Interorganellar Membrane Microdomains: Dynamic Platforms in the Control of Calcium Signaling and Apoptosis

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. ER-PM Junctions
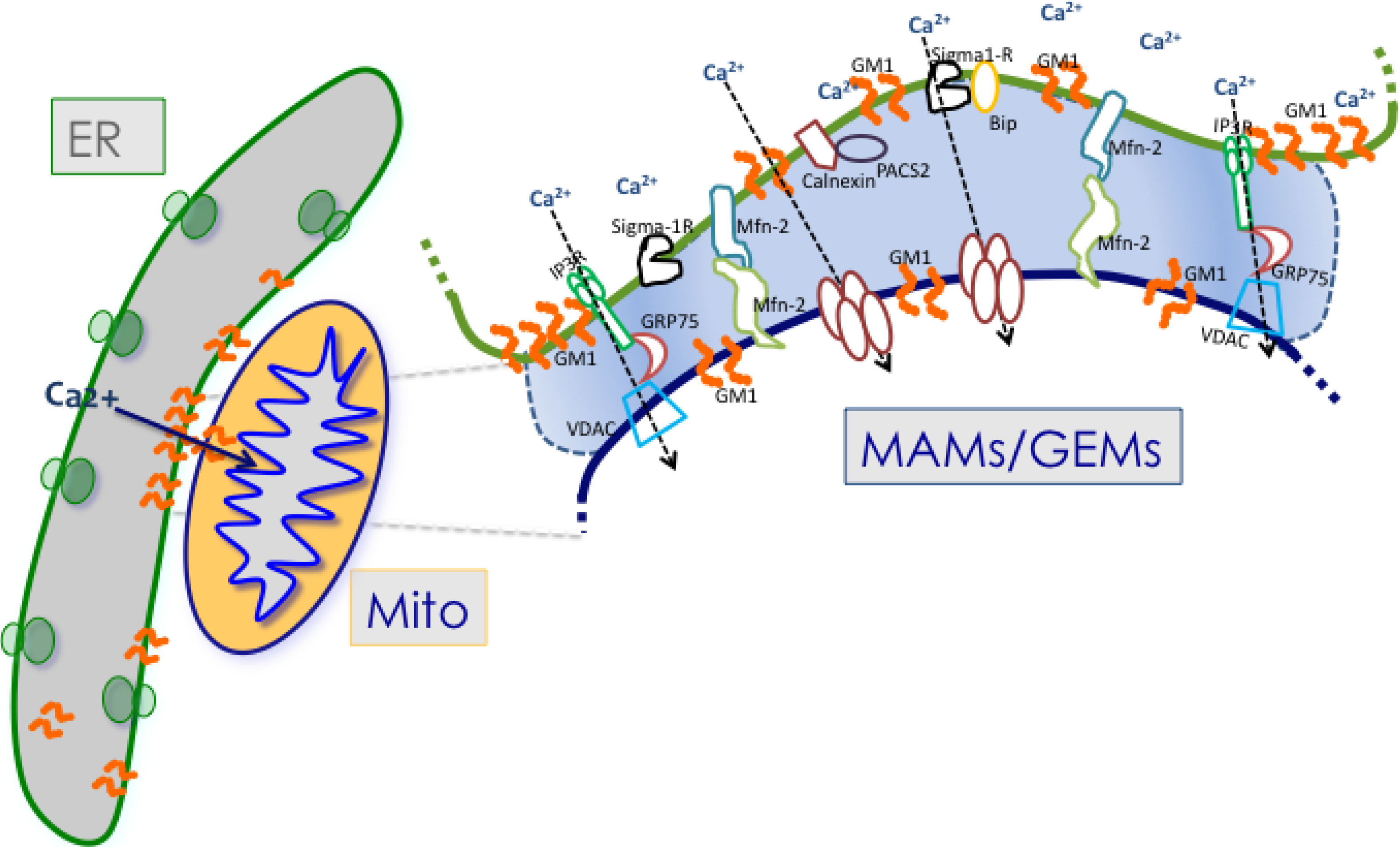
3. Mitochondria Associated ER Membranes or MAMs
3.1. Lipid Biosynthesis and Transport at the MAMs
3.2. Ca2+ Signaling between ER and Mitochondria at the MAMs
4. Protein Components of the MAMs

5. Lipid Components of the MAMs
6. GM1-Ganglioside and Ca2+ Regulation
7. GM1 at MAMs/GEMs and Activation of Cell Death

8. GM1 at the PM and its Relocalization in Intracellular Membranes
9. Conclusion
Acknowledgments
Conflict of Interest
References and Notes
- Carrasco, S.; Meyer, T. STIM proteins and the endoplasmic reticulum-plasma membrane junctions. Annu. Rev. Biochem. 2011, 80, 973–1000. [Google Scholar] [CrossRef]
- Toulmay, A.; Prinz, W.A. Lipid transfer and signaling at organelle contact sites: The tip of the iceberg. Curr. Opin. Cell. Biol. 2011, 23, 458–463. [Google Scholar] [CrossRef]
- Toulmay, A.; Prinz, W.A. A conserved membrane-binding domain targets proteins to organelle contact sites. J. Cell. Sci. 2012, 125, 49–58. [Google Scholar] [CrossRef]
- Kvam, E.; Goldfarb, D.S. Nucleus-vacuole junctions in yeast: Anatomy of a membrane contact site. Biochem. Soc. Trans. 2006, 34, 340–342. [Google Scholar] [CrossRef]
- Dolman, N.J.; Gerasimenko, J.V.; Gerasimenko, O.V.; Voronina, S.G.; Petersen, O.H.; Tepikin, A.V. Stable Golgi-mitochondria complexes and formation of Golgi Ca(2+) gradients in pancreatic acinar cells. J. Biol. Chem. 2005, 280, 15794–15799. [Google Scholar]
- Levine, T.; Loewen, C. Inter-organelle membrane contact sites: Through a glass, darkly. Curr. Opin. Cell Biol. 2006, 18, 371–378. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell. Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [Green Version]
- d'Azzo, A.; Tessitore, A.; Sano, R. Gangliosides as apoptotic signals in ER stress response. Cell. Death Differ. 2006, 13, 404–414. [Google Scholar] [CrossRef]
- Porter, K.R.; Palade, G.E. Studies on the endoplasmic reticulum. III. Its form and distribution in striated muscle cells. J. Biophys. Biochem. Cytol. 1957, 3, 269–300. [Google Scholar] [CrossRef]
- Pichler, H.; Gaigg, B.; Hrastnik, C.; Achleitner, G.; Kohlwein, S.D.; Zellnig, G.; Perktold, A.; Daum, G. A subfraction of the yeast endoplasmic reticulum associates with the plasma membrane and has a high capacity to synthesize lipids. Eur. J. Biochem. 2001, 268, 2351–2361. [Google Scholar] [CrossRef]
