DNA Methylation and Apoptosis Resistance in Cancer Cells


Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
| Gene methylated | Cancer | Ref |
|---|---|---|
| APAF-1 | Melanoma, Leukemia, Testicular (100 vs. 60), Bladder (11), RCC (100) | [14], [15], [16], [17], [18] |
| Bad | Myeloma | [19] |
| Bak | Myeloma | [19] |
| Bax | GBM, Myeloma | [20], [19] |
| Bcl2L10 | Gastric (38), Leukemia (12-45) | [21], [22] |
| Bim | CML | [23] |
| Bik | Glioma (30), HCC, RCC, Prostate, Myeloma | [24], [25], [26], [27] |
| BNIP3 | Pancreatic, Gastric (39), Breast, Colorectal, Leukemia, Myeloma, HCC | [28], [29], [30], [31] |
| Casp-8 | Medulloblastoma (62–81), Pituitary tract (54), Rhabdosarcoma (83 vs 0), Phaeochromocytoma (31), Neuroblastoma (35-52 vs 0), Retinoblastoma (59 vs. 0), HCC (34), GBM (30), Bladder (19), Lung (0–45 vs. 0), Rectal, Breast, Prostate, Gastric | [32], [33], [34], [35], [36], [37], [38], [39], [40], [41] |
| DAPK | Mesothelioma (20), Testicular (20–50 vs. 6), Nasopharyngial (76 vs. 0), Pituitary (43), Colorectal (81), ACC (27), Lung (25–44), Biliary tract (21), Lymphoma (71–85), GBM (14), Gastric (22–70), Leukemia (36), Breast (13–88), CXCA (56–79), Cholangiocarcinoma (31), Bladder (74–77), RCC (33–55), Head and Neck (11–33), Myeloma (40), Oesophageal (50–60 vs. 20), Ovarian | [42], [43], [44], [45], [46], [47], [48], [49], [16], [50], [36], [51], [52], [53] |
| DcR1-2 | Glioma (60), Neuroblatoma (11–25), Prostate (50), Breast, Prostate (37–45), Ovarian (31–43), Breast (70), Lung (31), Mesothelioma (63), Bladder (42), CXCA (100), Lymphoma (41), Leukemia (26), Myeloma (56), Phaeochromocytoma (23–26) | [54], [32], [55], [56], [57], [58], [37] |
| DR4 or 5 | Breast, Melanoma, Ovarian (10–28), Phaeochromocytoma (41), Neuroblastoma | [54], [59], [60], [37], [61] |
| Fas | Lymphomas, CXCA, Colon, Prostatic (12), Lung | [62], [63], [64], [65] |
| Hrk | Colorectal (36), Gastric (32), GBM (27–43), PCNSL (31), Prostate (38) | [66], [67], [68], [69] |
| Puma | Lymphoma | [12] |
| RASSF1a | ACC (42–45), Biliary tract (27), CRCC (45), Nasopharyngeal (71–84 vs. 0), Ovarian (26), Gastric, Bladder (48–60 vs. 42), Thyroid, Neuoblastoma (83–93), Osteosarcoma (14 vs. 1), Lung (43 vs. 5), Breast (63–74), , HCC Hepatoblastoma (39–44), CXCA (26 vs. 0), Mesothelioma, Glioma (12–82), Endometrial, Liver (38), Prostate, Parathiroid (98), Lung (26), Testis (78 vs. 0), Melanoma (69), Colorectal (31), Retinoblastoma (98) | [70], [71], [72], [73], [74], [75], [76], [32], [77], [78], [79], [80], [81], [82], [56], [83] |
| SARP2 | Pancreatic (90–90 vs. 0–5), Colorectal, Oesophageal | [84] |
| TMS1 | Cholangiocarcinoma (36), Prostatic (47–65), Colorectal (25–41 vs. 8), Ovarian (19 vs. 0), Thyroid (33), Breast (24–46), Gastric (32), GBM (21–57), Lung (41–70), Melanoma, Colorectal, Neuoblastoma, HCC, Pancreatic | [85], [86], [87], [88], [89], [55], [90], [91], [92], [93], [94] |
| TNFR10c | Pancreatic (54–97), Choroid plexus (50), Neuroblastoma (21), Breast (48), Lung (37), Mesothelioma (43), Ependymomas (50) | [95] |
| XAF-1 | Colorectal (40), Gastric, Bladder, RCC, Prostatic (35 vs. 0), Lung, GBM (22), Oesophageal | [96], [97], [98], [99], [34], [100] |
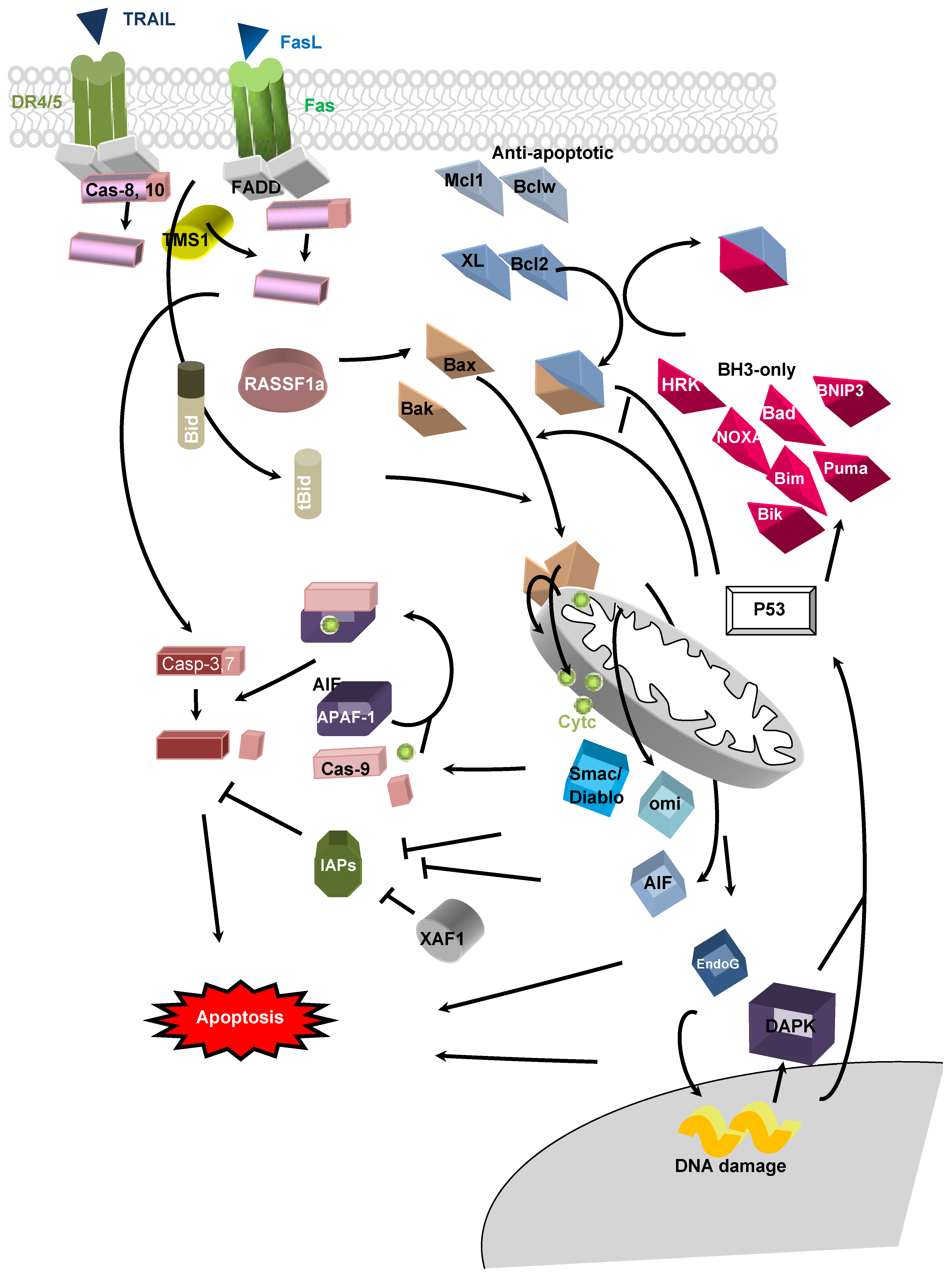
2. Apoptotic Pathways
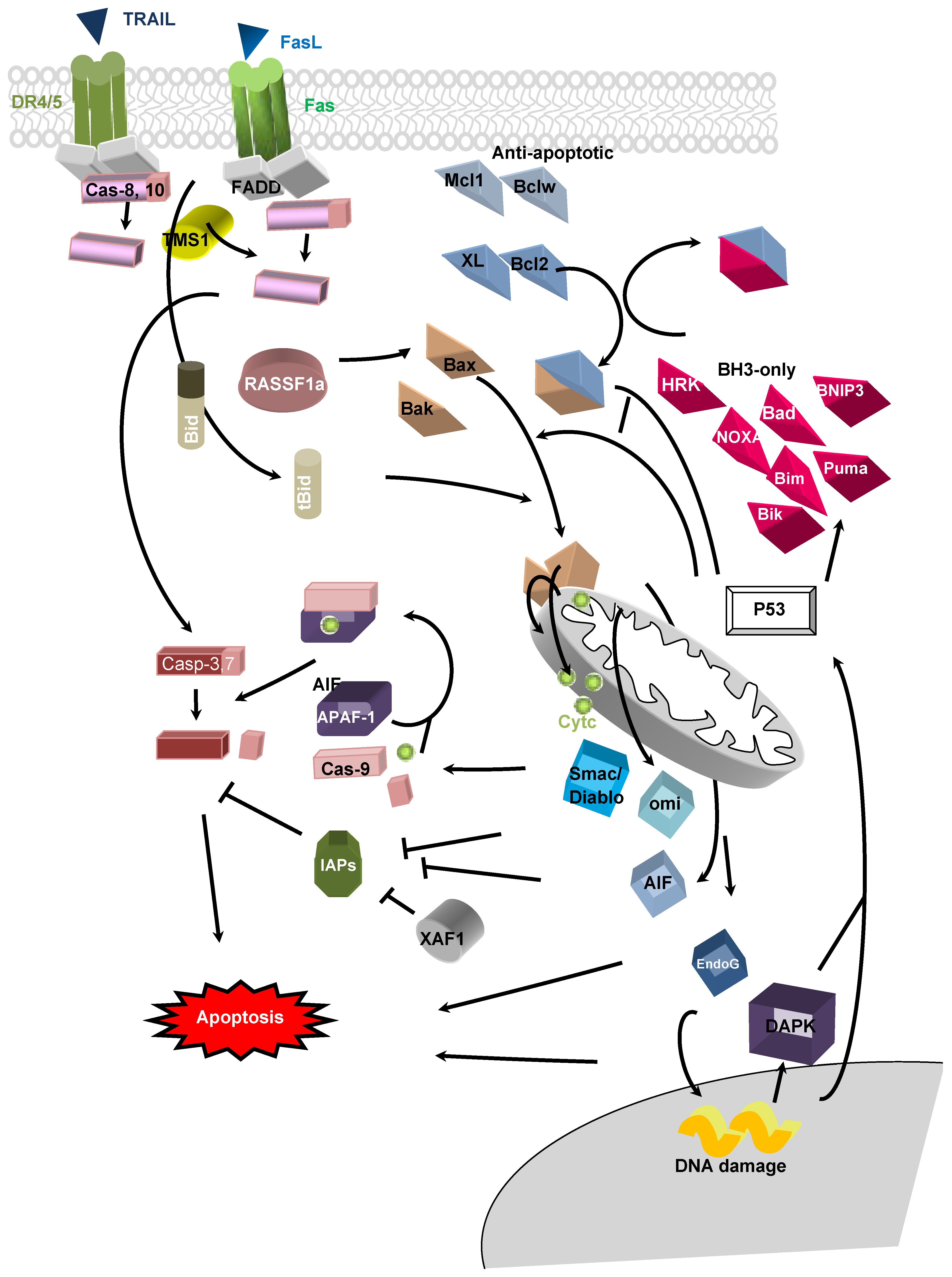
3. DNA Hypo/Hypermethylations in Apoptosis-Related Genes in Cancer
3.1. Extrinsic Pathway
3.1.1. Death Receptor
3.1.2. Caspase 8 (Casp 8)
3.1.3. TMS1/ASC (target for methylation-induced silencing-1/apoptosis-associated speck-like protein containing CARD-ASC).
