Google Scholar] [CrossRef] [PubMed]
- Caimmi, C.; Crowson, C.S.; Smith, W.M.; Matteson, E.L.; Makol, A. Clinical Correlates, Outcomes, and Predictors of Inflammatory Ocular Disease Associated with Rheumatoid Arthritis in the Biologic Era. J. Rheumatol. 2018, 45, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, H.; Panlaqui, O.M. Systematic Review and Meta-Analysis of the Risk of Rheumatoid Arthritis-Associated Interstitial Lung Disease Related to Anti-Cyclic Citrullinated Peptide (CCP) Antibody. BMJ Open 2021, 11, e040465. [Google Scholar] [CrossRef] [PubMed]
- Ambrosino, P.; Tasso, M.; Lupoli, R.; Di Minno, A.; Baldassarre, D.; Tremoli, E.; Di Minno, M.N.D. Non-Invasive Assessment of Arterial Stiffness in Patients with Rheumatoid Arthritis: A Systematic Review and Meta-Analysis of Literature Studies. Ann. Med. 2015, 47, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Aslan, A.N.; Şirin Özcan, A.N.; Erten, Ş.; Alsancak, Y.; Durmaz, T. Assessment of Local Carotid Stiffness in Seronegative and Seropositive Rheumatoid Arthritis. Scand. Cardiovasc. J. 2017, 51, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, L.B.; Lillegraven, S.; Aga, A.-B.; Sexton, J.; Olsen, I.C.; Lie, E.; Berner Hammer, H.; Uhlig, T.; van der Heijde, D.; Kvien, T.K.; et al. Comparing the Disease Course of Patients with Seronegative and Seropositive Rheumatoid Arthritis Fulfilling the 2010 ACR/EULAR Classification Criteria in a Treat-to-Target Setting: 2-Year Data from the ARCTIC Trial. RMD Open 2018, 4, e000752. [Google Scholar] [CrossRef] [PubMed]
- Wevers-de Boer, K.; Visser, K.; Heimans, L.; Ronday, H.K.; Molenaar, E.; Groenendael, J.H.L.M.; Peeters, A.J.; Westedt, M.-L.; Collée, G.; de Sonnaville, P.B.J.; et al. Remission Induction Therapy with Methotrexate and Prednisone in Patients with Early Rheumatoid and Undifferentiated Arthritis (the IMPROVED Study). Ann. Rheum. Dis. 2012, 71, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Bird, P.; Hall, S.; Nash, P.; Connell, C.A.; Kwok, K.; Witcombe, D.; Thirunavukkarasu, K. Treatment Outcomes in Patients with Seropositive versus Seronegative Rheumatoid Arthritis in Phase III Randomised Clinical Trials of Tofacitinib. RMD Open 2019, 5, e000742. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Breijo, B.; Brenis, C.M.; Plasencia-Rodríguez, C.; Martínez-Feito, A.; Novella-Navarro, M.; Pascual-Salcedo, D.; Balsa, A. Methotrexate Reduces the Probability of Discontinuation of TNF Inhibitors in Seropositive Patients with Rheumatoid Arthritis. A Real-World Data Analysis. Front. Med. 2021, 8, 692557. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Liu, J.; Desai, R.J.; Kim, S.C. Real-World Treatment Effectiveness of Disease-Modifying Antirheumatic Drugs by Serostatus Among Patients with Rheumatoid Arthritis. ACR Open Rheumatol. 2023, 5, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Matthijssen, X.M.E.; Niemantsverdriet, E.; Huizinga, T.W.J.; Mil, A.H.M.v.d.H. Enhanced Treatment Strategies and Distinct Disease Outcomes among Autoantibody-Positive and -Negative Rheumatoid Arthritis Patients over 25 Years: A Longitudinal Cohort Study in the Netherlands. PLoS Med. 2020, 17, e1003296. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Gravallese, E. Bone Erosion in Rheumatoid Arthritis: Mechanisms, Diagnosis and Treatment. Nat. Rev. Rheumatol. 2012, 8, 656–664. [Google Scholar] [CrossRef] [PubMed]
- van der Heijde, D.M. Joint Erosions and Patients with Early Rheumatoid Arthritis. Br. J. Rheumatol. 1995, 34 (Suppl. 2), 74–78. [Google Scholar] [CrossRef] [PubMed]
- Machold, K.P.; Stamm, T.A.; Nell, V.P.K.; Pflugbeil, S.; Aletaha, D.; Steiner, G.; Uffmann, M.; Smolen, J.S. Very Recent Onset Rheumatoid Arthritis: Clinical and Serological Patient Characteristics Associated with Radiographic Progression over the First Years of Disease. Rheumatol. Oxf. Engl. 2007, 46, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Berglin, E.; Johansson, T.; Sundin, U.; Jidell, E.; Wadell, G.; Hallmans, G.; Rantapää-Dahlqvist, S. Radiological Outcome in Rheumatoid Arthritis Is Predicted by Presence of Antibodies against Cyclic Citrullinated Peptide before and at Disease Onset, and by IgA-RF at Disease Onset. Ann. Rheum. Dis. 2006, 65, 453–458. [Google Scholar] [CrossRef]
- Lindqvist, E.; Eberhardt, K.; Bendtzen, K.; Heinegård, D.; Saxne, T. Prognostic Laboratory Markers of Joint Damage in Rheumatoid Arthritis. Ann. Rheum. Dis. 2005, 64, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Forslind, K.; Ahlmén, M.; Eberhardt, K.; Hafström, I.; Svensson, B.; BARFOT Study Group. Prediction of Radiological Outcome in Early Rheumatoid Arthritis in Clinical Practice: Role of Antibodies to Citrullinated Peptides (Anti-CCP). Ann. Rheum. Dis. 2004, 63, 1090–1095. [Google Scholar] [CrossRef] [PubMed]
- Geusens, P.; van den Bergh, J. Bone Erosions in Rheumatoid Arthritis. Rheumatology 2014, 53, 4–5. [Google Scholar] [CrossRef] [PubMed]
- Di Matteo, A.; Mankia, K.; Nam, J.L.; Cipolletta, E.; Garcia-Montoya, L.; Duquenne, L.; Rowbotham, E.; Emery, P. In Anti-CCP+ at-Risk Individuals, Radiographic Bone Erosions Are Uncommon and Are Not Associated with the Development of Clinical Arthritis. Rheumatology 2020, 60, 3156–3164. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, M.-A.; Terslev, L.; Aegerter, P.; Backhaus, M.; Balint, P.; Bruyn, G.A.; Filippucci, E.; Grassi, W.; Iagnocco, A.; Jousse-Joulin, S.; et al. Scoring Ultrasound Synovitis in Rheumatoid Arthritis: A EULAR-OMERACT Ultrasound Taskforce—Part 1: Definition and Development of a Standardised, Consensus-Based Scoring System. RMD Open 2017, 3, e000428. [Google Scholar] [CrossRef]