- Stefan, C.J.; Manford, A.G.; Emr, S.D. ER-PM connections: Sites of information transfer and inter-organelle communication. Curr. Opin. Cell. Biol. 2013, 25, 434–442. [Google Scholar] [CrossRef]
- Golovina, V.A. Visualization of localized store-operated calcium entry in mouse astrocytes. Close proximity to the endoplasmic reticulum. J. Physiol. 2005, 564, 737–749. [Google Scholar] [CrossRef]
- Manford, A.G.; Stefan, C.J.; Yuan, H.L.; Macgurn, J.A.; Emr, S.D. ER-to-plasma membrane tethering proteins regulate cell signaling and ER morphology. Dev. Cell. 2012, 23, 1129–1140. [Google Scholar] [CrossRef]
- Takeshima, H.; Komazaki, S.; Nishi, M.; Iino, M.; Kangawa, K. Junctophilins: A novel family of junctional membrane complex proteins. Mol. Cell. 2000, 6, 11–22. [Google Scholar]
- Stefan, C.J.; Manford, A.G.; Baird, D.; Yamada-Hanff, J.; Mao, Y.; Emr, S.D. Osh proteins regulate phosphoinositide metabolism at ER-plasma membrane contact sites. Cell 2011, 144, 389–401. [Google Scholar] [CrossRef]
- Raturi, A.; Simmen, T. Where the endoplasmic reticulum and the mitochondrion tie the knot: The mitochondria-associated membrane (MAM). Biochim. Biophys. Acta 2012, 1833, 213–224. [Google Scholar] [CrossRef]
- Copeland, D.E.; Dalton, A.J. An association between mitochondria and the endoplasmic reticulum in cells of the pseudobranch gland of a teleost. J. Biophys. Biochem. Cytol. 1959, 5, 393–396. [Google Scholar] [CrossRef]
- Vance, J.E. Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 1990, 265, 7248–7256. [Google Scholar]
- Ardail, D.; Popa., I.; Bodennec, J.; Louisot, P.; Schmitt, D.; Portoukalian, J. The mitochondria-associated endoplasmic-reticulum subcompartment (MAM fraction) of rat liver contains highly active sphingolipid-specific glycosyltransferases. Biochem. J. 2003, 371, 1013–1019. [Google Scholar] [CrossRef]
- Osman, C.; Voelker, D.R.; Langer, T. Making heads or tails of phospholipids in mitochondria. J. Cell. Biol. 2011, 192, 7–16. [Google Scholar] [CrossRef]
- Voelker, D.R. Phosphatidylserine translocation to the mitochondrion is an ATP-dependent process in permeabilized animal cells. Proc. Natl. Acad. Sci. USA 1989, 86, 9921–9925. [Google Scholar] [CrossRef]
- Bionda, C.; Portoukalian, J.; Schmitt, D.; Rodriguez-Lafrasse, C.; Ardail, D. Subcellular compartmentalization of ceramide metabolism: MAM (mitochondria-associated membrane) and/or mitochondria? Biochem. J. 2004, 382, 527–533. [Google Scholar] [CrossRef]