3.2. Intrinsic Pathway
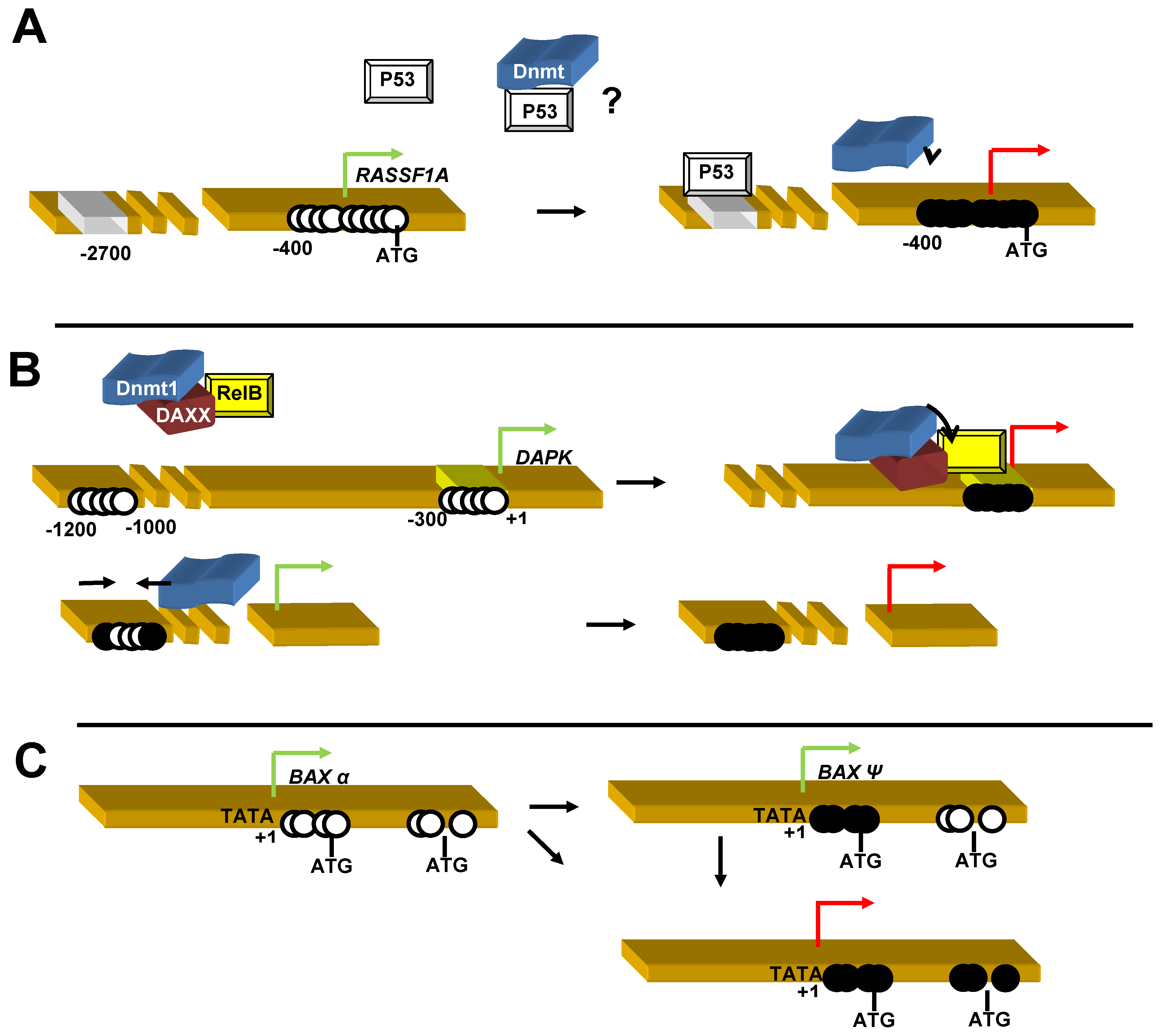
3.2.1. Bax
3.2.2. Bcl2
3.2.3. BH3-only
3.2.4. BCL2L10
3.2.5. APAF-1 (Apoptotic Protease-Activating Factor 1)
3.2.6. Inhibitor Apoptis Proteins (IAPs)
3.2.7. BNIP3
3.2.8. XAF-1
3.2.9. Death Associate Protein Kinase (DAPK)
3.2.10. Ras Association Doman Family 1A (RASSF1A)
3.2.11. Secreted Apoptos-Related Protein 2 (SARP2)
3.2.12. Interferon Regulatory Factor 8 (IRF8)
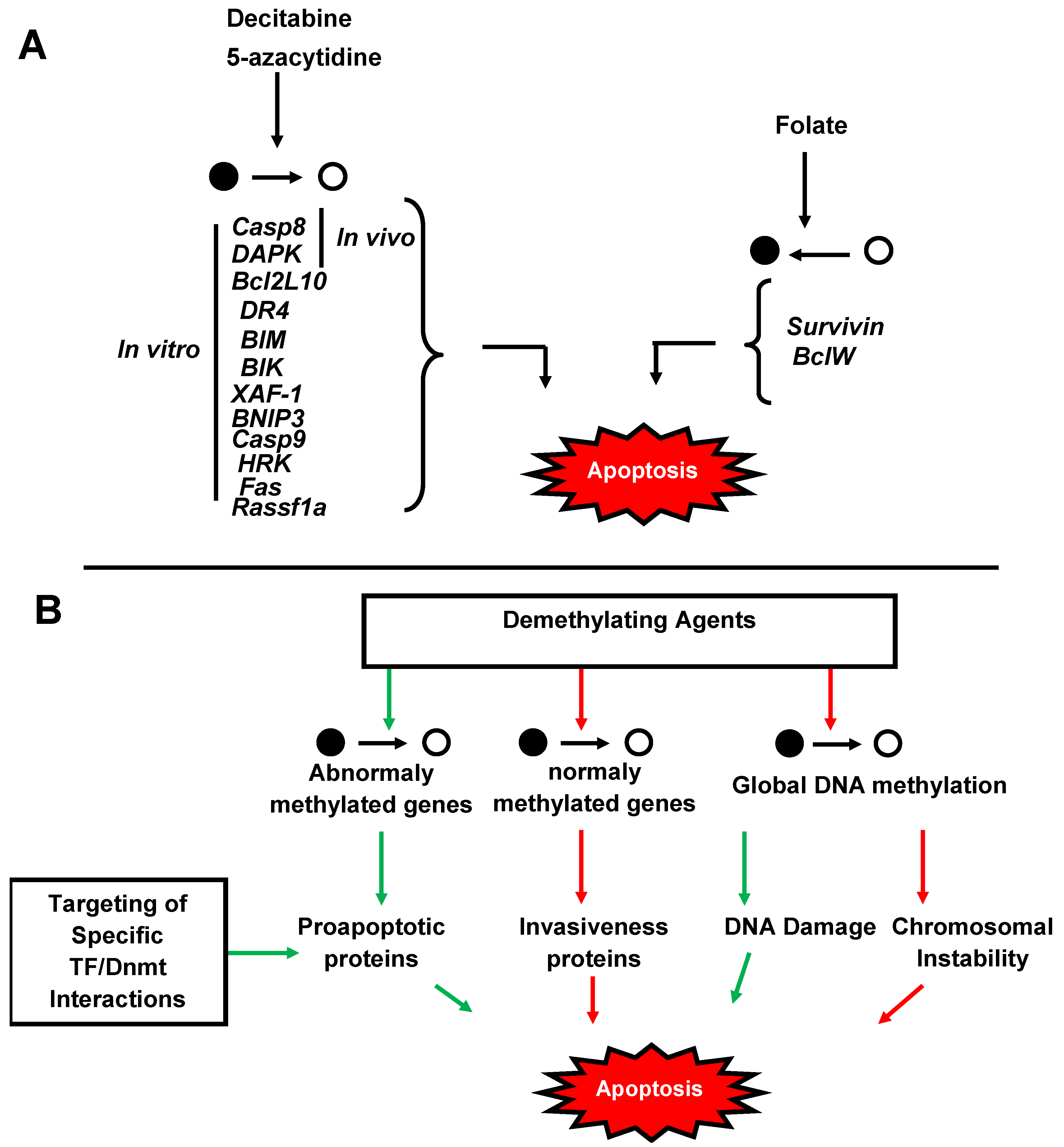
4. Significance of Methylation in Cancers
5. Therapeutical Strategies
6. Conclusions
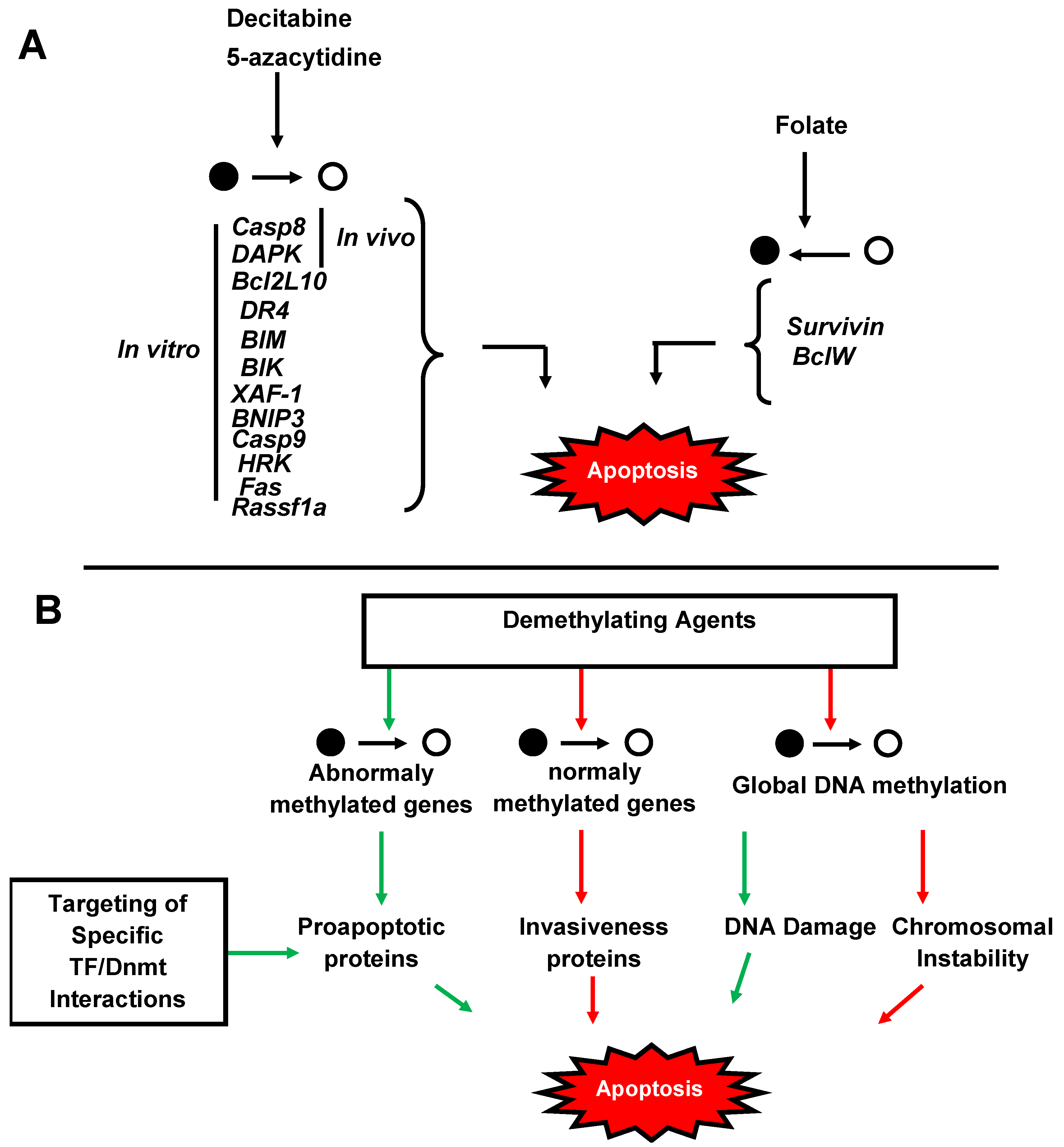
Acknowledgments
Conflict of Interest
References
- Kuribara, R.; Honda, H.; Matsui, H.; Shinjyo, T.; Inukai, T.; Sugita, K.; Nakazawa, S.; Hirai, H.; Ozawa, K.; Inaba, T. Roles of Bim in apoptosis of normal and Bcr-Abl-expressing hematopoietic progenitors. Mol. Cell. Biol. 2004, 24, 6172–6183. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev 2002, 16, 6–21. [Google Scholar] [CrossRef]
- Chen, T.; Ueda, Y.; Dodge, J.E.; Wang, Z.; Li, E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol. Cell. Biol. 2003, 23, 5594–5605. [Google Scholar] [CrossRef]
- Gama-Sosa, M.A.; Slagel, V.A.; Trewyn, R.W.; Oxenhandler, R.; Kuo, K.C.; Gehrke, C.W.; Ehrlich, M. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983, 11, 6883–6894. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef]
- Hoffmann, M.J.; Schulz, W.A. Causes and consequences of DNA hypomethylation in human cancer. Biochem. Cell. Biol. 2005, 83, 296–321. [Google Scholar] [CrossRef]
- Ehrlich, M.; Woods, C.B.; Yu, M.C.; Dubeau, L.; Yang, F.; Campan, M.; Weisenberger, D.J.; Long, T.; Youn, B.; Fiala, E.S.; et al. Quantitative analysis of associations between DNA hypermethylation, hypomethylation, and DNMT RNA levels in ovarian tumors. Oncogene 2006, 25, 2636–2645. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef]
- Hatziapostolou, M.; Iliopoulos, D. Epigenetic aberrations during oncogenesis. Cell. Mol. Life Sci. 2011, 68, 1681–1702. [Google Scholar] [CrossRef]
- Garrison, S.P.; Jeffers, J.R.; Yang, C.; Nilsson, J.A.; Hall, M.A.; Rehg, J.E.; Yue, W.; Yu, J.; Zhang, L.; Onciu, M.; et al. Selection against PUMA gene expression in Myc-driven B-cell lymphomagenesis. Mol. Cell. Biol. 2008, 28, 5391–5402. [Google Scholar] [CrossRef]
- Saldana-Meyer, R.; Recillas-Targa, F. Transcriptional and epigenetic regulation of the p53 tumor suppressor gene. Epigenetics 2011, 6, 1068–1077. [Google Scholar] [CrossRef]
- Christoph, F.; Hinz, S.; Weikert, S.; Kempkensteffen, C.; Schostak, M.; Miller, K.; Schrader, M. Comparative promoter methylation analysis of p53 target genes in urogenital cancers. Urol. Int. 2008, 80, 398–404. [Google Scholar] [CrossRef]
- Hinz, S.; Kempkensteffen, C.; Weikert, S.; Schostak, M.; Schrader, M.; Miller, K.; Christoph, F. EZH2 polycomb transcriptional repressor expression correlates with methylation of the APAF-1 gene in superficial transitional cell carcinoma of the bladder. Tumour Biol. 2007, 28, 151–157. [Google Scholar] [CrossRef]
- Christoph, F.; Kempkensteffen, C.; Weikert, S.; Kollermann, J.; Krause, H.; Miller, K.; Schostak, M.; Schrader, M. Methylation of tumour suppressor genes APAF-1 and DAPK-1 and in vitro effects of demethylating agents in bladder and kidney cancer. Br. J. Cancer. 2006, 95, 1701–1707. [Google Scholar] [CrossRef]
- Furukawa, Y.; Sutheesophon, K.; Wada, T.; Nishimura, M.; Saito, Y.; Ishii, H. Methylation silencing of the Apaf-1 gene in acute leukemia. Mol. Cancer Res. 2005, 3, 325–334. [Google Scholar] [CrossRef]
- Soengas, M.S.; Capodieci, P.; Polsky, D.; Mora, J.; Esteller, M.; Opitz-Araya, X.; McCombie, R.; Herman, J.G.; Gerald, W.L.; Lazebnik, Y.A.; et al. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature 2001, 409, 207–211. [Google Scholar] [CrossRef]
- Pompeia, C.; Hodge, D.R.; Plass, C.; Wu, Y.Z.; Marquez, V.E.; Kelley, J.A.; Farrar, W.L. Microarray analysis of epigenetic silencing of gene expression in the KAS-6/1 multiple myeloma cell line. Cancer Res. 2004, 64, 3465–3473. [Google Scholar] [CrossRef]
- Cartron, P.F.; Oliver, L.; Martin, S.; Moreau, C.; LeCabellec, M.T.; Jezequel, P.; Meflah, K.; Vallette, F.M. The expression of a new variant of the pro-apoptotic molecule Bax, Baxpsi, is correlated with an increased survival of glioblastoma multiforme patients. Hum. Mol. Genet. 2002, 11, 675–687. [Google Scholar] [CrossRef]
- Xu, J.D.; Cao, X.X.; Long, Z.W.; Liu, X.P.; Furuya, T.; Xu, J.W.; Liu, X.L.; De Xu, Z.; Sasaki, K.; Li, Q.Q. BCL2L10 protein regulates apoptosis/proliferation through differential pathways in gastric cancer cells. J. Pathol. 2011, 223, 400–409. [Google Scholar] [CrossRef]
- Fabiani, E.; Leone, G.; Giachelia, M.; D'Alo, F.; Greco, M.; Criscuolo, M.; Guidi, F.; Rutella, S.; Hohaus, S.; Voso, M.T. Analysis of genome-wide methylation and gene expression induced by 5-aza-2'-deoxycytidine identifies BCL2L10 as a frequent methylation target in acute myeloid leukemia. Leuk. Lymphoma 2010, 51, 2275–2284. [Google Scholar] [CrossRef]
- San Jose-Eneriz, E.; Agirre, X.; Jimenez-Velasco, A.; Cordeu, L.; Martin, V.; Arqueros, V.; Garate, L.; Fresquet, V.; Cervantes, F.; Martinez-Climent, J.A.; et al. Epigenetic down-regulation of BIM expression is associated with reduced optimal responses to imatinib treatment in chronic myeloid leukaemia. Eur. J. Cancer. 2009, 45, 1877–1889. [Google Scholar] [CrossRef]
- Kim, T.Y.; Zhong, S.; Fields, C.R.; Kim, J.H.; Robertson, K.D. Epigenomic profiling reveals novel and frequent targets of aberrant DNA methylation-mediated silencing in malignant glioma. Cancer Res. 2006, 66, 7490–7501. [Google Scholar] [CrossRef]
- Sturm, I.; Stephan, C.; Gillissen, B.; Siebert, R.; Janz, M.; Radetzki, S.; Jung, K.; Loening, S.; Dorken, B.; Daniel, P.T. Loss of the tissue-specific proapoptotic BH3-only protein Nbk/Bik is a unifying feature of renal cell carcinoma. Cell Death Differ. 2006, 13, 619–627. [Google Scholar] [CrossRef]