- Wang, J.; Wang, M.; Qi, Q.; Wu, Z.; Wen, J. High-Frequency Ultrasound in Patients with Seronegative Rheumatoid Arthritis. Sci. Rep. 2022, 12, 21372. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Zhao, M.; Zhang, Y.; Xie, Y.; Cao, J.; Pan, Y. Seronegative Rheumatic Arthritis Has Milder Inflammation and Bone Erosion in an Ultrasound Study of Disease-Modifying Anti-Rheumatic Drugs (DMARDs)-Naïve Chinese Cohort. Ann. Transl. Med. 2022, 10, 661. [Google Scholar] [CrossRef] [PubMed]
- Ruta, S.; Sanchez Prado, E.; Salvatori, F.; Arguello, J.; Aguerre, D.; Magri, S.; García Salinas, R. Ultrasound Tenosynovitis: A Differential Feature of Patients with Seronegative Rheumatoid Arthritis. Reumatol. Clin. 2023, 19, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Gandjbakhch, F.; Haavardsholm, E.A.; Conaghan, P.G.; Ejbjerg, B.; Foltz, V.; Brown, A.K.; Døhn, U.M.; Lassere, M.; Freeston, J.E.; Olsen, I.C.; et al. Determining a Magnetic Resonance Imaging Inflammatory Activity Acceptable State without Subsequent Radiographic Progression in Rheumatoid Arthritis: Results from a Followup MRI Study of 254 Patients in Clinical Remission or Low Disease Activity. J. Rheumatol. 2014, 41, 398–406. [Google Scholar] [CrossRef] [PubMed]
- den Hollander, N.K.; Verstappen, M.; Sidhu, N.; van Mulligen, E.; Reijnierse, M.; van der Helm-van Mil, A.H.M. Hand and Foot MRI in Contemporary Undifferentiated Arthritis: In Which Patients Is MRI Valuable to Detect Rheumatoid Arthritis Early? A Large Prospective Study. Rheumatology 2022, 61, 3963–3973. [Google Scholar] [CrossRef] [PubMed]
- Hassanzadeh, T.; Shamonin, D.P.; Li, Y.; Krijbolder, D.I.; Reijnierse, M.; van der Helm-van Mil, A.H.M.; Stoel, B.C. A Deep Learning-Based Comparative MRI Model to Detect Inflammatory Changes in Rheumatoid Arthritis. Biomed. Signal Process. Control 2024, 88, 105612. [Google Scholar] [CrossRef]
- Folle, L.; Bayat, S.; Kleyer, A.; Fagni, F.; Kapsner, L.A.; Schlereth, M.; Meinderink, T.; Breininger, K.; Tascilar, K.; Krönke, G.; et al. Advanced Neural Networks for Classification of MRI in Psoriatic Arthritis, Seronegative, and Seropositive Rheumatoid Arthritis. Rheumatology 2022, 61, 4945–4951. [Google Scholar] [CrossRef] [PubMed]
- Zendman, A.J.W.; Vossenaar, E.R.; van Venrooij, W.J. Autoantibodies to Citrullinated (Poly)Peptides: A Key Diagnostic and Prognostic Marker for Rheumatoid Arthritis. Autoimmunity 2004, 37, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Ingegnoli, F.; Castelli, R.; Gualtierotti, R. Rheumatoid Factors: Clinical Applications. Dis. Markers 2013, 35, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, C.; Legouffe, M.C.; Bologna, C.; Brochier, J.; Sany, J. IgA Isotype Rheumatoid Factor in Rheumatoid Arthritis: Clinical Implications. Clin. Exp. Rheumatol. 1996, 14, 301–304. [Google Scholar]
- de Brito Rocha, S.; Baldo, D.C.; Andrade, L.E.C. Clinical and Pathophysiologic Relevance of Autoantibodies in Rheumatoid Arthritis. Adv. Rheumatol. 2019, 59, 2. [Google Scholar] [CrossRef] [PubMed]
- Schellekens, G.A.; de Jong, B.A.; van den Hoogen, F.H.; van de Putte, L.B.; van Venrooij, W.J. Citrulline Is an Essential Constituent of Antigenic Determinants Recognized by Rheumatoid Arthritis-Specific Autoantibodies. J. Clin. Investig. 1998, 101, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Van Schaardenburg, D.; Lagaay, A.M.; Otten, H.G.; Breedveld, F.C. The Relation Between Class-specific Serum Rheumatoid Factors and Age in the General Population. Rheumatology 1993, 32, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Waaler, E. On the Occurrence of a Factor in Human Serum Activating the Specific Agglutination of Sheep Blood Corpuscles. Acta Pathol. Microbiol. Scand. 1940, 17, 172–188. [Google Scholar] [CrossRef]
- Falkenburg, W.J.J.; Oskam, N.; Koers, J.; van Boheemen, L.; Ooijevaar-de Heer, P.; Verstappen, G.M.; Bootsma, H.; Kroese, F.G.M.; van Schaardenburg, D.; Wolbink, G.; et al. Identification of Clinically and Pathophysiologically Relevant Rheumatoid Factor Epitopes by Engineered IgG Targets. Arthritis Rheumatol. 2020, 72, 2005–2016. [Google Scholar] [CrossRef] [PubMed]
- Laurent, L.; Anquetil, F.; Clavel, C.; Ndongo-Thiam, N.; Offer, G.; Miossec, P.; Pasquali, J.-L.; Sebbag, M.; Serre, G. IgM Rheumatoid Factor Amplifies the Inflammatory Response of Macrophages Induced by the Rheumatoid Arthritis-Specific Immune Complexes Containing Anticitrullinated Protein Antibodies. Ann. Rheum. Dis. 2015, 74, 1425–1431. [Google Scholar] [CrossRef] [PubMed]
- Nielen, M.M.J.; van der Horst, A.R.; van Schaardenburg, D.; van der Horst-Bruinsma, I.E.; van de Stadt, R.J.; Aarden, L.; Dijkmans, B.a.C.; Hamann, D. Antibodies to Citrullinated Human Fibrinogen (ACF) Have Diagnostic and Prognostic Value in Early Arthritis. Ann. Rheum. Dis. 2005, 64, 1199–1204. [Google Scholar] [CrossRef] [PubMed]
- Braschi, E.; Shojania, K.; Allan, G.M. Anti-CCP: A Truly Helpful Rheumatoid Arthritis Test? Can. Fam. Physician 2016, 62, 234. [Google Scholar] [PubMed]
- Lundberg, K.; Kinloch, A.; Fisher, B.A.; Wegner, N.; Wait, R.; Charles, P.; Mikuls, T.R.; Venables, P.J. Antibodies to Citrullinated Alpha-Enolase Peptide 1 Are Specific for Rheumatoid Arthritis and Cross-React with Bacterial Enolase. Arthritis Rheum. 2008, 58, 3009–3019. [Google Scholar] [CrossRef] [PubMed]