- Kornmann, B.; Currie, E.; Collins, S.R.; Schuldiner, M.; Nunnari, J.; Weissman, J.S.; Walter, P. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 2009, 325, 477–481. [Google Scholar] [CrossRef]
- Helle, S.C.; Kanfer, G.; Kolar, K.; Lang, A.; Michel, A.H.; Kornmann, B. Organization and function of membrane contact sites. Biochim. Biophys. Acta 2013, 1833, 2526–2541. [Google Scholar] [CrossRef]
- Michalak, M.; Robert Parker, J.M.; Opas, M. Ca2+ signaling and calcium binding chaperones of the endoplasmic reticulum. Cell. Calcium 2002, 32, 269–278. [Google Scholar] [CrossRef]
- Chouhan, A.K.; Ivannikov, M.V.; Lu, Z.; Sugimori, M.; Llinas, R.R.; Macleod, G.T. Cytosolic calcium coordinates mitochondrial energy metabolism with presynaptic activity. J. Neurosci. 2012, 32, 1233–1243. [Google Scholar] [CrossRef]
- Glancy, B.; Willis, W.T.; Chess, D.J.; Balaban, R.S. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry 2013, 52, 2793–2809. [Google Scholar] [CrossRef]
- McCormack, J.G.; Halestrap, A.P.; Denton, R.M. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol. Rev. 1990, 70, 391–425. [Google Scholar]
- Cali, T.; Ottolini, D.; Brini, M. Mitochondrial Ca(2+) as a key regulator of mitochondrial activities. Adv. Exp. Med. Biol. 2012, 942, 53–73. [Google Scholar] [CrossRef]
- Csordas, G.; Varnai, P.; Golenar, T.; Roy, S.; Purkins, G.; Schneider, T.G.; Balla, T.; Hajnoczky, G. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cell. 2010, 39, 121–132. [Google Scholar] [CrossRef]
- Rizzuto, R.; Brini, M.; Murgia, M.; Pozzan, T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 1993, 262, 744–747. [Google Scholar]
- Rapizzi, E.; Pinton, P.; Szabadkai, G.; Wieckowski, M.R.; Vandecasteele, G.; Baird, G.; Tuft, R.A.; Fogarty, K.E.; Rizzuto, R. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell. Biol. 2002, 159, 613–624. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pozzan, T. Microdomains of intracellular Ca2+: Molecular determinants and functional consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef]
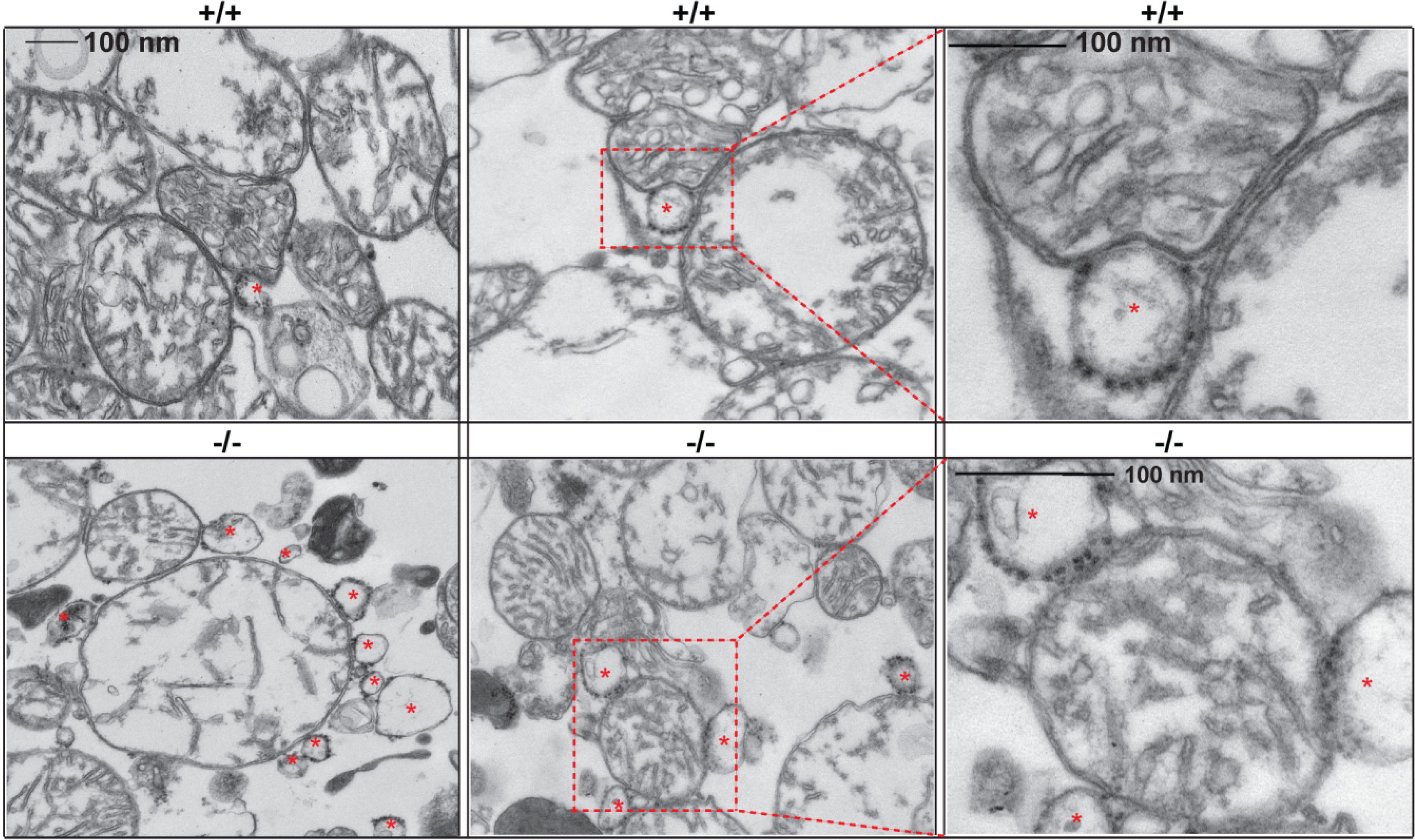
- Sano, R.; Annunziata, I.; Patterson, A.; Moshiach, S.; Gomero, E.; Opferman, J.; Forte, M.; d'Azzo, A. GM1-ganglioside accumulation at the mitochondria-associated ER membranes links ER stress to Ca(2+)-dependent mitochondrial apoptosis. Mol. Cell. 2009, 36, 500–511. [Google Scholar] [CrossRef]