- Murphy, T.M.; Sullivan, L.; Lane, C.; O'Connor, L.; Barrett, C.; Hollywood, D.; Lynch, T.; Lawler, M.; Perry, A.S. In silico analysis and DHPLC screening strategy identifies novel apoptotic gene targets of aberrant promoter hypermethylation in prostate cancer. Prostate 2011, 71, 1–17. [Google Scholar] [CrossRef]
- Hatzimichael, E.; Dasoula, A.; Kounnis, V.; Benetatos, L.; Lo Nigro, C.; Lattanzio, L.; Papoudou-Bai, A.; Dranitsaris, G.; Briasoulis, E.; Crook, T. Bcl2-interacting killer CpG methylation in multiple myeloma: a potential predictor of relapsed/refractory disease with therapeutic implications. Leuk. Lymphoma. 2012, 53, 1709–1713. [Google Scholar] [CrossRef]
- Sugita, H.; Iida, S.; Inokuchi, M.; Kato, K.; Ishiguro, M.; Ishikawa, T.; Takagi, Y.; Enjoji, M.; Yamada, H.; Uetake, H.; et al. Methylation of BNIP3 and DAPK indicates lower response to chemotherapy and poor prognosis in gastric cancer. Oncol. Rep. 2011, 25, 513–518. [Google Scholar]
- Hiraki, M.; Kitajima, Y.; Nakafusa, Y.; Nakamura, J.; Hashiguchi, K.; Sumi, K.; Noshiro, H.; Miyazaki, K. CpG island methylation of BNIP3 predicts resistance against S-1/CPT-11 combined therapy in colorectal cancer patients. Oncol. Rep. 2010, 23, 191–197. [Google Scholar]
- Pike, B.L.; Greiner, T.C.; Wang, X.; Weisenburger, D.D.; Hsu, Y.H.; Renaud, G.; Wolfsberg, T.G.; Kim, M.; Weisenberger, D.J.; Siegmund, K.D.; et al. DNA methylation profiles in diffuse large B-cell lymphoma and their relationship to gene expression status. Leukemia 2008, 22, 1035–1043. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Ladu, S.; Gorden, A.; Farina, M.; Lee, J.S.; Conner, E.A.; Schroeder, I.; Factor, V.M.; Thorgeirsson, S.S. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. J. Clin. Invest. 2007, 117, 2713–2722. [Google Scholar] [CrossRef]
- Michalowski, M.B.; de Fraipont, F.; Plantaz, D.; Michelland, S.; Combaret, V.; Favrot, M.C. Methylation of tumor-suppressor genes in neuroblastoma: The RASSF1A gene is almost always methylated in primary tumors. Pediatr. Blood Cancer 2008, 50, 29–32. [Google Scholar] [CrossRef]
- Cho, S.; Lee, J.H.; Cho, S.B.; Yoon, K.W.; Park, S.Y.; Lee, W.S.; Park, C.H.; Joo, Y.E.; Kim, H.S.; Choi, S.K.; et al. Epigenetic methylation and expression of caspase 8 and survivin in hepatocellular carcinoma. Pathol. Int. 2010, 60, 203–211. [Google Scholar] [CrossRef]
- Hervouet, E.; Vallette, F.M.; Cartron, P.F. Impact of the DNA methyltransferases expression on the methylation status of apoptosis-associated genes in glioblastoma multiforme. Cell Death Dis. 2010, 1, e8. [Google Scholar] [CrossRef]
- Malekzadeh, K.; Sobti, R.C.; Nikbakht, M.; Shekari, M.; Hosseini, S.A.; Tamandani, D.K.; Singh, S.K. Methylation patterns of Rb1 and Casp-8 promoters and their impact on their expression in bladder cancer. Cancer Invest. 2009, 27, 70–80. [Google Scholar] [CrossRef]
- Bello, M.J.; De Campos, J.M.; Isla, A.; Casartelli, C.; Rey, J.A. Promoter CpG methylation of multiple genes in pituitary adenomas: frequent involvement of caspase-8. Oncol. Rep. 2006, 15, 443–448. [Google Scholar]
- Margetts, C.D.; Astuti, D.; Gentle, D.C.; Cooper, W.N.; Cascon, A.; Catchpoole, D.; Robledo, M.; Neumann, H.P.; Latif, F.; Maher, E.R. Epigenetic analysis of HIC1, CASP8, FLIP, TSP1, DCR1, DCR2, DR4, DR5, KvDMR1, H19 and preferential 11p15.5 maternal-allele loss in von Hippel-Lindau and sporadic phaeochromocytomas. Endocr. Relat. Cancer 2005, 12, 161–172. [Google Scholar] [CrossRef]
- Gonzalez-Gomez, P.; Bello, M.J.; Inda, M.M.; Alonso, M.E.; Arjona, D.; Aminoso, C.; Lopez-Marin, I.; de Campos, J.M.; Sarasa, J.L.; Castresana, J.S.; et al. Deletion and aberrant CpG island methylation of Caspase 8 gene in medulloblastoma. Oncol. Rep. 2004, 12, 663–666. [Google Scholar]
- Harada, K.; Toyooka, S.; Shivapurkar, N.; Maitra, A.; Reddy, J.L.; Matta, H.; Miyajima, K.; Timmons, C.F.; Tomlinson, G.E.; Mastrangelo, D.; et al. Deregulation of caspase 8 and 10 expression in pediatric tumors and cell lines. Cancer Res. 2002, 62, 5897–5901. [Google Scholar]
- Shivapurkar, N.; Toyooka, S.; Eby, M.T.; Huang, C.X.; Sathyanarayana, U.G.; Cunningham, H.T.; Reddy, J.L.; Brambilla, E.; Takahashi, T.; Minna, J.D.; et al. Differential inactivation of caspase-8 in lung cancers. Cancer Biol. Ther. 2002, 1, 65–69. [Google Scholar] [CrossRef]
- Wu, Y.; Alvarez, M.; Slamon, D.J.; Koeffler, P.; Vadgama, J.V. Caspase 8 and maspin are downregulated in breast cancer cells due to CpG site promoter methylation. BMC Cancer 2010, 10, 32. [Google Scholar] [CrossRef]
- Fischer, J.R.; Ohnmacht, U.; Rieger, N.; Zemaitis, M.; Stoffregen, C.; Kostrzewa, M.; Buchholz, E.; Manegold, C.; Lahm, H. Promoter methylation of RASSF1A, RARbeta and DAPK predict poor prognosis of patients with malignant mesothelioma. Lung Cancer 2006, 54, 109–116. [Google Scholar] [CrossRef]
- Zou, X.P.; Zhang, B.; Zhang, X.Q.; Chen, M.; Cao, J.; Liu, W.J. Promoter hypermethylation of multiple genes in early gastric adenocarcinoma and precancerous lesions. Hum. Pathol. 2009, 40, 1534–1542. [Google Scholar] [CrossRef]
- Yuregir, O.O.; Yurtcu, E.; Kizilkilic, E.; Kocer, N.E.; Ozdogu, H.; Sahin, F.I. Detecting methylation patterns of p16, MGMT, DAPK and E-cadherin genes in multiple myeloma patients. Int. J. Lab. Hematol. 2010, 32, 142–149. [Google Scholar] [CrossRef]
- De Schutter, H.; Geeraerts, H.; Verbeken, E.; Nuyts, S. Promoter methylation of TIMP3 and CDH1 predicts better outcome in head and neck squamous cell carcinoma treated by radiotherapy only. Oncol. Rep. 2009, 21, 507–513. [Google Scholar]
- Leung, R.C.; Liu, S.S.; Chan, K.Y.; Tam, K.F.; Chan, K.L.; Wong, L.C.; Ngan, H.Y. Promoter methylation of death-associated protein kinase and its role in irradiation response in cervical cancer. Oncol. Rep. 2008, 19, 1339–1345. [Google Scholar]
- Kato, K.; Iida, S.; Uetake, H.; Takagi, Y.; Yamashita, T.; Inokuchi, M.; Yamada, H.; Kojima, K.; Sugihara, K. Methylated TMS1 and DAPK genes predict prognosis and response to chemotherapy in gastric cancer. Int. J. Cancer 2008, 122, 603–608. [Google Scholar] [CrossRef]