- Vossenaar, E.R.; Després, N.; Lapointe, E.; van der Heijden, A.; Lora, M.; Senshu, T.; van Venrooij, W.J.; Ménard, H.A. Rheumatoid Arthritis Specific Anti-Sa Antibodies Target Citrullinated Vimentin. Arthritis Res. Ther. 2004, 6, R142–R150. [Google Scholar] [CrossRef] [PubMed]
- Klareskog, L.; Stolt, P.; Lundberg, K.; Källberg, H.; Bengtsson, C.; Grunewald, J.; Rönnelid, J.; Erlandsson Harris, H.; Ulfgren, A.-K.; Rantapää-Dahlqvist, S.; et al. A New Model for an Etiology of Rheumatoid Arthritis: Smoking May Trigger HLA–DR (Shared Epitope)–Restricted Immune Reactions to Autoantigens Modified by Citrullination. Arthritis Rheum. 2006, 54, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Regueiro, C.; Rodriguez-Rodriguez, L.; Lopez-Mejias, R.; Nuño, L.; Triguero-Martinez, A.; Perez-Pampin, E.; Corrales, A.; Villalba, A.; Lopez-Golan, Y.; Abasolo, L.; et al. A Predominant Involvement of the Triple Seropositive Patients and Others with Rheumatoid Factor in the Association of Smoking with Rheumatoid Arthritis. Sci. Rep. 2020, 10, 3355. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.-C.; Lee, Y.H. Association between Anti-Porphyromonas Gingivalis Antibody, Anti-Citrullinated Protein Antibodies, and Rheumatoid Arthritis. Z. Für Rheumatol. 2018, 77, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Ge, C.; Holmdahl, R. The Structure, Specificity and Function of Anti-Citrullinated Protein Antibodies. Nat. Rev. Rheumatol. 2019, 15, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Barra, L.; Pope, J.; Bessette, L.; Haraoui, B.; Bykerk, V. Lack of Seroconversion of Rheumatoid Factor and Anti-Cyclic Citrullinated Peptide in Patients with Early Inflammatory Arthritis: A Systematic Literature Review. Rheumatology 2011, 50, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Burr, M.L.; Viatte, S.; Bukhari, M.; Plant, D.; Symmons, D.P.; Thomson, W.; Barton, A. Long-Term Stability of Anti-Cyclic Citrullinated Peptide Antibody Status in Patients with Early Inflammatory Polyarthritis. Arthritis Res. Ther. 2012, 14, R109. [Google Scholar] [CrossRef] [PubMed]
- Amezcua-Guerra, L.M.; Carbonell-Bobadilla, N.; Soto-Fajardo, C.; Vargas, A.; Batres-Marroquín, A.B.; Vargas, T.; Medina-García, A.C.; Hernández-Diazcouder, A.; Jiménez-Rojas, V.; Pineda, C.; et al. Influence of Anti-Carbamylated Protein Antibodies on Disease Activity and Joint Erosions in Seronegative and Seropositive Rheumatoid Arthritis. Rheumatol. Int. 2023, 43, 2245–2250. [Google Scholar] [CrossRef] [PubMed]
- Brevet, P.; Lattard, C.; Guillou, C.; Rottenberg, P.; Fardellone, P.; Le-Loët, X.; Lequerré, T.; Cosette, P.; Boyer, O.; Fréret, M.; et al. Anti-Carbamylated Fibrinogen Antibodies Might Be Associated with a Specific Rheumatoid Phenotype and Include a Subset Recognizing In Vivo Epitopes of Its γ Chain One of Which Is Not Cross Reactive with Anti-Citrullinated Protein Antibodies. Front. Immunol. 2021, 12, 733511. [Google Scholar] [CrossRef] [PubMed]
- Scinocca, M.; Bell, D.A.; Racapé, M.; Joseph, R.; Shaw, G.; McCormick, J.K.; Gladman, D.D.; Pope, J.; Barra, L.; Cairns, E. Antihomocitrullinated Fibrinogen Antibodies Are Specific to Rheumatoid Arthritis and Frequently Bind Citrullinated Proteins/Peptides. J. Rheumatol. 2014, 41, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.D.; Hamilton, B.J.; Rigby, W.F.C. Brief Report: Anti-Carbamylated Protein Antibodies in Rheumatoid Arthritis Patients Are Reactive with Specific Epitopes of the Human Fibrinogen β-Chain. Arthritis Rheumatol. 2017, 69, 1381–1386. [Google Scholar] [CrossRef] [PubMed]
- van Delft, M.A.M.; Verheul, M.K.; Burgers, L.E.; Derksen, V.F.A.M.; van der Helm-van Mil, A.H.M.; van der Woude, D.; Huizinga, T.W.J.; Toes, R.E.M.; Trouw, L.A. The Isotype and IgG Subclass Distribution of Anti-Carbamylated Protein Antibodies in Rheumatoid Arthritis Patients. Arthritis Res. Ther. 2017, 19, 190. [Google Scholar] [CrossRef] [PubMed]
- Gan, R.W.; Trouw, L.A.; Shi, J.; Toes, R.E.M.; Huizinga, T.W.J.; Demoruelle, M.K.; Kolfenbach, J.R.; Zerbe, G.O.; Deane, K.D.; Edison, J.D.; et al. Anti-Carbamylated Protein Antibodies Are Present Prior to Rheumatoid Arthritis and Are Associated with Its Future Diagnosis. J. Rheumatol. 2015, 42, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Brink, M.; Verheul, M.K.; Rönnelid, J.; Berglin, E.; Holmdahl, R.; Toes, R.E.M.; Klareskog, L.; Trouw, L.A.; Rantapää-Dahlqvist, S. Anti-Carbamylated Protein Antibodies in the Pre-Symptomatic Phase of Rheumatoid Arthritis, Their Relationship with Multiple Anti-Citrulline Peptide Antibodies and Association with Radiological Damage. Arthritis Res. Ther. 2015, 17, 25. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Knevel, R.; Suwannalai, P.; van der Linden, M.P.; Janssen, G.M.; van Veelen, P.A.; Levarht, N.E.; van der Helm-van Mil, A.H.; Cerami, A.; Huizinga, T.W.; et al. Autoantibodies Recognizing Carbamylated Proteins Are Present in Sera of Patients with Rheumatoid Arthritis and Predict Joint Damage. Proc. Natl. Acad. Sci. USA 2011, 108, 17372–17377. [Google Scholar] [CrossRef] [PubMed]
