Emerin Is Required for Proper Nucleus Reassembly after Mitosis: Implications for New Pathogenetic Mechanisms for Laminopathies Detected in EDMD1 Patients

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids
2.2. Cell Culture and Transfection
2.3. Immunofluorescence
2.4. SDS-PAGE and Western Blotting
2.5. Antibodies
2.6. Laser Scanning Confocal Microscopy Analyses
2.7. In Vitro Nuclear Assembly and Chromatin Binding Assay
2.8. Phenotype Analysis
2.9. Phenotype Quantitative and Statistical Analysis
2.10. Cell Cycle Analysis
3. Results
3.1. Emerin and Interacting Protein BAF Transiently Associate with Mitotic Spindle Microtubules, Centrosomes, and Associated Membranes during Mitosis
3.2. Lamin A/C Transiently Associates with Emerin and Mitotic Spindle Microtubules and Membranes
3.3. Emerin, Unlike another LEM Domain Protein, LAP2β, Follows the Mitotic Spindle and Not the Membrane
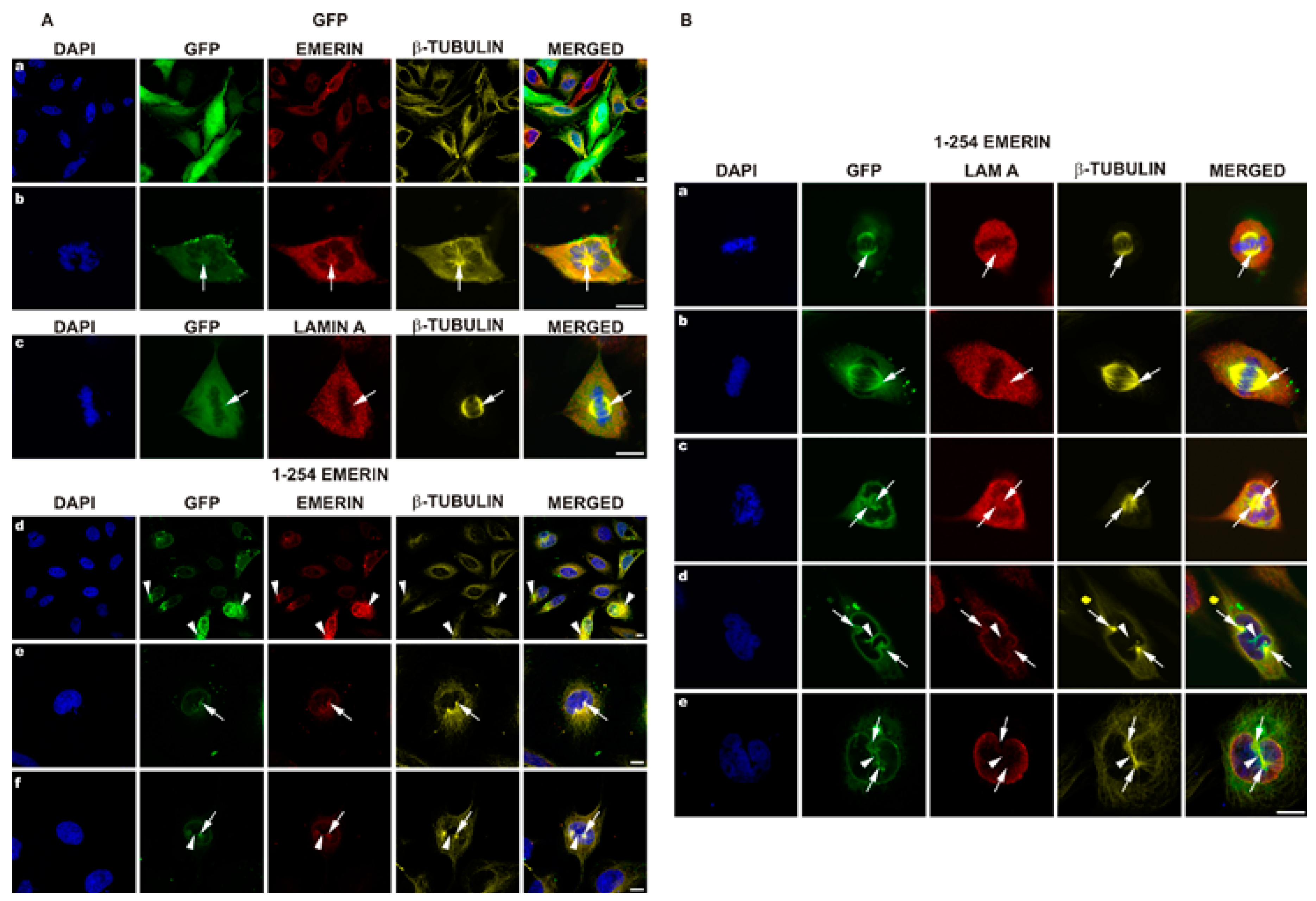
3.4. EGFP-Emerin Transfection Studies Confirm Antibodies-Based Observations
3.5. Transient Transfection of Emerin Deletion Mutants Results in a Large Variety of Cellular Phenotypes
3.6. Full Nucleoplasmic Domain of Emerin can Bind Chromatin Condensed Chromatin Independently of BAF, Membranes, and Tubulin
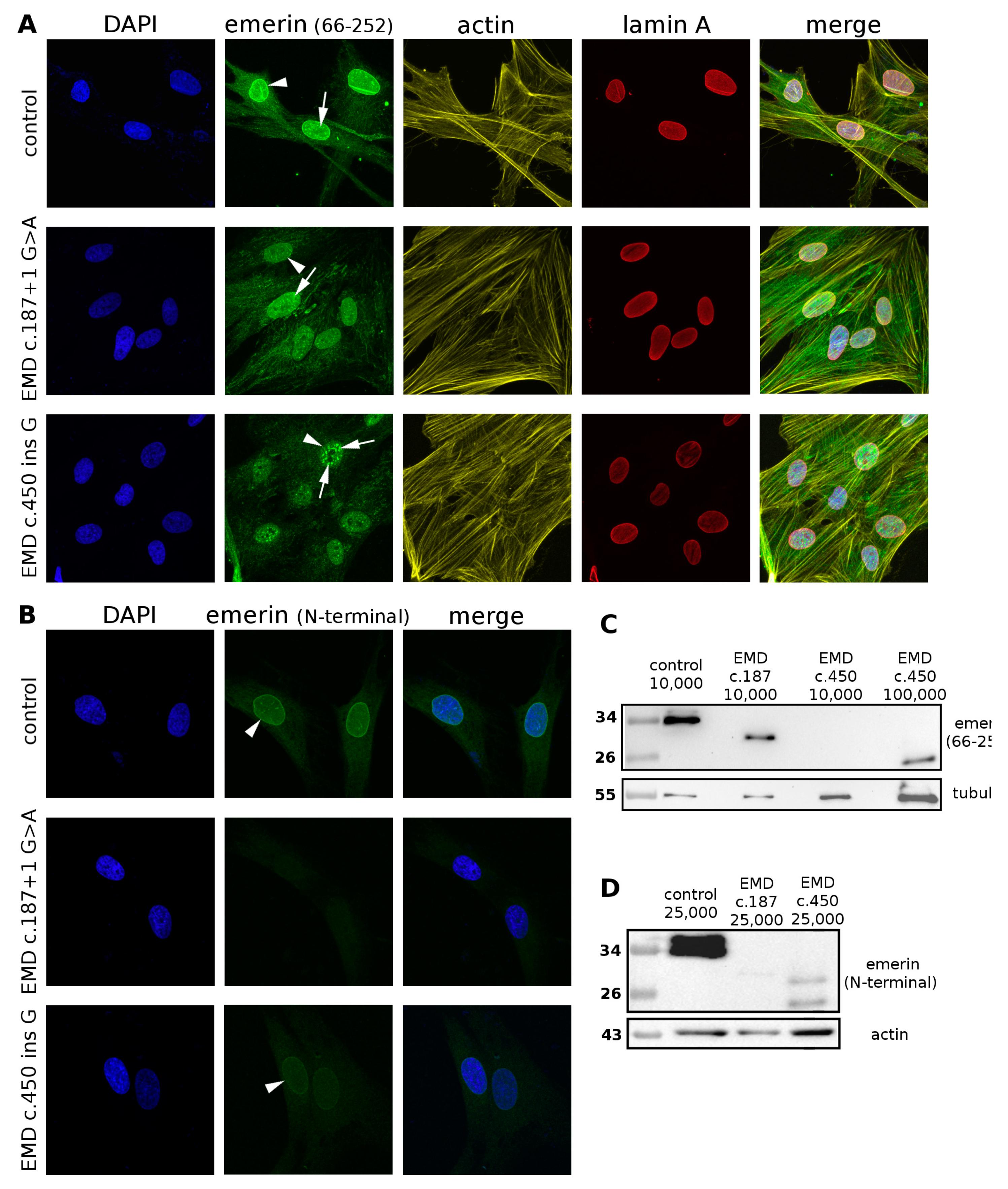
3.7. Detailed Analyses of Emerin Protein Expression in Patient Cells Reveal Truncated Emerin Protein Presence and an Epitope Masking Effect in Immunofluorescent Analyses
4. Discussion
4.1. Subcellular Location of Emerin during Mitosis: Disassembly and Reassembly
4.2. Emerin, LAP2β, Lamin A/C, and Membrane Location of Mitotic Spindle Microtubules
4.3. Role of Emerin Domains
4.4. Pathogenetic Mechanisms in EDMD1 Patients
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Leeuw, R.; Gruenbaum, Y.; Medalia, O. Nuclear Lamins: Thin Filaments with Major Functions. Trends Cell Biol. 2018, 28, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.J.; Holaska, J.M. Emerin in health and disease. Semin. Cell Dev. Biol. 2014, 29, 95–106. [Google Scholar] [CrossRef]
- Tran, J.R.; Chen, H.Y.; Zheng, X.B.; Zheng, Y.X. Lamin in inflammation and aging. Curr. Opin. Cell Biol. 2016, 40, 124–130. [Google Scholar] [CrossRef] [Green Version]
- Meier, J.; Campbell, K.H.S.; Ford, C.C.; Stick, R.; Hutchison, C.J. The role of lamin LIII in nuclear assembly and DNA replication, in cell-free extracts of Xenopus eggs. J. Cell Sci. 1991, 98, 271–279. [Google Scholar] [PubMed]