- Liu, X.F.; Kong, F.M.; Xu, Z.; Yu, S.P.; Sun, F.B.; Zhang, C.S.; Huang, Q.X.; Zhou, X.T.; Song, Z.W. Promoter hypermethylation of death-associated protein kinase gene in cholangiocarcinoma. Hepatobiliary Pancreat. Dis. Int. 2007, 6, 407–411. [Google Scholar]
- Kuester, D.; Dar, A.A.; Moskaluk, C.C.; Krueger, S.; Meyer, F.; Hartig, R.; Stolte, M.; Malfertheiner, P.; Lippert, H.; Roessner, A.; et al. Early involvement of death-associated protein kinase promoter hypermethylation in the carcinogenesis of Barrett’s esophageal adenocarcinoma and its association with clinical progression. Neoplasia 2007, 9, 236–245. [Google Scholar] [CrossRef]
- Kong, W.J.; Zhang, S.; Guo, C.K.; Wang, Y.J.; Chen, X.; Zhang, S.L.; Zhang, D.; Liu, Z.; Kong, W. Effect of methylation-associated silencing of the death-associated protein kinase gene on nasopharyngeal carcinoma. Anticancer Drugs 2006, 17, 251–259. [Google Scholar] [CrossRef]
- Mittag, F.; Kuester, D.; Vieth, M.; Peters, B.; Stolte, B.; Roessner, A.; Schneider-Stock, R. DAPK promotor methylation is an early event in colorectal carcinogenesis. Cancer Lett. 2006, 240, 69–75. [Google Scholar] [CrossRef]
- Bai, T.; Tanaka, T.; Yukawa, K.; Maeda, M.; Umesaki, N. Reduced expression of death-associated protein kinase in human uterine and ovarian carcinoma cells. Oncol. Rep. 2004, 11, 661–665. [Google Scholar]
- Toyooka, S.; Toyooka, K.O.; Miyajima, K.; Reddy, J.L.; Toyota, M.; Sathyanarayana, U.G.; Padar, A.; Tockman, M.S.; Lam, S.; Shivapurkar, N.; et al. Epigenetic down-regulation of death-associated protein kinase in lung cancers. Clin. Cancer Res. 2003, 9, 3034–3041. [Google Scholar]
- Pal, R.; Srivastava, N.; Chopra, R.; Gochhait, S.; Gupta, P.; Prakash, N.; Agarwal, G.; Bamezai, R.N. Investigation of DNA damage response and apoptotic gene methylation pattern in sporadic breast tumors using high throughput quantitative DNA methylation analysis technology. Mol. Cancer 2010, 9, 303. [Google Scholar] [CrossRef]
- Suzuki, M.; Shigematsu, H.; Shivapurkar, N.; Reddy, J.; Miyajima, K.; Takahashi, T.; Gazdar, A.F.; Frenkel, E.P. Methylation of apoptosis related genes in the pathogenesis and prognosis of prostate cancer. Cancer Lett. 2006, 242, 222–230. [Google Scholar] [CrossRef]
- Teodoridis, J.M.; Hall, J.; Marsh, S.; Kannall, H.D.; Smyth, C.; Curto, J.; Siddiqui, N.; Gabra, H.; McLeod, H.L.; Strathdee, G.; et al. CpG island methylation of DNA damage response genes in advanced ovarian cancer. Cancer Res. 2005, 65, 8961–8967. [Google Scholar] [CrossRef]
- Shivapurkar, N.; Toyooka, S.; Toyooka, K.O.; Reddy, J.; Miyajima, K.; Suzuki, M.; Shigematsu, H.; Takahashi, T.; Parikh, G.; Pass, H.I.; et al. Aberrant methylation of trail decoy receptor genes is frequent in multiple tumor types. Int. J. Cancer 2004, 109, 786–792. [Google Scholar] [CrossRef]
- Van Noesel, M.M.; Van Bezouw, S.; Salomons, G.S.; Voute, P.A.; Pieters, R.; Baylin, S.B.; Herman, J.G.; Versteeg, R. Tumor-specific down-regulation of the tumor necrosis factor-related apoptosis-inducing ligand decoy receptors DcR1 and DcR2 is associated with dense promoter hypermethylation. Cancer Res. 2002, 62, 2157–2161. [Google Scholar]
- Bae, S.I.; Cheriyath, V.; Jacobs, B.S.; Reu, F.J.; Borden, E.C. Reversal of methylation silencing of Apo2L/TRAIL receptor 1 (DR4) expression overcomes resistance of SK-MEL-3 and SK-MEL-28 melanoma cells to interferons (IFNs) or Apo2L/TRAIL. Oncogene 2008, 27, 490–498. [Google Scholar] [CrossRef]
- Horak, P.; Pils, D.; Haller, G.; Pribill, I.; Roessler, M.; Tomek, S.; Horvat, R.; Zeillinger, R.; Zielinski, C.; Krainer, M. Contribution of epigenetic silencing of tumor necrosis factor-related apoptosis inducing ligand receptor 1 (DR4) to TRAIL resistance and ovarian cancer. Mol. Cancer Res. 2005, 3, 335–343. [Google Scholar] [CrossRef]
- Van Noesel, M.M.; Van Bezouw, S.; Voute, P.A.; Herman, J.G.; Pieters, R.; Versteeg, R. Clustering of hypermethylated genes in neuroblastoma. Genes Chromosomes Cancer 2003, 38, 226–233. [Google Scholar] [CrossRef]
- Wu, J.; Wood, G.S. Reduction of Fas/CD95 promoter methylation, upregulation of Fas protein, and enhancement of sensitivity to apoptosis in cutaneous T-cell lymphoma. Arch. Dermatol. 2011, 147, 443–449. [Google Scholar] [CrossRef]
- Chaopatchayakul, P.; Jearanaikoon, P.; Yuenyao, P.; Limpaiboon, T. Aberrant DNA methylation of apoptotic signaling genes in patients responsive and nonresponsive to therapy for cervical carcinoma. Am. J. Obstet. Gynecol. 2010, 202, e1–e9. [Google Scholar]
- Petak, I.; Danam, R.P.; Tillman, D.M.; Vernes, R.; Howell, S.R.; Berczi, L.; Kopper, L.; Brent, T.P.; Houghton, J.A. Hypermethylation of the gene promoter and enhancer region can regulate Fas expression and sensitivity in colon carcinoma. Cell Death. Differ. 2003, 10, 211–217. [Google Scholar] [CrossRef]
- Hopkins-Donaldson, S.; Ziegler, A.; Kurtz, S.; Bigosch, C.; Kandioler, D.; Ludwig, C.; Zangemeister-Wittke, U.; Stahel, R. Silencing of death receptor and caspase-8 expression in small cell lung carcinoma cell lines and tumors by DNA methylation. Cell Death Differ. 2003, 10, 356–364. [Google Scholar] [CrossRef]
- Obata, T.; Toyota, M.; Satoh, A.; Sasaki, Y.; Ogi, K.; Akino, K.; Suzuki, H.; Murai, M.; Kikuchi, T.; Mita, H.; et al. Identification of HRK as a target of epigenetic inactivation in colorectal and gastric cancer. Clin. Cancer Res. 2003, 9, 6410–6418. [Google Scholar]
- Nakamura, M.; Ishida, E.; Shimada, K.; Nakase, H.; Sakaki, T.; Konishi, N. Frequent HRK inactivation associated with low apoptotic index in secondary glioblastomas. Acta Neuropathol. 2005, 110, 402–410. [Google Scholar] [CrossRef]
- Nakamura, M.; Ishida, E.; Shimada, K.; Nakase, H.; Sakaki, T.; Konishi, N. Defective expression of HRK is associated with promoter methylation in primary central nervous system lymphomas. Oncology 2006, 70, 212–221. [Google Scholar] [CrossRef]