- Pacher, P.; Hajnoczky, G. Propagation of the apoptotic signal by mitochondrial waves. EMBO J. 2001, 20, 4107–4121. [Google Scholar] [CrossRef]
- Santo-Domingo, J.; Demaurex, N. Calcium uptake mechanisms of mitochondria. Biochim. Biophys. Acta 2010, 1797, 907–912. [Google Scholar] [CrossRef]
- Perocchi, F.; Gohil, V.M.; Girgis, H.S.; Bao, X.R.; McCombs, J.E.; Palmer, A.E.; Mootha, V.K. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature 2010, 467, 291–296. [Google Scholar] [CrossRef]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabo, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef]
- Jiang, D.; Zhao, L.; Clapham, D.E. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 2009, 326, 144–147. [Google Scholar] [CrossRef]
- Pizzo, P.; Pozzan, T. Mitochondria-endoplasmic reticulum choreography: structure and signaling dynamics. Trends Cell. Biol. 2007, 17, 511–517. [Google Scholar] [CrossRef]
- Szabadkai, G.; Bianchi, K.; Varnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell. Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef]
- Simmen, T.; Aslan, J.E.; Blagoveshchenskaya, A.D.; Thomas, L.; Wan, L.; Xiang, Y.; Feliciangeli, S.F.; Hung, C.H.; Crump, C.M.; Thomas, G. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J. 2005, 24, 717–729. [Google Scholar] [CrossRef]
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef]
- Myhill, N.; Lynes, E.M.; Nanji, J.A.; Blagoveshchenskaya, A.D.; Fei, H.; Carmine Simmen, K.; Cooper, T.J.; Thomas, G.; Simmen, T. The subcellular distribution of calnexin is mediated by PACS-2. Mol. Biol. Cell. 2008, 19, 2777–2788. [Google Scholar] [CrossRef]
- Ottolini, D.; Cali, T.; Negro, A.; Brini, M. The Parkinson disease-related protein DJ-1 counteracts mitochondrial impairment induced by the tumour suppressor protein p53 by enhancing endoplasmic reticulum-mitochondria tethering. Hum. Mol. Genet. 2013, 22, 2152–2168. [Google Scholar]
- Bui, M.; Gilady, S.Y.; Fitzsimmons, R.E.; Benson, M.D.; Lynes, E.M.; Gesson, K.; Alto, N.M.; Strack, S.; Scott, J.D.; Simmen, T. Rab32 modulates apoptosis onset and mitochondria-associated membrane (MAM) properties. J. Biol. Chem. 2010, 285, 31590–31602. [Google Scholar] [CrossRef]
- Giorgi, C.; Ito, K.; Lin, H.K.; Santangelo, C.; Wieckowski, M.R.; Lebiedzinska, M.; Bononi, A.; Bonora, M.; Duszynski, J.; Bernardi, R.; et al. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science 2010, 330, 1247–1251. [Google Scholar] [CrossRef]