- Ulitzur, N.; Harel, A.; Feinstein, N.; Gruenbaum, Y. Lamin activity is essential for nuclear envelope assembly in a Drosophila embryo cell-free extract. J. Cell Biol. 1992, 119, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugh, G.E.; Coates, P.J.; Lane, E.B.; Raymond, Y.; Quinlan, R.A. Distinct nuclear assembly pathways for lamins A and C lead to their increase during quiescence in Swiss 3T3 cells. J. Cell Sci. 1997, 110, 2483–2493. [Google Scholar] [PubMed]
- Holaska, J.M.; Wilson, K.L.; Mansharamani, M. The nuclear envelope, lamins and nuclear assembly. Curr. Opin. Cell Biol. 2002, 14, 357–364. [Google Scholar] [CrossRef]
- Shumaker, D.K.; Lopez-Soler, R.I.; Adam, S.A.; Herrmann, H.; Moir, R.D.; Spann, T.P.; Goldman, R.D. Functions and dysfunctions of the nuclear lamin Ig-fold domain in nuclear assembly, growth, and Emery-Dreifuss muscular dystrophy. Proc. Natl. Acad. Sci. USA 2005, 102, 15494–15499. [Google Scholar] [CrossRef] [Green Version]
- Grossman, E.; Dahan, I.; Stick, R.; Goldberg, M.W.; Gruenbaum, Y.; Medalia, O. Filaments assembly of ectopically expressed Caenorhabditis elegans lamin within Xenopus oocytes. J. Struct. Biol. 2012, 177, 113–118. [Google Scholar] [CrossRef]
- Rzepecki, R. The nuclear lamins and the nuclear envelope. Cell. Mol. Biol. Lett. 2002, 7, 1019–1035. [Google Scholar] [PubMed]
- Haraguchi, T.; Koujin, T.; Segura-Totten, M.; Lee, K.K.; Matsuoka, Y.; Yoneda, Y.; Wilson, K.L.; Hiraoka, Y. BAF is required for emerin assembly into the reforming nuclear envelope. J. Cell Sci. 2001, 114, 4575–4585. [Google Scholar] [PubMed]
- Hawryluk-Gara, L.A.; Shibuya, E.K.; Wozniak, R.W. Vertebrate Nup53 interacts with the nuclear lamina and is required for the assembly of a Nup93-containing complex. Mol. Biol. Cell 2005, 16, 2382–2394. [Google Scholar] [CrossRef]
- Ma, L.; Tsai, M.Y.; Wang, S.; Lu, B.; Chen, R.; Iii, J.R.; Zhu, X.; Zheng, Y. Requirement for Nudel and dynein for assembly of the lamin B spindle matrix. Nat. Cell Biol. 2009, 11, 247–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Shi, Y.; Lu, Q.; Ma, Y.; Luo, J.; Wang, Q.; Ji, J.; Jiang, Q.; Zhang, C. Requirement for lamin B receptor and its regulation by importin and phosphorylation in nuclear envelope assembly during mitotic exit. J. Biol. Chem. 2010, 285, 33281–33293. [Google Scholar] [CrossRef] [PubMed]