- Higuchi, T.; Nakamura, M.; Shimada, K.; Ishida, E.; Hirao, K.; Konishi, N. HRK inactivation associated with promoter methylation and LOH in prostate cancer. Prostate 2008, 68, 105–113. [Google Scholar] [CrossRef]
- Zheng, S.; Houseman, E.A.; Morrison, Z.; Wrensch, M.R.; Patoka, J.S.; Ramos, C.; Haas-Kogan, D.A.; McBride, S.; Marsit, C.J.; Christensen, B.C.; et al. DNA hypermethylation profiles associated with glioma subtypes and EZH2 and IGFBP2 mRNA expression. Neuro. Oncol. 2011, 13, 280–289. [Google Scholar] [CrossRef]
- Velasco, A.; Pallares, J.; Santacana, M.; Gatius, S.; Fernandez, M.; Domingo, M.; Valls, J.; Yeramian, A.; Encinas, M.; Dolcet, X.; et al. Promoter hypermethylation and expression of sprouty 2 in endometrial carcinoma. Hum. Pathol. 2011, 42, 185–193. [Google Scholar] [CrossRef]
- Trankenschuh, W.; Puls, F.; Christgen, M.; Albat, C.; Heim, A.; Poczkaj, J.; Fleming, P.; Kreipe, H.; Lehmann, U. Frequent and distinct aberrations of DNA methylation patterns in fibrolamellar carcinoma of the liver. PLoS One 2010, 5, e13688. [Google Scholar]
- Jing, F.; Yuping, W.; Yong, C.; Jie, L.; Jun, L.; Xuanbing, T.; Lihua, H. CpG island methylator phenotype of multigene in serum of sporadic breast carcinoma. Tumour Biol. 2010, 31, 321–331. [Google Scholar] [CrossRef]
- Juhlin, C.C.; Kiss, N.B.; Villablanca, A.; Haglund, F.; Nordenstrom, J.; Hoog, A.; Larsson, C. Frequent promoter hypermethylation of the APC and RASSF1A tumour suppressors in parathyroid tumours. PLoS One 2010, 5, e9472. [Google Scholar]
- Wang, T.; Liu, H.; Chen, Y.; Liu, W.; Yu, J.; Wu, G. Methylation associated inactivation of RASSF1A and its synergistic effect with activated K-Ras in nasopharyngeal carcinoma. J. Exp. Clin. Cancer Res. 2009, 28, 160. [Google Scholar] [CrossRef]
- Niklinska, W.; Naumnik, W.; Sulewska, A.; Kozlowski, M.; Pankiewicz, W.; Milewski, R. Prognostic significance of DAPK and RASSF1A promoter hypermethylation in non-small cell lung cancer (NSCLC). Folia Histochem. Cytobiol. 2009, 47, 275–280. [Google Scholar]
- Tian, Y.; Hou, Y.; Zhou, X.; Cheng, H.; Zhou, R. Tumor suppressor RASSF1A promoter: p53 binding and methylation. PLoS One 2011, 6, e17017. [Google Scholar]
- Tellez, C.S.; Shen, L.; Estecio, M.R.; Jelinek, J.; Gershenwald, J.E.; Issa, J.P. CpG island methylation profiling in human melanoma cell lines. Melanoma Res. 2009, 19, 146–155. [Google Scholar] [CrossRef]
- Ahlquist, T.; Bottillo, I.; Danielsen, S.A.; Meling, G.I.; Rognum, T.O.; Lind, G.E.; Dallapiccola, B.; Lothe, R.A. RAS signaling in colorectal carcinomas through alteration of RAS, RAF, NF1, and/or RASSF1A. Neoplasia 2008, 10, 680–686. [Google Scholar]
- Honda, S.; Haruta, M.; Sugawara, W.; Sasaki, F.; Ohira, M.; Matsunaga, T.; Yamaoka, H.; Horie, H.; Ohnuma, N.; Nakagawara, A.; et al. The methylation status of RASSF1A promoter predicts responsiveness to chemotherapy and eventual cure in hepatoblastoma patients. Int. J. Cancer 2008, 123, 1117–1125. [Google Scholar]
- Cohen, Y.; Merhavi-Shoham, E.; Avraham, R.B.; Frenkel, S.; Pe'er, J.; Goldenberg-Cohen, N. Hypermethylation of CpG island loci of multiple tumor suppressor genes in retinoblastoma. Exp. Eye Res. 2008, 86, 201–206. [Google Scholar] [CrossRef]
- Lai, H.C.; Lin, Y.W.; Chang, C.C.; Wang, H.C.; Chu, T.W.; Yu, M.H.; Chu, T.Y. Hypermethylation of two consecutive tumor suppressor genes, BLU and RASSF1A, located at 3p21.3 in cervical neoplasias. Gynecol. Oncol. 2007, 104, 629–635. [Google Scholar] [CrossRef]
- Pfeifer, G.P.; Dammann, R. Methylation of the tumor suppressor gene RASSF1A in human tumors. Biochemistry (Mosc) 2005, 70, 576–583. [Google Scholar] [CrossRef]
- Watanabe, H.; Okada, G.; Ohtsubo, K.; Yao, F.; Jiang, P.H.; Mouri, H.; Wakabayashi, T.; Sawabu, N. Aberrant methylation of secreted apoptosis-related protein 2 (SARP2) in pure pancreatic juice in diagnosis of pancreatic neoplasms. Pancreas 2006, 32, 382–389. [Google Scholar] [CrossRef]
- Grau, E.; Martinez, F.; Orellana, C.; Canete, A.; Yanez, Y.; Oltra, S.; Noguera, R.; Hernandez, M.; Bermudez, J.D.; Castel, V. Hypermethylation of apoptotic genes as independent prognostic factor in neuroblastoma disease. Mol. Carcinog. 2011, 50, 153–162. [Google Scholar] [CrossRef]
- Mirza, S.; Sharma, G.; Prasad, C.P.; Parshad, R.; Srivastava, A.; Gupta, S.D.; Ralhan, R. Promoter hypermethylation of TMS1, BRCA1, ERalpha and PRB in serum and tumor DNA of invasive ductal breast carcinoma patients. Life Sci. 2007, 81, 280–287. [Google Scholar] [CrossRef]
- Zhang, C.; Li, H.; Zhou, G.; Zhang, Q.; Zhang, T.; Li, J.; Zhang, J.; Hou, J.; Liew, C.T.; Yin, D. Transcriptional silencing of the TMS1/ASC tumour suppressor gene by an epigenetic mechanism in hepatocellular carcinoma cells. J. Pathol. 2007, 212, 134–142. [Google Scholar] [CrossRef]
- Liu, X.F.; Zhu, S.G.; Zhang, H.; Xu, Z.; Su, H.L.; Li, S.J.; Zhou, X.T. The methylation status of the TMS1/ASC gene in cholangiocarcinoma and its clinical significance. Hepatobiliary Pancreat. Dis. Int. 2006, 5, 449–453. [Google Scholar]
- Martinez, R.; Schackert, G.; Esteller, M. Hypermethylation of the proapoptotic gene TMS1/ASC: prognostic importance in glioblastoma multiforme. J. Neurooncol. 2007, 82, 133–139. [Google Scholar] [CrossRef]
- Terasawa, K.; Sagae, S.; Toyota, M.; Tsukada, K.; Ogi, K.; Satoh, A.; Mita, H.; Imai, K.; Tokino, T.; Kudo, R. Epigenetic inactivation of TMS1/ASC in ovarian cancer. Clin. Cancer. Res. 2004, 10, 2000–2006. [Google Scholar] [CrossRef]
- Yokoyama, T.; Sagara, J.; Guan, X.; Masumoto, J.; Takeoka, M.; Komiyama, Y.; Miyata, K.; Higuchi, K.; Taniguchi, S. Methylation of ASC/TMS1, a proapoptotic gene responsible for activating procaspase-1, in human colorectal cancer. Cancer Lett. 2003, 202, 101–108. [Google Scholar] [CrossRef]
- Guan, X.; Sagara, J.; Yokoyama, T.; Koganehira, Y.; Oguchi, M.; Saida, T.; Taniguchi, S. ASC/TMS1, a caspase-1 activating adaptor, is downregulated by aberrant methylation in human melanoma. Int. J. Cancer 2003, 107, 202–208. [Google Scholar] [CrossRef]