- Su, T.P.; Hayashi, T.; Maurice, T.; Buch, S.; Ruoho, A.E. The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharmacol. Sci. 2010, 31, 557–566. [Google Scholar] [CrossRef]
- Zhang, H.; Cuevas, J. sigma Receptor activation blocks potassium channels and depresses neuroexcitability in rat intracardiac neurons. J. Pharmacol. Exp. Ther. 2005, 313, 1387–1396. [Google Scholar] [CrossRef]
- Kennedy, C.; Henderson, G. Inhibition of potassium currents by the sigma receptor ligand (+)-3-(3-hydroxyphenyl)-N-(1-propyl)piperidine in sympathetic neurons of the mouse isolated hypogastric ganglion. Neuroscience 1990, 35, 725–733. [Google Scholar] [CrossRef]
- Monnet, F.P.; Debonnel, G.; Junien, J.L.; De Montigny, C. N-methyl-D-aspartate-induced neuronal activation is selectively modulated by sigma receptors. Eur. J. Pharmacol. 1990, 179, 441–445. [Google Scholar] [CrossRef]
- Martina, M.; Turcotte, M.E.; Halman, S.; Bergeron, R. The sigma-1 receptor modulates NMDA receptor synaptic transmission and plasticity via SK channels in rat hippocampus. J. Physiol. 2007, 578, 143–157. [Google Scholar]
- Grimm, S. The ER-mitochondria interface: the social network of cell death. Biochim. Biophys. Acta 2011, 1823, 327–334. [Google Scholar] [CrossRef]
- Wang, H.J.; Guay, G.; Pogan, L.; Sauve, R.; Nabi, I.R. Calcium regulates the association between mitochondria and a smooth subdomain of the endoplasmic reticulum. J. Cell. Biol. 2000, 150, 1489–1498. [Google Scholar] [CrossRef]
- Csordas, G.; Renken, C.; Varnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnoczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell. Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef]
- Zuchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef]
- de Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Hayashi, T.; Fujimoto, M. Detergent-resistant microdomains determine the localization of sigma-1 receptors to the endoplasmic reticulum-mitochondria junction. Mol. Pharmacol. 2010, 77, 517–528. [Google Scholar] [CrossRef]
- Hayashi, T.; Su, T.P. Cholesterol at the endoplasmic reticulum: roles of the sigma-1 receptor chaperone and implications thereof in human diseases. Subcell. Biochem. 2010, 51, 381–398. [Google Scholar] [CrossRef]
- Brown, D.A.; Rose, J.K. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell 1992, 68, 533–544. [Google Scholar] [CrossRef]