- Sidiras, P.; Lechanteur, J.; Imbault, V.; Sokolova, T.; Durez, P.; Gangji, V.; Communi, D.; Rasschaert, J. Human Carbamylome Description Identifies Carbamylated A2-Macroglobulin and Hemopexin as Two Novel Autoantigens in Early Rheumatoid Arthritis. Rheumatology 2022, 61, 2826–2834. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.L.; Darrah, E.; Lam, G.K.; Bartlett, S.J.; Giles, J.T.; Grant, A.V.; Gao, P.; Scott, W.W.; El-Gabalawy, H.; Casciola-Rosen, L.; et al. Association of Autoimmunity to Peptidyl Arginine Deiminase Type 4 with Genotype and Disease Severity in Rheumatoid Arthritis. Arthritis Rheum. 2008, 58, 1958–1967. [Google Scholar] [CrossRef] [PubMed]
- Halvorsen, E.H.; Pollmann, S.; Gilboe, I.-M.; van der Heijde, D.; Landewé, R.; Ødegård, S.; Kvien, T.K.; Molberg, Ø. Serum IgG Antibodies to Peptidylarginine Deiminase 4 in Rheumatoid Arthritis and Associations with Disease Severity. Ann. Rheum. Dis. 2008, 67, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Halvorsen, E.H.; Haavardsholm, E.A.; Pollmann, S.; Boonen, A.; van der Heijde, D.; Kvien, T.K.; Molberg, Ø. Serum IgG Antibodies to Peptidylarginine Deiminase 4 Predict Radiographic Progression in Patients with Rheumatoid Arthritis Treated with Tumour Necrosis Factor-Alpha Blocking Agents. Ann. Rheum. Dis. 2009, 68, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Millán, I.; Darrah, E.; Westfall, A.O.; Mikuls, T.R.; Reynolds, R.J.; Danila, M.I.; Curtis, J.R.; CLEAR Investigators; Rosen, A.; Bridges, S.L. Association of Anti-Peptidyl Arginine Deiminase Antibodies with Radiographic Severity of Rheumatoid Arthritis in African Americans. Arthritis Res. Ther. 2016, 18, 241. [Google Scholar] [CrossRef] [PubMed]
- Darrah, E.; Giles, J.T.; Ols, M.L.; Bull, H.G.; Andrade, F.; Rosen, A. Erosive Rheumatoid Arthritis Is Associated with Antibodies That Activate PAD4 by Increasing Calcium Sensitivity. Sci. Transl. Med. 2013, 5, 186ra65. [Google Scholar] [CrossRef] [PubMed]
- Lamacchia, C.; Courvoisier, D.S.; Jarlborg, M.; Bas, S.; Roux-Lombard, P.; Möller, B.; Ciurea, A.; Finckh, A.; Bentow, C.; Martinez-Prat, L.; et al. Predictive Value of Anti-CarP and Anti-PAD3 Antibodies Alone or in Combination with RF and ACPA on the Severity of Rheumatoid Arthritis. Rheumatology 2021, 60, 4598–4608. [Google Scholar] [CrossRef] [PubMed]
- Kolfenbach, J.R.; Deane, K.D.; Derber, L.A.; O’Donnell, C.I.; Gilliland, W.R.; Edison, J.D.; Rosen, A.; Darrah, E.; Norris, J.M.; Holers, V.M. Autoimmunity to Peptidyl Arginine Deiminase Type 4 Precedes Clinical Onset of Rheumatoid Arthritis. Arthritis Rheum. 2010, 62, 2633–2639. [Google Scholar] [CrossRef] [PubMed]
- Darrah, E.; Giles, J.T.; Davis, R.L.; Naik, P.; Wang, H.; Konig, M.F.; Cappelli, L.C.; Bingham, C.O.; Danoff, S.K.; Andrade, F. Autoantibodies to Peptidylarginine Deiminase 2 Are Associated with Less Severe Disease in Rheumatoid Arthritis. Front. Immunol. 2018, 9, 2696. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.E.; Wang, T.; Duvvuri, B.; Feser, M.L.; Deane, K.D.; Solomon, J.J.; Nelson, J.L.; Demoruelle, M.K.; Lood, C. Prediction of Erosive Disease Development by Antimitochondrial Antibodies in Rheumatoid Arthritis. Arthritis Rheumatol. 2023, 75, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Rykova, E.; Sizikov, A.; Roggenbuck, D.; Antonenko, O.; Bryzgalov, L.; Morozkin, E.; Skvortsova, K.; Vlassov, V.; Laktionov, P.; Kozlov, V. Circulating DNA in Rheumatoid Arthritis: Pathological Changes and Association with Clinically Used Serological Markers. Arthritis Res. Ther. 2017, 19, 85. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.M.; Yan, S.D.; Yan, S.F.; Stern, D.M. The Multiligand Receptor RAGE as a Progression Factor Amplifying Immune and Inflammatory Responses. J. Clin. Investig. 2001, 108, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Thiele, G.M.; Duryee, M.J.; Anderson, D.R.; Klassen, L.W.; Mohring, S.M.; Young, K.A.; Benissan-Messan, D.; Sayles, H.; Dusad, A.; Hunter, C.D.; et al. Malondialdehyde-Acetaldehyde Adducts (MAA) and Anti-MAA Antibody in Rheumatoid Arthritis. Arthritis Rheumatol. 2015, 67, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Nikolov, A.; Blazhev, A.; Tzekova, M.; Kostov, K.; Popovski, N. Serum Levels of Antibodies to Advanced Glycation End Products in Patients with Type 2 Diabetes Mellitus and Hypertension. Folia Med. 2020, 62, 295–301. [Google Scholar] [CrossRef] [PubMed]
- van den Beukel, M.D.; van Wesemael, T.J.; Hoogslag, A.T.W.; Borggreven, N.V.; Huizinga, T.W.; van der Helm-van Mil, A.H.; Toes, R.E.; van der Woude, D.; Trouw, L.A. Antibodies against Advanced Glycation End-Products and Malondialdehyde-Acetaldehyde Adducts Identify a New Specific Subgroup of Hitherto Patients with Seronegative Arthritis with a Distinct Clinical Phenotype and an HLA Class II Association. RMD Open 2023, 9, e003480. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Mo, W.; Wu, L.; Wu, X.; Luo, C.; Xiao, X.; Jia, X.; Yang, H.; Fei, Y.; Chen, H.; et al. Novel Autoantibodies Identified in ACPA-Negative Rheumatoid Arthritis. Ann. Rheum. Dis. 2021, 80, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Cappelli, L.C.; Gutierrez, A.K.; Bingham, C.O.; Shah, A.A. Rheumatic and Musculoskeletal Immune-Related Adverse Events Due to Immune Checkpoint Inhibitors: A Systematic Review of the Literature. Arthritis Care Res. 2017, 69, 1751–1763. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, N.; Tiongson, M.D.; Stewart, C.; Chan, K.K.; Jivanelli, B.; Cappelli, L.; Bass, A.R. Checkpoint Inhibitor–Associated Arthritis. J. Clin. Rheumatol. Pract. Rep. Rheum. Musculoskelet. Dis. 2021, 27, e317–e322. [Google Scholar] [CrossRef] [PubMed]