- Talamas, J.A.; Hetzer, M.W. POM121 and Sun1 play a role in early steps of interphase NPC assembly. J. Cell Biol. 2011, 194, 27–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, L.C.; Chen, R.H. Temporal control of nuclear envelope assembly by phosphorylation of lamin B receptor. Mol. Biol. Cell 2011, 22, 3306–3317. [Google Scholar] [CrossRef] [Green Version]
- Gruenbaum, Y.; Foisner, R. Lamins: Nuclear Intermediate Filament Proteins with Fundamental Functions in Nuclear Mechanics and Genome Regulation. Annu. Rev. Biochem. 2015, 84, 131–164. [Google Scholar] [CrossRef]
- Braun, S.; Barrales, R.R. Beyond Tethering and the LEM domain: MSCellaneous functions of the inner nuclear membrane Lem2. Nucleus 2016, 7, 523–531. [Google Scholar] [CrossRef] [Green Version]
- Rzepecki, R.; Gruenbaum, Y. Invertebrate models of lamin diseases. Nucleus 2018, 9, 227–234. [Google Scholar] [CrossRef] [Green Version]
- Palka, M.; Tomczak, A.; Grabowska, K.; Machowska, M.; Piekarowicz, K.; Rzepecka, D.; Rzepecki, R. Laminopathies: What can humans learn from fruit flies. Cell. Mol. Biol. Lett. 2018, 23, 32. [Google Scholar] [CrossRef]
- Nagano, A.; Arahata, K. Nuclear envelope proteins and associated diseases. Curr. Opin. Neurol. 2000, 13, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Butin-Israeli, V.; Adam, S.A.; Goldman, A.E.; Goldman, R.D. Nuclear lamin functions and disease. Trends Genet. 2012, 28, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Dubinska-Magiera, M.; Zaremba-Czogalla, M.; Rzepecki, R. Muscle development, regeneration and laminopathies: How lamins or lamina-associated proteins can contribute to muscle development, regeneration and disease. Cell Mol. Life Sci. 2013, 70, 2713–2741. [Google Scholar] [CrossRef] [PubMed]
- Bonne, G. Laminopathies: Why make it simple when it can be complex? Neuromuscul. Disord. 2016, 26, S150–S151. [Google Scholar] [CrossRef]
- Piekarowicz, K.; Machowska, M.; Dzianisava, V.; Rzepecki, R. Hutchinson-Gilford Progeria Syndrome-Current Status and Prospects for Gene Therapy Treatment. Cells 2019, 8, 88. [Google Scholar] [CrossRef]
- Astejada, M.N.; Goto, K.; Nagano, A.; Ura, S.; Noguchi, S.; Nonaka, I.; Nishino, I.; Hayashi, Y.K. Emerinopathy and laminopathy clinical, pathological and molecular features of muscular dystrophy with nuclear envelopathy in Japan. Acta Myol. 2007, 26, 159–164. [Google Scholar]