- Sonnino, S.; Prinetti, A. Membrane domains and the “lipid raft” concept. Curr. Med. Chem. 2012, 20, 4–21. [Google Scholar]
- Sonnino, S.; Prinetti, A.; Mauri, L.; Chigorno, V.; Tettamanti, G. Dynamic and structural properties of sphingolipids as driving forces for the formation of membrane domains. Chem. Rev. 2006, 106, 2111–2125. [Google Scholar] [CrossRef]
- Sonnino, S.; Prinetti, A. Gangliosides as regulators of cell membrane organization and functions. Adv. Exp. Med. Biol. 2010, 688, 165–184. [Google Scholar] [CrossRef]
- Ledeen, R.; Wu, G. New findings on nuclear gangliosides: Overview on metabolism and function. J. Neurochem. 2011, 116, 714–720. [Google Scholar] [CrossRef]
- Yu, R.K.; Tsai, Y.T.; Ariga, T. Functional roles of gangliosides in neurodevelopment: An overview of recent advances. Neurochem. Res. 2012, 37, 1230–1244. [Google Scholar] [CrossRef]
- Todeschini, R.A.; Hakomori, S.I. Functional role of glycosphingolipids and gangliosides in control of cell adhesion, motility, and growth, through glycosynaptic microdomains. Biochim. Biophys. Acta 2008, 1780, 421–433. [Google Scholar] [CrossRef]
- Lopez, P.H.; Schnaar, R.L. Gangliosides in cell recognition and membrane protein regulation. Curr. Opin. Struct. Biol. 2009, 19, 549–557. [Google Scholar] [CrossRef]
- Miljan, E.A.; Bremer, E.G. Regulation of growth factor receptors by gangliosides. Sci. STKE 2002, 2002, re15. [Google Scholar]
- Fujinaga, Y.; Wolf, A.; Rodighiero, C.; Wheeler, H.; Tsai, B.; Allen, L.; Jobling, M.G.; Rapoport, T.; Holmes, R.K.; Lencer, W.I. Gangliosides that associate with lipid rafts mediate transport of cholera and related toxins from the plasma membrane to endoplasmic reticulm. Mol. Biol. Cell. 2003, 14, 4783–4793. [Google Scholar] [CrossRef]
- Ledeen, R.W.; Wu, G. Ganglioside function in calcium homeostasis and signaling. Neurochem. Res. 2002, 27, 637–647. [Google Scholar] [CrossRef]
- Ledeen, R.W.; Wu, G. GM1 ganglioside: another nuclear lipid that modulates nuclear calcium. GM1 potentiates the nuclear sodium-calcium exchanger. Can. J. Physiol. Pharmacol. 2006, 84, 393–402. [Google Scholar]
- Tessitore, A.; del Pilar Martin, M.; Sano, R.; Ma, Y.; Mann, L.; Ingrassia, A.; Laywell, E.D.; Steindler, D.A.; Hendershot, L.M.; d'Azzo, A. GM1-ganglioside-mediated activation of the unfolded protein response causes neuronal death in a neurodegenerative gangliosidosis. Mol. Cell. 2004, 15, 753–766. [Google Scholar] [CrossRef]