- Cappelli, L.C.; Bingham, C.O.; Forde, P.M.; Anagnostou, V.; Brahmer, J.; Lipson, E.J.; Mammen, J.; Schollenberger, M.; Shah, A.A.; Darrah, E. Anti-RA33 Antibodies Are Present in a Subset of Patients with Immune Checkpoint Inhibitor-Induced Inflammatory Arthritis. RMD Open 2022, 8, e002511. [Google Scholar] [CrossRef] [PubMed]
- Boutet, M.-A.; Nerviani, A.; Lliso-Ribera, G.; Leone, R.; Sironi, M.; Hands, R.; Rivellese, F.; Del Prete, A.; Goldmann, K.; Lewis, M.J.; et al. Circulating and Synovial Pentraxin-3 (PTX3) Expression Levels Correlate with Rheumatoid Arthritis Severity and Tissue Infiltration Independently of Conventional Treatments Response. Front. Immunol. 2021, 12, 686795. [Google Scholar] [CrossRef] [PubMed]
- Deban, L.; Jaillon, S.; Garlanda, C.; Bottazzi, B.; Mantovani, A. Pentraxins in Innate Immunity: Lessons from PTX3. Cell Tissue Res. 2011, 343, 237–249. [Google Scholar] [CrossRef]
- Balbaloglu, O.; Ozcan, S.S. Is Pentraxin 3 Level an Effective Biomarker in Disease Activity in Patients with Rheumatoid Arthritis? Arch. Med. Sci. AMS 2019, 16, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Luchetti, M.M.; Piccinini, G.; Mantovani, A.; Peri, G.; Matteucci, C.; Pomponio, G.; Fratini, M.; Fraticelli, P.; Sambo, P.; Loreto, C.D.; et al. Expression and Production of the Long Pentraxin PTX3 in Rheumatoid Arthritis (RA). Clin. Exp. Immunol. 2000, 119, 196. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, Y.F.; Aizaki, Y.; Noma, H.; Yokota, K.; Matsuda, M.; Kozu, N.; Takebayashi, Y.; Nakatani, H.; Hasunuma, T.; Kawai, S.; et al. Plasma Pentraxin 3 Is Associated with Progression of Radiographic Joint Damage, but Not Carotid Atherosclerosis, in Female Rheumatoid Arthritis Patients: 3-Year Prospective Study. Mod. Rheumatol. 2020, 30, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Khan, R.; Gupta, N.; Sharma, A.; Zaheer, M.S.; Abbas, M.; Khan, S.A. Acute Phase Reactant, Pentraxin 3, as a Novel Marker for the Diagnosis of Rheumatoid Arthritis. Clin. Chim. Acta Int. J. Clin. Chem. 2018, 480, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Liou, L.; Tsai, W.; Chang, C.J.; Chao, W.; Chen, M. Blood Monocyte Chemotactic Protein-1 (MCP-1) and Adapted Disease Activity Score28-MCP-1: Favorable Indicators for Rheumatoid Arthritis Activity. PLoS ONE 2013, 8, e55346. [Google Scholar] [CrossRef] [PubMed]
- Tekeoğlu, İ.; Harman, H.; Sağ, S.; Altındiş, M.; Kamanlı, A.; Nas, K. Levels of Serum Pentraxin 3, IL-6, Fetuin A and Insulin in Patients with Rheumatoid Arthritis. Cytokine 2016, 83, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Weitoft, T.; Larsson, A.; Saxne, T.; Manivel, V.A.; Lysholm, J.; Knight, A.; Rönnelid, J. Pentraxin 3 in Serum and Synovial Fluid of Patients with Rheumatoid Arthritis with and without Autoantibodies. Scand. J. Rheumatol. 2017, 46, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Alivernini, S.; Gremese, E.; McSharry, C.; Tolusso, B.; Ferraccioli, G.; McInnes, I.B.; Kurowska-Stolarska, M. MicroRNA-155-at the Critical Interface of Innate and Adaptive Immunity in Arthritis. Front. Immunol. 2017, 8, 1932. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, S. Posttranscriptional Upregulation by microRNAs. Wiley Interdiscip. Rev. RNA 2012, 3, 311–330. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most Mammalian mRNAs Are Conserved Targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Kurowska-Stolarska, M.; Alivernini, S.; Ballantine, L.E.; Asquith, D.L.; Millar, N.L.; Gilchrist, D.S.; Reilly, J.; Ierna, M.; Fraser, A.R.; Stolarski, B.; et al. MicroRNA-155 as a Proinflammatory Regulator in Clinical and Experimental Arthritis. Proc. Natl. Acad. Sci. USA 2011, 108, 11193–11198. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Jin, L.; Yan, L.; Shi, J.; Wang, H.; Zhou, B.; Wu, X. Comprehensive Review of Genetic Association Studies and Meta-Analysis on miRNA Polymorphisms and Rheumatoid Arthritis and Systemic Lupus Erythematosus Susceptibility. Hum. Immunol. 2016, 77, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Stanczyk, J.; Ospelt, C.; Karouzakis, E.; Filer, A.; Raza, K.; Kolling, C.; Gay, R.; Buckley, C.D.; Tak, P.P.; Gay, S.; et al. Altered Expression of microRNA-203 in Rheumatoid Arthritis Synovial Fibroblasts and Its Role in Fibroblast Activation. Arthritis Rheum. 2011, 63, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Bogunia-Kubik, K.; Wysoczańska, B.; Piątek, D.; Iwaszko, M.; Ciechomska, M.; Świerkot, J. Significance of Polymorphism and Expression of miR-146a and NFkB1 Genetic Variants in Patients with Rheumatoid Arthritis. Arch. Immunol. Ther. Exp. 2016, 64, 131–136. [Google Scholar] [CrossRef] [PubMed]
- He, X.-H.; Xiao, Y.-T.; Chen, W.-Y.; Wang, M.-J.; Wu, X.-D.; Mei, L.-Y.; Gao, K.-X.; Huang, Q.-C.; Huang, R.-Y.; Chen, X.-M. In Silico Analysis of Serum miRNA Profiles in Seronegative and Seropositive Rheumatoid Arthritis Patients by Small RNA Sequencing. PeerJ 2023, 11, e15690. [Google Scholar] [CrossRef] [PubMed]
- Bresnihan, B.; Baeten, D.; Firestein, G.S.; Fitzgerald, O.M.; Gerlag, D.M.; Haringman, J.J.; McInnes, I.B.; Reece, R.J.; Smith, M.D.; Ulfgren, A.-K.; et al. Synovial Tissue Analysis in Clinical Trials. J. Rheumatol. 2005, 32, 2481–2484. [Google Scholar] [PubMed]