- Virmani, A.; Rathi, A.; Sugio, K.; Sathyanarayana, U.G.; Toyooka, S.; Kischel, F.C.; Tonk, V.; Padar, A.; Takahashi, T.; Roth, J.A.; et al. Aberrant methylation of TMS1 in small cell, non small cell lung cancer and breast cancer. Int. J. Cancer 2003, 106, 198–204. [Google Scholar] [CrossRef]
- Ramachandran, K.; Miller, H.; Gordian, E.; Rocha-Lima, C.; Singal, R. Methylation-mediated silencing of TMS1 in pancreatic cancer and its potential contribution to chemosensitivity. Anticancer Res. 2010, 30, 3919–3925. [Google Scholar]
- Cai, H.H.; Sun, Y.M.; Miao, Y.; Gao, W.T.; Peng, Q.; Yao, J.; Zhao, H.L. Aberrant methylation frequency of TNFRSF10C promoter in pancreatic cancer cell lines. Hepatobiliary Pancreat. Dis. Int. 2011, 10, 95–100. [Google Scholar] [CrossRef]
- Chung, S.K.; Lee, M.G.; Ryu, B.K.; Lee, J.H.; Han, J.; Byun, D.S.; Chae, K.S.; Lee, K.Y.; Jang, J.Y.; Kim, H.J.; Chi, S.G. Frequent alteration of XAF1 in human colorectal cancers: implication for tumor cell resistance to apoptotic stresses. Gastroenterology 2007, 132, 2459–2477. [Google Scholar] [CrossRef]
- Murphy, T.M.; Perry, A.S.; Lawler, M. The emergence of DNA methylation as a key modulator of aberrant cell death in prostate cancer. Endocr. Relat. Cancer 2008, 15, 11–25. [Google Scholar] [CrossRef]
- Kempkensteffen, C.; Hinz, S.; Schrader, M.; Christoph, F.; Magheli, A.; Krause, H.; Schostak, M.; Miller, K.; Weikert, S. Gene expression and promoter methylation of the XIAP-associated Factor 1 in renal cell carcinomas: correlations with pathology and outcome. Cancer Lett. 2007, 254, 227–235. [Google Scholar] [CrossRef]
- Byun, D.S.; Cho, K.; Ryu, B.K.; Lee, M.G.; Kang, M.J.; Kim, H.R.; Chi, S.G. Hypermethylation of XIAP-associated factor 1, a putative tumor suppressor gene from the 17p13.2 locus, in human gastric adenocarcinomas. Cancer Res. 2003, 63, 7068–7075. [Google Scholar]
- Chen, X.Y.; He, Q.Y.; Guo, M.Z. XAF1 is frequently methylated in human esophageal cancer. World J. Gastroenterol. 2012, 18, 2844–2849. [Google Scholar] [CrossRef]
- Butler, L.M.; Dobrovic, A.; Bianco, T.; Cowled, P.A. Promoter region methylation does not account for the frequent loss of expression of the Fas gene in colorectal carcinoma. Br. J. Cancer. 2000, 82, 131–135. [Google Scholar] [CrossRef]
- Kurita, S.; Higuchi, H.; Saito, Y.; Nakamoto, N.; Takaishi, H.; Tada, S.; Saito, H.; Gores, G.J.; Hibi, T. DNMT1 and DNMT3b silencing sensitizes human hepatoma cells to TRAIL-mediated apoptosis via up-regulation of TRAIL-R2/DR5 and caspase-8. Cancer Sci. 2010, 101, 1431–1439. [Google Scholar] [CrossRef]
- Liedtke, C.; Zschemisch, N.H.; Cohrs, A.; Roskams, T.; Borlak, J.; Manns, M.P.; Trautwein, C. Silencing of caspase-8 in murine hepatocellular carcinomas is mediated via methylation of an essential promoter element. Gastroenterology 2005, 129, 1602–1615. [Google Scholar] [CrossRef]
- Teitz, T.; Wei, T.; Valentine, M.B.; Vanin, E.F.; Grenet, J.; Valentine, V.A.; Behm, F.G.; Look, A.T.; Lahti, J.M.; Kidd, V.J. Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat. Med. 2000, 6, 529–535. [Google Scholar] [CrossRef]
- Stone, A.R.; Bobo, W.; Brat, D.J.; Devi, N.S.; Van Meir, E.G.; Vertino, P.M. Aberrant methylation and down-regulation of TMS1/ASC in human glioblastoma. Am. J. Pathol. 2004, 165, 1151–1161. [Google Scholar] [CrossRef]
- Siraj, A.K.; Hussain, A.R.; Al-Rasheed, M.; Ahmed, M.; Bavi, P.; Alsobhi, S.A.; Al-Nuaim, A.; Uddin, S.; Al-Kuraya, K. Demethylation of TMS1 gene sensitizes thyroid cancer cells to TRAIL-induced apoptosis. J. Clin. Endocrinol. Metab. 2011, 96, E215–E224. [Google Scholar] [CrossRef]
- Collard, R.L.; Harya, N.S.; Monzon, F.A.; Maier, C.E.; O'Keefe, D.S. Methylation of the ASC gene promoter is associated with aggressive prostate cancer. Prostate 2006, 66, 687–695. [Google Scholar] [CrossRef]
- Das, P.M.; Ramachandran, K.; Vanwert, J.; Ferdinand, L.; Gopisetty, G.; Reis, I.M.; Singal, R. Methylation mediated silencing of TMS1/ASC gene in prostate cancer. Mol. Cancer. 2006, 5, 28. [Google Scholar] [CrossRef]
- Conway, K.E.; McConnell, B.B.; Bowring, C.E.; Donald, C.D.; Warren, S.T.; Vertino, P.M. TMS1, a novel proapoptotic caspase recruitment domain protein, is a target of methylation-induced gene silencing in human breast cancers. Cancer Res. 2000, 60, 6236–6242. [Google Scholar]
- Kim, S.J.; Kelly, W.K.; Fu, A.; Haines, K.; Hoffman, A.; Zheng, T.; Zhu, Y. Genome-wide methylation analysis identifies involvement of TNF-alpha mediated cancer pathways in prostate cancer. Cancer Lett. 2011, 302, 47–53. [Google Scholar] [CrossRef]
- Nakamura, M.; Shimada, K.; Konishi, N. The role of HRK gene in human cancer. Oncogene 2008, 27 Suppl 1, S105–S113. [Google Scholar] [CrossRef]
- Dai, Z.; Liu, S.; Marcucci, G.; Sadee, W. 5-Aza-2'-deoxycytidine and depsipeptide synergistically induce expression of BIK (BCL2-interacting killer). Biochem. Biophys. Res. Commun. 2006, 351, 455–461. [Google Scholar] [CrossRef]
- Christoph, F.; Hinz, S.; Kempkensteffen, C.; Weikert, S.; Krause, H.; Schostak, M.; Schrader, M.; Miller, K. A gene expression profile of tumor suppressor genes commonly methylated in bladder cancer. J. Cancer Res. Clin. Oncol. 2007, 133, 343–349. [Google Scholar] [CrossRef]
- Fu, W.N.; Bertoni, F.; Kelsey, S.M.; McElwaine, S.M.; Cotter, F.E.; Newland, A.C.; Jia, L. Role of DNA methylation in the suppression of Apaf-1 protein in human leukaemia. Oncogene 2003, 22, 451–455. [Google Scholar] [CrossRef]
- Chen, Y.K.; Huse, S.S.; Lin, L.M. Expression of inhibitor of apoptosis family proteins in human oral squamous cell carcinogenesis. Head Neck 2011, 33, 985–998. [Google Scholar] [CrossRef]
- Esteve, P.O.; Chin, H.G.; Pradhan, S. Molecular mechanisms of transactivation and doxorubicin-mediated repression of survivin gene in cancer cells. J. Biol. Chem. 2007, 282, 2615–2625. [Google Scholar] [CrossRef]
- Okami, J.; Simeone, D.M.; Logsdon, C.D. Silencing of the hypoxia-inducible cell death protein BNIP3 in pancreatic cancer. Cancer Res. 2004, 64, 5338–5346. [Google Scholar] [CrossRef]