- Nagano, A.; Koga, R.; Ogawa, M.; Kurano, Y.; Kawada, J.; Okada, R.; Hayashi, Y.K.; Tsukahara, T.; Arahata, K. Emerin deficiency at the nuclear membrane in patients with Emery-Dreifuss muscular dystrophy. Nat. Genet. 1996, 12, 254–259. [Google Scholar] [CrossRef]
- Brown, C.A.; Scharner, J.; Felice, K.; Meriggioli, M.N.; Tarnopolsky, M.; Bower, M.; Zammit, P.S.; Mendell, J.R.; Ellis, J.A. Novel and recurrent EMD mutations in patients with Emery-Dreifuss muscular dystrophy, identify exon 2 as a mutation hot spot. J. Hum. Genet. 2011, 56, 589–594. [Google Scholar] [CrossRef]
- Melcon, G.; Kozlov, S.; Cutler, D.A.; Sullivan, T.; Hernandez, L.; Zhao, P.; Mitchell, S.; Nader, G.; Bakay, M.; Rottman, J.N.; et al. Loss of emerin at the nuclear envelope disrupts the Rb1/E2F and MyoD pathways during muscle regeneration. Hum. Mol. Genet. 2006, 15, 637–651. [Google Scholar] [CrossRef] [Green Version]
- Ozawa, R.; Hayashi, Y.K.; Ogawa, M.; Kurokawa, R.; Matsumoto, H.; Noguchi, S.; Nonaka, I.; Nishino, I. Emerin-lacking mice show minimal motor and cardiac dysfunctions with nuclear-associated vacuoles. Am. J. Pathol. 2006, 168, 907–917. [Google Scholar] [CrossRef]
- Semenova, E.; Wang, X.F.; Jablonski, M.M.; Levorse, J.; Tilghman, S.M. An engineered 800 kilobase deletion of Uchl3 and Lmo7 on mouse chromosome 14 causes defects in viability, postnatal growth and degeneration of muscle and retina. Hum. Mol. Genet. 2003, 12, 1301–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolff, N.; Gilquin, B.; Courchay, K.; Callebaut, I.; Worman, H.J.; Zinn-Justin, S. Structural analysis of emerin, an inner nuclear membrane protein mutated in X-linked Emery-Dreifuss muscular dystrophy. FEBS Lett. 2001, 501, 171–176. [Google Scholar] [CrossRef] [Green Version]
- Laguri, C.; Gilquin, B.; Wolff, N.; Romi-Lebrun, R.; Courchay, K.; Callebaut, I.; Worman, H.J.; Zinn-Justin, S. Structural characterization of the LEM motif common to three human inner nuclear membrane proteins. Structure 2001, 9, 503–511. [Google Scholar] [CrossRef]
- Berk, J.M.; Tifft, K.E.; Wilson, K.L. The nuclear envelope LEM-domain protein emerin. Nucleus 2013, 4, 298–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holaska, J.M.; Wilson, K.L. An emerin “proteome”: Purification of distinct emerin-containing complexes from HeLa cells suggests molecular basis for diverse roles including gene regulation, mRNA splicing, signaling, mechanosensing, and nuclear architecture. Biochemistry 2007, 46, 8897–8908. [Google Scholar] [CrossRef]