- Kelly, S.; Humby, F.; Filer, A.; Ng, N.; Cicco, M.D.; Hands, R.E.; Rocher, V.; Bombardieri, M.; D’Agostino, M.A.; McInnes, I.B.; et al. Ultrasound-Guided Synovial Biopsy: A Safe, Well-Tolerated and Reliable Technique for Obtaining High-Quality Synovial Tissue from Both Large and Small Joints in Early Arthritis Patients. Ann. Rheum. Dis. 2015, 74, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Humby, F.; Lewis, M.; Ramamoorthi, N.; Hackney, J.A.; Barnes, M.R.; Bombardieri, M.; Setiadi, A.F.; Kelly, S.; Bene, F.; DiCicco, M.; et al. Synovial Cellular and Molecular Signatures Stratify Clinical Response to csDMARD Therapy and Predict Radiographic Progression in Early Rheumatoid Arthritis Patients. Ann. Rheum. Dis. 2019, 78, 761–772. [Google Scholar] [CrossRef]
- Lliso-Ribera, G.; Humby, F.; Lewis, M.; Nerviani, A.; Mauro, D.; Rivellese, F.; Kelly, S.; Hands, R.; Bene, F.; Ramamoorthi, N.; et al. Synovial Tissue Signatures Enhance Clinical Classification and Prognostic/Treatment Response Algorithms in Early Inflammatory Arthritis and Predict Requirement for Subsequent Biological Therapy: Results from the Pathobiology of Early Arthritis Cohort (PEAC). Ann. Rheum. Dis. 2019, 78, 1642–1652. [Google Scholar] [CrossRef] [PubMed]
- Nerviani, A.; Di Cicco, M.; Mahto, A.; Lliso-Ribera, G.; Rivellese, F.; Thorborn, G.; Hands, R.; Bellan, M.; Mauro, D.; Boutet, M.-A.; et al. A Pauci-Immune Synovial Pathotype Predicts Inadequate Response to TNFα-Blockade in Rheumatoid Arthritis Patients. Front. Immunol. 2020, 11, 845. [Google Scholar] [CrossRef] [PubMed]
- Bombardieri, M.; Lewis, M.; Pitzalis, C. Ectopic Lymphoid Neogenesis in Rheumatic Autoimmune Diseases. Nat. Rev. Rheumatol. 2017, 13, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Aloisi, F.; Pujol-Borrell, R. Lymphoid Neogenesis in Chronic Inflammatory Diseases. Nat. Rev. Immunol. 2006, 6, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Bugatti, S.; Caporali, R.; Manzo, A.; Vitolo, B.; Pitzalis, C.; Montecucco, C. Involvement of Subchondral Bone Marrow in Rheumatoid Arthritis: Lymphoid Neogenesis and in Situ Relationship to Subchondral Bone Marrow Osteoclast Recruitment. Arthritis Rheum. 2005, 52, 3448–3459. [Google Scholar] [CrossRef] [PubMed]
- Aziz, K.E.; McCluskey, P.J.; Wakefield, D. Characterisation of Follicular Dendritic Cells in Labial Salivary Glands of Patients with Primary Sjögren Syndrome: Comparison with Tonsillar Lymphoid Follicles. Ann. Rheum. Dis. 1997, 56, 140–143. [Google Scholar] [CrossRef]
- Chang, A.; Henderson, S.G.; Brandt, D.; Liu, N.; Guttikonda, R.; Hsieh, C.; Kaverina, N.; Utset, T.O.; Meehan, S.M.; Quigg, R.J.; et al. In Situ B Cell-Mediated Immune Responses and Tubulointerstitial Inflammation in Human Lupus Nephritis. J. Immunol. 2011, 186, 1849–1860. [Google Scholar] [CrossRef] [PubMed]
- Boutet, M.-A.; Nerviani, A.; Fossati-Jimack, L.; Hands-Greenwood, R.; Ahmed, M.; Rivellese, F.; Pitzalis, C. Comparative Analysis of Late-Stage Rheumatoid Arthritis and Osteoarthritis Reveals Shared Histopathological Features. Osteoarthr. Cartil. 2024, 32, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.; Pitcher, L.A.; Sullivan, J.M.; Mitsdoerffer, M.; Acton, S.E.; Franz, B.; Wucherpfennig, K.; Turley, S.; Carroll, M.C.; Sobel, R.A.; et al. Th17 Cells Induce Ectopic Lymphoid Follicles in Central Nervous System Tissue Inflammation. Immunity 2011, 35, 986–996. [Google Scholar] [CrossRef] [PubMed]
- Cañete, J.D.; Celis, R.; Yeremenko, N.; Sanmartí, R.; van Duivenvoorde, L.; Ramírez, J.; Blijdorp, I.; García-Herrero, C.M.; Pablos, J.L.; Baeten, D.L. Ectopic Lymphoid Neogenesis Is Strongly Associated with Activation of the IL-23 Pathway in Rheumatoid Synovitis. Arthritis Res. Ther. 2015, 17, 173. [Google Scholar] [CrossRef] [PubMed]
- Corsiero, E.; Bombardieri, M.; Manzo, A.; Bugatti, S.; Uguccioni, M.; Pitzalis, C. Role of Lymphoid Chemokines in the Development of Functional Ectopic Lymphoid Structures in Rheumatic Autoimmune Diseases. Immunol. Lett. 2012, 145, 62–67. [Google Scholar] [CrossRef] [PubMed]
- van de Pavert, S.A.; Mebius, R.E. New Insights into the Development of Lymphoid Tissues. Nat. Rev. Immunol. 2010, 10, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Comerford, I.; Harata-Lee, Y.; Bunting, M.D.; Gregor, C.; Kara, E.E.; McColl, S.R. A Myriad of Functions and Complex Regulation of the CCR7/CCL19/CCL21 Chemokine Axis in the Adaptive Immune System. Cytokine Growth Factor Rev. 2013, 24, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Yanni, G.; Whelan, A.; Feighery, C.; Bresnihan, B. Analysis of Cell Populations in Rheumatoid Arthritis Synovial Tissues. Semin. Arthritis Rheum. 1992, 21, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Thurlings, R.M.; Wijbrandts, C.A.; Mebius, R.E.; Cantaert, T.; Dinant, H.J.; van der Pouw-Kraan, T.C.T.M.; Verweij, C.L.; Baeten, D.; Tak, P.P. Synovial Lymphoid Neogenesis Does Not Define a Specific Clinical Rheumatoid Arthritis Phenotype. Arthritis Rheum. 2008, 58, 1582–1589. [Google Scholar] [CrossRef] [PubMed]
- Vos, K.; Thurlings, R.M.; Wijbrandts, C.A.; van Schaardenburg, D.; Gerlag, D.M.; Tak, P.P. Early Effects of Rituximab on the Synovial Cell Infiltrate in Patients with Rheumatoid Arthritis. Arthritis Rheum. 2007, 56, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Humby, F.; Bombardieri, M.; Manzo, A.; Kelly, S.; Blades, M.C.; Kirkham, B.; Spencer, J.; Pitzalis, C. Ectopic Lymphoid Structures Support Ongoing Production of Class-Switched Autoantibodies in Rheumatoid Synovium. PLoS Med. 2009, 6, e1. [Google Scholar] [CrossRef] [PubMed]