- Hansson, H.A.; Holmgren, J.; Svennerholm, L. Ultrastructural localization of cell membrane GM1 ganglioside by cholera toxin. Proc. Natl. Acad. Sci. USA 1977, 74, 3782–3786. [Google Scholar] [CrossRef]
- Fang, Y.; Wu, G.; Xie, X.; Lu, Z.H.; Ledeen, R.W. Endogenous GM1 ganglioside of the plasma membrane promotes neuritogenesis by two mechanisms. Neurochem. Res. 2000, 25, 931–940. [Google Scholar] [CrossRef]
- Wu, G.; Lu, Z.H.; Ledeen, R.W. GM1 ganglioside in the nuclear membrane modulates nuclear calcium homeostasis during neurite outgrowth. J. Neurochem. 1995, 65, 1419–1422. [Google Scholar]
- Ledeen, R.; Wu, G. GM1 in the nuclear envelope regulates nuclear calcium through association with a nuclear sodium-calcium exchanger. J. Neurochem. 2007, 103 (Suppl. 1), 126–134. [Google Scholar]
- Ledeen, R.; Wu, G. New findings on nuclear gangliosides: overview on metabolism and function. J. Neurochem. 2011, 116, 714–720. [Google Scholar] [CrossRef]
- Xie, X.; Wu, G.; Lu, Z.H.; Rohowsky-Kochan, C.; Ledeen, R.W. Presence of sodium-calcium exchanger/GM1 complex in the nuclear envelope of non-neural cells: nature of exchanger-GM1 interaction. Neurochem. Res. 2004, 29, 2135–2146. [Google Scholar] [CrossRef]
- Wu, G.; Xie, X.; Lu, Z.H.; Ledeen, R.W. Sodium-calcium exchanger complexed with GM1 ganglioside in nuclear membrane transfers calcium from nucleoplasm to endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2009, 106, 10829–10834. [Google Scholar] [CrossRef]
- Wu, G.; Xie, X.; Lu, Z.H.; Ledeen, R.W. Cerebellar neurons lacking complex gangliosides degenerate in the presence of depolarizing levels of potassium. Proc. Natl. Acad. Sci. USA 2001, 98, 307–312. [Google Scholar] [CrossRef]
- Wu, G.; Lu, Z.H.; Xie, X.; Ledeen, R.W. Susceptibility of cerebellar granule neurons from GM2/GD2 synthase-null mice to apoptosis induced by glutamate excitotoxicity and elevated KCl: Rescue by GM1 and LIGA20. Glycoconj. J. 2004, 21, 305–313. [Google Scholar] [CrossRef]
- Suzuki, Y.A.O.; Namba, E. b-Galactosidase Deficiency (b-Galactosialidosis): GM1 Gangliosidosis and Morquio B Disease. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C., Sly, W., Childs, B., Beaudet, A., Valle, D., Kinzler, K., Vogelstein, B., Eds.; McGraw-Hill Publishing Co.: New York, NY, USA, 2001; pp. 3775–3810. [Google Scholar]
- Hahn, C.N.; del Pilar Martin, M.; Schröder, M.; Vanier, M.T.; Hara, Y.; Suzuki, K.; Suzuki, K.; d'Azzo, A. Generalized CNS disease and massive GM1-ganglioside accumulation in mice defective in lysosomal acid beta-galactosidase. Hum. Mol. Genet. 1997, 6, 205–211. [Google Scholar] [CrossRef]