- Shimizu, S.; Iida, S.; Ishiguro, M.; Uetake, H.; Ishikawa, T.; Takagi, Y.; Kobayashi, H.; Higuchi, T.; Enomoto, M.; Mogushi, K.; Mizushima, H.; Tanaka, H.; Sugihara, K. Methylated BNIP3 gene in colorectal cancer prognosis. Oncol. Lett. 2010, 1, 865–872. [Google Scholar]
- Liu, F.; Liu, Q.; Yang, D.; Bollag, W.B.; Robertson, K.; Wu, P.; Liu, K. Verticillin A overcomes apoptosis resistance in human colon carcinoma through DNA methylation-dependent upregulation of BNIP3. Cancer Res. 2011, 71, 6807–6816. [Google Scholar] [CrossRef]
- Deng, Q.; Huang, C.M.; Chen, N.; Li, L.; Wang, X.D.; Zhang, W.; Bi, F.; Tang, Q.L.; Li, Z.P.; Wang, W. Chemotherapy and Radiotherapy Downregulate the Activity and Expression of DNA Methyltransferase and Enhance Bcl-2/E1B-19-kDa Interacting Protein-3-Induced Apoptosis in Human Colorectal Cancer Cells. Chemotherapy 2012, 58, 445–453. [Google Scholar] [CrossRef]
- Zou, B.; Chim, C.S.; Zeng, H.; Leung, S.Y.; Yang, Y.; Tu, S.P.; Lin, M.C.; Wang, J.; He, H.; Jiang, S.H.; et al. Correlation between the single-site CpG methylation and expression silencing of the XAF1 gene in human gastric and colon cancers. Gastroenterology 2006, 131, 1835–1843. [Google Scholar] [CrossRef]
- Lee, M.G.; Huh, J.S.; Chung, S.K.; Lee, J.H.; Byun, D.S.; Ryu, B.K.; Kang, M.J.; Chae, K.S.; Lee, S.J.; Lee, C.H.; et al. Promoter CpG hypermethylation and downregulation of XAF1 expression in human urogenital malignancies: implication for attenuated p53 response to apoptotic stresses. Oncogene 2006, 25, 5807–5822. [Google Scholar] [CrossRef]
- Tada, Y.; Wada, M.; Taguchi, K.; Mochida, Y.; Kinugawa, N.; Tsuneyoshi, M.; Naito, S.; Kuwano, M. The association of death-associated protein kinase hypermethylation with early recurrence in superficial bladder cancers. Cancer Res. 2002, 62, 4048–4053. [Google Scholar]
- Satoh, A.; Toyota, M.; Itoh, F.; Kikuchi, T.; Obata, T.; Sasaki, Y.; Suzuki, H.; Yawata, A.; Kusano, M.; Fujita, M.; et al. DNA methylation and histone deacetylation associated with silencing DAP kinase gene expression in colorectal and gastric cancers. Br. J. Cancer 2002, 86, 1817–1823. [Google Scholar] [CrossRef]
- Pulling, L.C.; Grimes, M.J.; Damiani, L.A.; Juri, D.E.; Do, K.; Tellez, C.S.; Belinsky, S.A. Dual promoter regulation of death-associated protein kinase gene leads to differentially silenced transcripts by methylation in cancer. Carcinogenesis 2009, 30, 2023–2030. [Google Scholar]
- Puto, L.A.; Reed, J.C. Daxx represses RelB target promoters via DNA methyltransferase recruitment and DNA hypermethylation. Genes Dev. 2008, 22, 998–1010. [Google Scholar] [CrossRef]
- Kawai, Y.; Sakano, S.; Suehiro, Y.; Okada, T.; Korenaga, Y.; Hara, T.; Naito, K.; Matsuyama, H.; Hinoda, Y. Methylation level of the RASSF1A promoter is an independent prognostic factor for clear-cell renal cell carcinoma. Ann. Oncol. 2010, 21, 1612–1617. [Google Scholar] [CrossRef]
- Friedrich, M.G.; Weisenberger, D.J.; Cheng, J.C.; Chandrasoma, S.; Siegmund, K.D.; Gonzalgo, M.L.; Toma, M.I.; Huland, H.; Yoo, C.; Tsai, Y.C.; et al. Detection of methylated apoptosis-associated genes in urine sediments of bladder cancer patients. Clin. Cancer Res. 2004, 10, 7457–7465. [Google Scholar] [CrossRef]
- Kwong, J.; Lo, K.W.; To, K.F.; Teo, P.M.; Johnson, P.J.; Huang, D.P. Promoter hypermethylation of multiple genes in nasopharyngeal carcinoma. Clin. Cancer Res. 2002, 8, 131–137. [Google Scholar]
- Li, H.; Rauch, T.; Chen, Z.X.; Szabo, P.E.; Riggs, A.D.; Pfeifer, G.P. The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J. Biol. Chem. 2006, 281, 19489–19500. [Google Scholar]
- Wang, J.; Bhutani, M.; Pathak, A.K.; Lang, W.; Ren, H.; Jelinek, J.; He, R.; Shen, L.; Issa, J.P.; Mao, L. Delta DNMT3B variants regulate DNA methylation in a promoter-specific manner. Cancer Res. 2007, 67, 10647–10652. [Google Scholar] [CrossRef]
- Cho, S.W.; Her, S.J.; Sun, H.J.; Choi, O.K.; Yang, J.Y.; Kim, S.W.; Kim, S.Y.; Shin, C.S. Differential effects of secreted frizzled-related proteins (sFRPs) on osteoblastic differentiation of mouse mesenchymal cells and apoptosis of osteoblasts. Biochem. Biophys. Res. Commun. 2008, 367, 399–405. [Google Scholar] [CrossRef]
- Yang, J.; Hu, X.; Zimmerman, M.; Torres, C.M.; Yang, D.; Smith, S.B.; Liu, K. Cutting edge: IRF8 regulates Bax transcription in vivo in primary myeloid cells. J. Immunol. 2011, 187, 4426–4430. [Google Scholar] [CrossRef]
- Yang, D.; Thangaraju, M.; Greeneltch, K.; Browning, D.D.; Schoenlein, P.V.; Tamura, T.; Ozato, K.; Ganapathy, V.; Abrams, S.I.; Liu, K. Repression of IFN regulatory factor 8 by DNA methylation is a molecular determinant of apoptotic resistance and metastatic phenotype in metastatic tumor cells. Cancer Res. 2007, 67, 3301–3309. [Google Scholar] [CrossRef]
- Lee, K.Y.; Geng, H.; Ng, K.M.; Yu, J.; van Hasselt, A.; Cao, Y.; Zeng, Y.X.; Wong, A.H.; Wang, X.; Ying, J.; et al. Epigenetic disruption of interferon-gamma response through silencing the tumor suppressor interferon regulatory factor 8 in nasopharyngeal, esophageal and multiple other carcinomas. Oncogene 2008, 27, 5267–5276. [Google Scholar] [CrossRef]
- Tshuikina, M.; Jernberg-Wiklund, H.; Nilsson, K.; Oberg, F. Epigenetic silencing of the interferon regulatory factor ICSBP/IRF8 in human multiple myeloma. Exp. Hematol. 2008, 36, 1673–1681. [Google Scholar] [CrossRef]
- Vinken, M.; Snykers, S.; Fraczek, J.; Decrock, E.; Leybaert, L.; Rogiers, V.; Vanhaecke, T. DNA methyltransferase 3a expression decreases during apoptosis in primary cultures of hepatocytes. Toxicol. In Vitro 2010, 24, 445–451. [Google Scholar] [CrossRef]
- Brenner, C.; Deplus, R.; Didelot, C.; Loriot, A.; Vire, E.; De Smet, C.; Gutierrez, A.; Danovi, D.; Bernard, D.; Boon, T.; et al. Myc represses transcription through recruitment of DNA methyltransferase corepressor. EMBO J. 2005, 24, 336–346. [Google Scholar] [CrossRef]
- Esteve, P.O.; Chin, H.G.; Pradhan, S. Human maintenance DNA (cytosine-5)-methyltransferase and p53 modulate expression of p53-repressed promoters. Proc. Natl. Acad. Sci. U S A 2005, 102, 1000–1005. [Google Scholar] [CrossRef]