- Cañete, J.D.; Celis, R.; Moll, C.; Izquierdo, E.; Marsal, S.; Sanmartí, R.; Palacín, A.; Lora, D.; de la Cruz, J.; Pablos, J.L. Clinical Significance of Synovial Lymphoid Neogenesis and Its Reversal after Anti-Tumour Necrosis Factor Alpha Therapy in Rheumatoid Arthritis. Ann. Rheum. Dis. 2009, 68, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.J.; Barnes, M.R.; Blighe, K.; Goldmann, K.; Rana, S.; Hackney, J.A.; Ramamoorthi, N.; John, C.R.; Watson, D.S.; Kummerfeld, S.K.; et al. Molecular Portraits of Early Rheumatoid Arthritis Identify Clinical and Treatment Response Phenotypes. Cell Rep. 2019, 28, 2455–2470.e5. [Google Scholar] [CrossRef] [PubMed]
- Humby, F.; Durez, P.; Buch, M.H.; Lewis, M.J.; Rizvi, H.; Rivellese, F.; Nerviani, A.; Giorli, G.; Mahto, A.; Montecucco, C.; et al. Rituximab versus Tocilizumab in Anti-TNF Inadequate Responder Patients with Rheumatoid Arthritis (R4RA): 16-Week Outcomes of a Stratified, Biopsy-Driven, Multicentre, Open-Label, Phase 4 Randomised Controlled Trial. Lancet 2021, 397, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Rivellese, F.; Surace, A.E.A.; Goldmann, K.; Sciacca, E.; Çubuk, C.; Giorli, G.; John, C.R.; Nerviani, A.; Fossati-Jimack, L.; Thorborn, G.; et al. Rituximab versus Tocilizumab in Rheumatoid Arthritis: Synovial Biopsy-Based Biomarker Analysis of the Phase 4 R4RA Randomized Trial. Nat. Med. 2022, 28, 1256–1268. [Google Scholar] [CrossRef]
- Zhang, F.; Jonsson, A.H.; Nathan, A.; Wei, K.; Millard, N.; Xiao, Q.; Gutierrez-Arcelus, M.; Apruzzese, W.; Watts, G.F.M.; Weisenfeld, D.; et al. Cellular Deconstruction of Inflamed Synovium Defines Diverse Inflammatory Phenotypes in Rheumatoid Arthritis. BioRxiv 2022. [Google Scholar] [CrossRef]
- Krenn, V.; Morawietz, L.; Burmester, G.-R.; Kinne, R.W.; Mueller-Ladner, U.; Muller, B.; Haupl, T. Synovitis Score: Discrimination between Chronic Low-Grade and High-Grade Synovitis. Histopathology 2006, 49, 358–364. [Google Scholar] [CrossRef]
- Paalanen, K.; Puolakka, K.; Nikiphorou, E.; Hannonen, P.; Sokka, T. Is Seronegative Rheumatoid Arthritis True Rheumatoid Arthritis? A Nationwide Cohort Study. Rheumatology 2021, 60, 2391–2395. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, F.; Romagnani, C.; Romagnani, S. The 3 Major Types of Innate and Adaptive Cell-Mediated Effector Immunity. J. Allergy Clin. Immunol. 2015, 135, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-W.; Kim, H.-R.; Kim, B.-M.; Cho, M.-L.; Lee, S.-H. Th17 Cytokines Regulate Osteoclastogenesis in Rheumatoid Arthritis. Am. J. Pathol. 2015, 185, 3011–3024. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.D.; Ryan, S.E.; Steel, K.J.A.; van den Beukel, M.D.; Trouw, L.A.; van Schie, K.A.J.; Toes, R.E.M.; Menon, B.; Kirkham, B.W.; Taams, L.S. Type 17-Specific Immune Pathways Are Active in Early Spondyloarthritis. RMD Open 2023, 9, e003328. [Google Scholar] [CrossRef] [PubMed]
- Azuaga, A.B.; Cuervo, A.; Celis, R.; Frade-Sosa, B.; Sarmiento-Monroy, J.C.; Ruiz-Esquide, V.; Gómez-Puerta, J.A.; Sanmartí, R.; Ramírez, J. Synovial Tissue Features Associated with Poor Prognosis in Inflammatory Arthritis. Arthritis Res. Ther. 2024, 26, 18. [Google Scholar] [CrossRef]
- Nakayama, T.; Hirahara, K.; Onodera, A.; Endo, Y.; Hosokawa, H.; Shinoda, K.; Tumes, D.J.; Okamoto, Y. Th2 Cells in Health and Disease. Annu. Rev. Immunol. 2017, 35, 53–84. [Google Scholar] [CrossRef] [PubMed]
- Bridgewood, C.; Wittmann, M.; Macleod, T.; Watad, A.; Newton, D.; Bhan, K.; Amital, H.; Damiani, G.; Giryes, S.; Bragazzi, N.L.; et al. T Helper 2 IL-4/IL-13 Dual Blockade with Dupilumab Is Linked to Some Emergent T Helper 17–Type Diseases, Including Seronegative Arthritis and Enthesitis/Enthesopathy, but Not to Humoral Autoimmune Diseases. J. Investig. Dermatol. 2022, 142, 2660–2667. [Google Scholar] [CrossRef] [PubMed]
- Jaulent, L.; Staumont-Sallé, D.; Tauber, M.; Paul, C.; Aubert, H.; Marchetti, A.; Sassolas, B.; Valois, A.; Nicolas, J.-F.; Nosbaum, A.; et al. De Novo Psoriasis in Atopic Dermatitis Patients Treated with Dupilumab: A Retrospective Cohort. J. Eur. Acad. Dermatol. Venereol. JEADV 2021, 35, e296–e297. [Google Scholar] [CrossRef]
- Bridgewood, C.; Sharif, K.; Freeston, J.; Saleem, B.; Russell, T.; Watad, A.; Khan, A.; Loughenbury, P.; Rao, A.; Wittmann, M.; et al. Regulation of Entheseal IL-23 Expression by IL-4 and IL-13 as an Explanation for Arthropathy Development under Dupilumab Therapy. Rheumatology 2021, 60, 2461–2466. [Google Scholar] [CrossRef]
- Bridgewood, C.; Watad, A.; Russell, T.; Palmer, T.M.; Marzo-Ortega, H.; Khan, A.; Millner, P.A.; Dunsmuir, R.; Rao, A.; Loughenbury, P.; et al. Identification of Myeloid Cells in the Human Enthesis as the Main Source of Local IL-23 Production. Ann. Rheum. Dis. 2019, 78, 929–933. [Google Scholar] [CrossRef] [PubMed]
- Bendrups, A.; Hilton, A.; Meager, A.; Hamilton, J.A. Reduction of Tumor Necrosis Factor Alpha and Interleukin-1 Beta Levels in Human Synovial Tissue by Interleukin-4 and Glucocorticoid. Rheumatol. Int. 1993, 12, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, N.; Bhatt, L.K.; Prabhavalkar, K.S. Experimental Animal Models for Rheumatoid Arthritis. Immunopharmacol. Immunotoxicol. 2018, 40, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Pearson: Development of Arthritis, Periarthritis—Google Scholar. Available online: https://scholar.google.com/scholar_lookup?author=CM+Pearson&publication_year=1956&title=Development%20of%20Arthritis%2C%20Periarthritis%20and%20Periostitis%20in%20Rats%20Given%20Adjuvants&journal=Proc+Soc+Exp+Biol+Med+Soc+Exp+Biol+Med&volume=91&pages=95 (accessed on 5 April 2024).