- Jeyakumar, M.; Thomas, R.; Elliot-Smith, E.; Smith, D.A.; van der Spoel, A.C.; d'Azzo, A.; Perry, V.H.; Butters, T.D.; Dwek, R.A.; Platt, F.M. Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain 2003, 126, 974–987. [Google Scholar] [CrossRef]
- Annunziata, I.; Patterson, A.; d'Azzo, A. Mitochondria-associated ER membranes (MAMs) and glycosphingolipid enriched microdomains (GEMs): isolation from mouse brain. J. Vis. Exp. 2013, e50215. [Google Scholar]
- Pizzo, P.; Giurisato, E.; Bigsten, A.; Tassi, M.; Tavano, R.; Shaw, A.; Viola, A. Physiological T cell activation starts and propagates in lipid rafts. Immunol. Lett. 2004, 91, 3–9. [Google Scholar] [CrossRef]
- Gupta, G.; Surolia, A. Glycosphingolipids in microdomain formation and their spatial organization. FEBS Lett. 2009, 584, 1634–1641. [Google Scholar]
- Garofalo, T.; Misasi, R.; Mattei, V.; Giammarioli, A.M.; Malorni, W.; Pontieri, G.M.; Pavan, A.; Sorice, M. Association of the death-inducing signaling complex with microdomains after triggering through CD95/Fas. Evidence for caspase-8-ganglioside interaction in T cells. J. Biol. Chem. 2003, 278, 8309–8315. [Google Scholar]
- Bathori, G.; Csordas, G.; Garcia-Perez, C.; Davies, E.; Hajnoczky, G. Ca2+-dependent control of the permeability properties of the mitochondrial outer membrane and voltage-dependent anion-selective channel (VDAC). J. Biol. Chem. 2006, 281, 17347–17358. [Google Scholar]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef]
- Chinnapen, D.J.; Chinnapen, H.; Saslowsky, D.; Lencer, W.I. Rafting with cholera toxin: Endocytosis and trafficking from plasma membrane to ER. FEMS Microbiol. Lett. 2007, 266, 129–137. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Annunziata, I.; D'Azzo, A. Interorganellar Membrane Microdomains: Dynamic Platforms in the Control of Calcium Signaling and Apoptosis. Cells 2013, 2, 574-590. https://doi.org/10.3390/cells2030574
Annunziata I, D'Azzo A. Interorganellar Membrane Microdomains: Dynamic Platforms in the Control of Calcium Signaling and Apoptosis. Cells. 2013; 2(3):574-590. https://doi.org/10.3390/cells2030574
Chicago/Turabian StyleAnnunziata, Ida, and Alessandra D'Azzo. 2013. "Interorganellar Membrane Microdomains: Dynamic Platforms in the Control of Calcium Signaling and Apoptosis" Cells 2, no. 3: 574-590. https://doi.org/10.3390/cells2030574