- Wang, Y.A.; Kamarova, Y.; Shen, K.C.; Jiang, Z.; Hahn, M.J.; Wang, Y.; Brooks, S.C. DNA methyltransferase-3a interacts with p53 and represses p53-mediated gene expression. Cancer Biol. Ther. 2005, 4, 1138–1143. [Google Scholar] [CrossRef]
- Hervouet, E.; Lalier, L.; Debien, E.; Cheray, M.; Geairon, A.; Rogniaux, H.; Loussouarn, D.; Martin, S.A.; Vallette, F.M.; Cartron, P.F. Disruption of Dnmt1/PCNA/UHRF1 interactions promotes tumorigenesis from human and mice glial cells. PLoS One 2010, 5, e11333. [Google Scholar] [CrossRef]
- Hervouet, E.; Vallette, F.M.; Cartron, P.F. Dnmt3/transcription factor interactions as crucial players in targeted DNA methylation. Epigenetics 2009, 4, 487–499. [Google Scholar] [CrossRef]
- Hervouet, E.; Nadaradjane, A.; Gueguen, M.; Vallette, F.M.; Cartron, P.F. Kinetics of DNA methylation inheritance by the Dnmt1-including complexes during the cell cycle. Cell. Div. 2012, 7, 5. [Google Scholar] [CrossRef]
- Chen, H.; Tu, S.W.; Hsieh, J.T. Down-regulation of human DAB2IP gene expression mediated by polycomb Ezh2 complex and histone deacetylase in prostate cancer. J. Biol. Chem. 2005, 280, 22437–22444. [Google Scholar] [CrossRef]
- Saito, Y.; Jones, P.A. Epigenetic activation of tumor suppressor microRNAs in human cancer cells. Cell Cycle 2006, 5, 2220–2222. [Google Scholar] [CrossRef]
- Bhatnagar, N.; Li, X.; Padi, S.K.; Zhang, Q.; Tang, M.S.; Guo, B. Downregulation of miR-205 and miR-31 confers resistance to chemotherapy-induced apoptosis in prostate cancer cells. Cell Death Dis. 2010, 1, e105. [Google Scholar] [CrossRef]
- Wang, F.; Liu, M.; Li, X.; Tang, H. MiR-214 reduces cell survival and enhances cisplatin-induced cytotoxicity via down-regulation of Bcl2l2 in cervical cancer cells. FEBS Lett. 2013, 587, 488–495. [Google Scholar] [CrossRef]
- Hermeking, H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010, 17, 193–199. [Google Scholar] [CrossRef]
- Lee, T.L.; Leung, W.K.; Chan, M.W.; Ng, E.K.; Tong, J.H.; Lo, K.W.; Chung, S.C.; Sung, J.J.; To, K.F. Detection of gene promoter hypermethylation in the tumor and serum of patients with gastric carcinoma. Clin. Cancer Res. 2002, 8, 1761–1766. [Google Scholar]
- Chen, D.; Wan, S.B.; Yang, H.; Yuan, J.; Chan, T.H.; Dou, Q.P. EGCG, green tea polyphenols and their synthetic analogs and prodrugs for human cancer prevention and treatment. Adv. Clin. Chem. 2011, 53, 155–177. [Google Scholar] [CrossRef]
- Li, Y.; Tollefsbol, T.O. Impact on DNA methylation in cancer prevention and therapy by bioactive dietary components. Curr. Med. Chem. 2010, 17, 2141–2151. [Google Scholar] [CrossRef]
- Khan, S.I.; Aumsuwan, P.; Khan, I.A.; Walker, L.A.; Dasmahapatra, A.K. Epigenetic events associated with breast cancer and their prevention by dietary components targeting the epigenome. Chem. Res. Toxicol. 2012, 25, 61–73. [Google Scholar] [CrossRef]
- Singh, V.; Sharma, P.; Capalash, N. DNA methyltransferase inhibitors as epigenetic therapy for cancer. Curr. Cancer Drug Targets 2013.
- Khan, R.; Schmidt-Mende, J.; Karimi, M.; Gogvadze, V.; Hassan, M.; Ekstrom, T.J.; Zhivotovsky, B.; Hellstrom-Lindberg, E. Hypomethylation and apoptosis in 5-azacytidine-treated myeloid cells. Exp. Hematol. 2008, 36, 149–157. [Google Scholar] [CrossRef]
- Eramo, A.; Pallini, R.; Lotti, F.; Sette, G.; Patti, M.; Bartucci, M.; Ricci-Vitiani, L.; Signore, M.; Stassi, G.; Larocca, L.M.; et al. Inhibition of DNA methylation sensitizes glioblastoma for tumor necrosis factor-related apoptosis-inducing ligand-mediated destruction. Cancer Res. 2005, 65, 11469–11477. [Google Scholar] [CrossRef]
- Yang, D.; Torres, C.M.; Bardhan, K.; Zimmerman, M.; McGaha, T.L.; Liu, K. Decitabine and vorinostat cooperate to sensitize colon carcinoma cells to Fas ligand-induced apoptosis in vitro and tumor suppression in vivo. J. Immunol. 2012, 188, 4441–4449. [Google Scholar] [CrossRef]
- Hervouet, E.; Debien, E.; Campion, L.; Charbord, J.; Menanteau, J.; Vallette, F.M.; Cartron, P.F. Folate supplementation limits the aggressiveness of glioma via the remethylation of DNA repeats element and genes governing apoptosis and proliferation. Clin. Cancer Res. 2009, 15, 3519–3529. [Google Scholar] [CrossRef]
- Park, C.S.; Cho, K.; Bae, D.R.; Joo, N.E.; Kim, H.H.; Mabasa, L.; Fowler, A.W. Methyl-donor nutrients inhibit breast cancer cell growth. In Vitro Cell Dev. Biol. Anim. 2008, 44, 268–272. [Google Scholar] [CrossRef]
- Cartron, P.F.; Hervouet, E.; Debien, E.; Olivier, C.; Pouliquen, D.; Menanteau, J.; Loussouarn, D.; Martin, S.A.; Campone, M.; Vallette, F.M. Folate supplementation limits the tumourigenesis in rodent models of gliomagenesis. Eur. J. Cancer 2012, 48, 2431–2441. [Google Scholar] [CrossRef]
- Gaudet, F.; Hodgson, J.G.; Eden, A.; Jackson-Grusby, L.; Dausman, J.; Gray, J.W.; Leonhardt, H.; Jaenisch, R. Induction of tumors in mice by genomic hypomethylation. Science 2003, 300, 489–492. [Google Scholar] [CrossRef]
- Ateeq, B.; Unterberger, A.; Szyf, M.; Rabbani, S.A. Pharmacological inhibition of DNA methylation induces proinvasive and prometastatic genes in vitro and in vivo. Neoplasia 2008, 10, 266–278. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hervouet, E.; Cheray, M.; Vallette, F.M.; Cartron, P.-F. DNA Methylation and Apoptosis Resistance in Cancer Cells. Cells 2013, 2, 545-573. https://doi.org/10.3390/cells2030545
Hervouet E, Cheray M, Vallette FM, Cartron P-F. DNA Methylation and Apoptosis Resistance in Cancer Cells. Cells. 2013; 2(3):545-573. https://doi.org/10.3390/cells2030545
Chicago/Turabian StyleHervouet, Eric, Mathilde Cheray, François Marie Vallette, and Pierre-François Cartron. 2013. "DNA Methylation and Apoptosis Resistance in Cancer Cells" Cells 2, no. 3: 545-573. https://doi.org/10.3390/cells2030545
APA StyleHervouet, E., Cheray, M., Vallette, F. M., & Cartron, P.-F. (2013). DNA Methylation and Apoptosis Resistance in Cancer Cells. Cells, 2(3), 545-573. https://doi.org/10.3390/cells2030545