- Nandakumar, K.S.; Holmdahl, R. Collagen Antibody Induced Arthritis. Methods Mol. Med. 2007, 136, 215–223. [Google Scholar] [CrossRef]
- Inglis, J.J.; Notley, C.A.; Essex, D.; Wilson, A.W.; Feldmann, M.; Anand, P.; Williams, R. Collagen-Induced Arthritis as a Model of Hyperalgesia: Functional and Cellular Analysis of the Analgesic Actions of Tumor Necrosis Factor Blockade. Arthritis Rheum. 2007, 56, 4015–4023. [Google Scholar] [CrossRef] [PubMed]
- Taneja, V.; Taneja, N.; Paisansinsup, T.; Behrens, M.; Griffiths, M.; Luthra, H.; David, C.S. CD4 and CD8 T Cells in Susceptibility/Protection to Collagen-Induced Arthritis in HLA-DQ8-Transgenic Mice: Implications for Rheumatoid Arthritis. J. Immunol. 2002, 168, 5867–5875. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.-S.; Jeong, G.H.; Yoo, S.-A. The Use of Animal Models in Rheumatoid Arthritis Research. J. Yeungnam Med. Sci. 2022, 40, 23–29. [Google Scholar] [CrossRef]
- Bitoun, S.; Roques, P.; Larcher, T.; Nocturne, G.; Serguera, C.; Chrétien, P.; Serre, G.; Grand, R.L.; Mariette, X. Both Systemic and Intra-Articular Immunization with Citrullinated Peptides Are Needed to Induce Arthritis in the Macaque. Front. Immunol. 2017, 8, 1816. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Xie, Z.; Xi, Y.; Liu, L.; Li, Z.; Qin, D. How to Model Rheumatoid Arthritis in Animals: From Rodents to Non-Human Primates. Front. Immunol. 2022, 13, 887460. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, G.; Ramesh, A.; Aishwarya, P.; Sally, J.; Ravi, M. Devices and Techniques Used to Obtain and Analyze Three-Dimensional Cell Cultures. Biotechnol. Prog. 2021, 37, e3126. [Google Scholar] [CrossRef] [PubMed]
- Rothbauer, M.; Höll, G.; Eilenberger, C.; Kratz, S.R.A.; Farooq, B.; Schuller, P.; Calvo, I.O.; Byrne, R.A.; Meyer, B.; Niederreiter, B.; et al. Monitoring Tissue-Level Remodelling during Inflammatory Arthritis Using a Three-Dimensional Synovium-on-a-Chip with Non-Invasive Light Scattering Biosensing. Lab Chip 2020, 20, 1461–1471. [Google Scholar] [CrossRef]
- Yao, Y.; Xu, X.; Yang, L.; Zhu, J.; Wan, J.; Shen, L.; Xia, F.; Fu, G.; Deng, Y.; Pan, M.; et al. Patient-Derived Organoids Predict Chemoradiation Responses of Locally Advanced Rectal Cancer. Cell Stem Cell 2020, 26, 17–26.e6. [Google Scholar] [CrossRef] [PubMed]
- Bhamidipati, K.; Wei, K. Precision Medicine in Rheumatoid Arthritis. Best Pract. Res. Clin. Rheumatol. 2022, 36, 101742. [Google Scholar] [CrossRef] [PubMed]
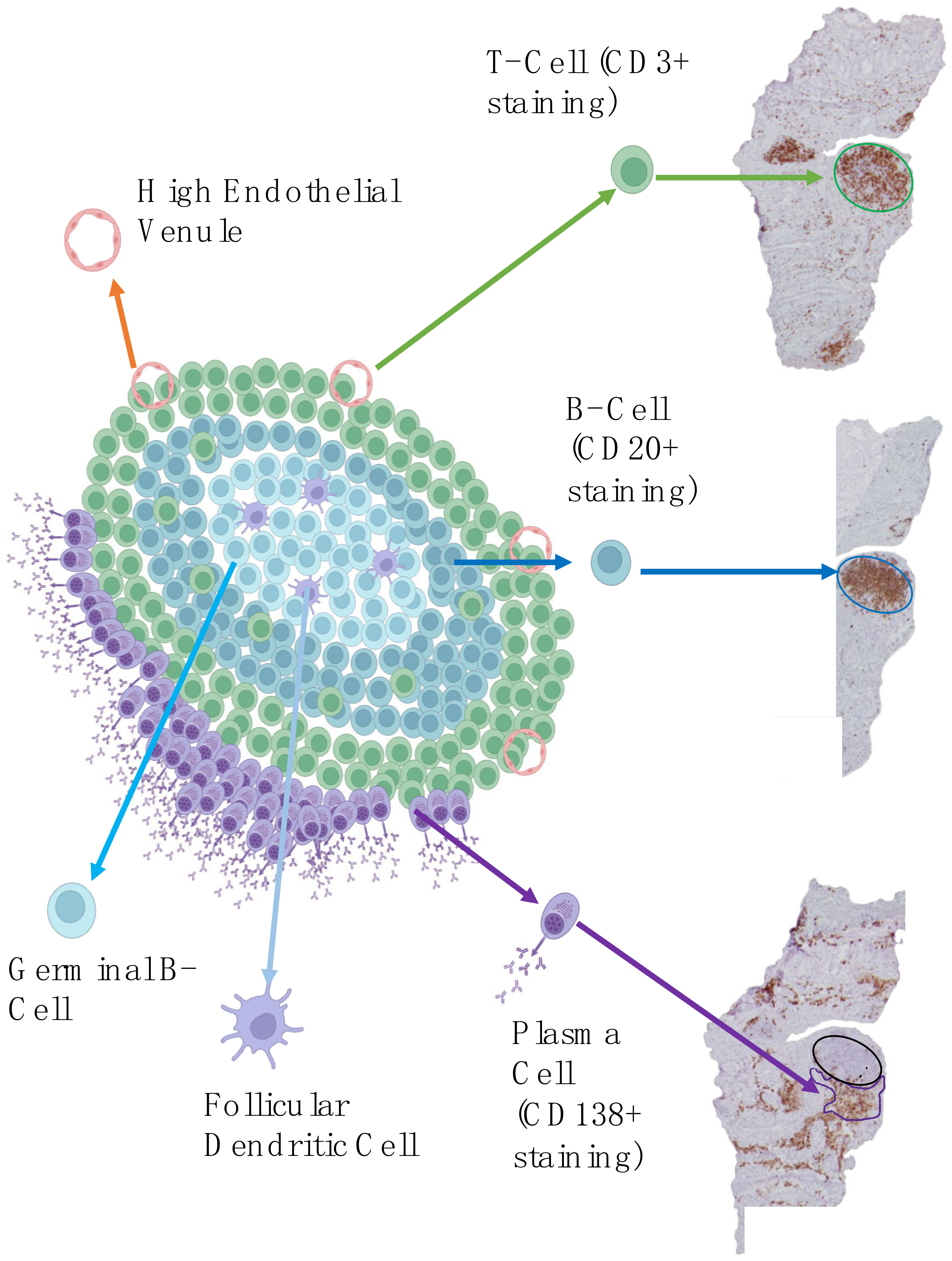
| Seropositive | Seronegative | |
|---|---|---|
| Age at diagnosis [20] | 43 ± 14 years | 54 ± 11 years |
| Genetic and environmental factor differences [25,26,27,28,29,30] |
|
|
| Immunopathogenesis [21,36,37,42,51] |
|
|
| Clinical features [61,62,63,64,68,84] |
|
|
| Synovial histological and molecular differences [15,155,174,177] |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perera, J.; Delrosso, C.A.; Nerviani, A.; Pitzalis, C. Clinical Phenotypes, Serological Biomarkers, and Synovial Features Defining Seropositive and Seronegative Rheumatoid Arthritis: A Literature Review. Cells 2024, 13, 743. https://doi.org/10.3390/cells13090743
Perera J, Delrosso CA, Nerviani A, Pitzalis C. Clinical Phenotypes, Serological Biomarkers, and Synovial Features Defining Seropositive and Seronegative Rheumatoid Arthritis: A Literature Review. Cells. 2024; 13(9):743. https://doi.org/10.3390/cells13090743
Chicago/Turabian StylePerera, James, Chiara Aurora Delrosso, Alessandra Nerviani, and Costantino Pitzalis. 2024. "Clinical Phenotypes, Serological Biomarkers, and Synovial Features Defining Seropositive and Seronegative Rheumatoid Arthritis: A Literature Review" Cells 13, no. 9: 743. https://doi.org/10.3390/cells13090743