- Berk, J.M.; Simon, D.N.; Jenkins-Houk, C.R.; Westerbeck, J.W.; Gronning-Wang, L.M.; Carlson, C.R.; Wilson, K.L. The molecular basis of emerin-emerin and emerin-BAF interactions. J. Cell Sci. 2014, 127, 3956–3969. [Google Scholar] [CrossRef]
- Berk, J.M.; Maitra, S.; Dawdy, A.W.; Shabanowitz, J.; Hunt, D.F.; Wilson, K.L. O-Linked beta-N-Acetylglucosamine (O-GlcNAc) Regulates Emerin Binding to Barrier to Autointegration Factor (BAF) in a Chromatin- and Lamin B-enriched “Niche”. J. Biol. Chem. 2013, 288, 30192–30209. [Google Scholar] [CrossRef]
- Bengtsson, L.; Wilson, K.L. Barrier-to-autointegration factor phosphorylation on Ser-4 regulates emerin binding to lamin A in vitro and emerin localization in vivo. Mol. Biol. Cell 2006, 17, 1154–1163. [Google Scholar] [CrossRef]
- Clements, L.; Manilal, S.; Love, D.R.; Morris, G.E. Direct interaction between emerin and lamin A. Biochem. Biophys. Res. Commun. 2000, 267, 709–714. [Google Scholar] [CrossRef]
- Samson, C.; Celli, F.; Hendriks, K.; Zinke, M.; Essawy, N.; Herrada, I.; Arteni, A.A.; Theillet, F.X.; Alpha-Bazin, B.; Armengaud, J.; et al. Emerin self-assembly mechanism: Role of the LEM domain. Febs J. 2017, 284, 338–352. [Google Scholar] [CrossRef]
- Bengtsson, L.; Otto, H. LUMA interacts with emerin and influences its distribution at the inner nuclear membrane. J. Cell Sci. 2008, 121, 536–548. [Google Scholar] [CrossRef] [Green Version]
- Demmerle, J.; Koch, A.J.; Holaska, J.M. The Nuclear Envelope Protein Emerin Binds Directly to Histone Deacetylase 3 (HDAC3) and Activates HDAC3 Activity. J. Biol. Chem. 2012, 287, 22080–22088. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, T.; Holaska, J.M.; Yamane, M.; Koujin, T.; Hashiguchi, N.; Mori, C.; Wilson, K.L.; Hiraoka, Y. Emerin binding to Btf, a death-promoting transcriptional repressor, is disrupted by a missense mutation that causes Emery-Dreifuss muscular dystrophy. Eur. J. Biochem. 2004, 271, 1035–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holaska, J.M.; Lee, K.K.; Kowalski, A.K.; Wilson, K.L. Transcriptional repressor germ cell-less (GCL) and barrier to autointegration factor (BAF) compete for binding to emerin in vitro. J. Biol. Chem. 2003, 278, 6969–6975. [Google Scholar] [CrossRef] [PubMed]
- Holaska, J.M.; Kowalski, A.K.; Wilson, K.L. Emerin caps the pointed end of actin filaments: Evidence for an actin cortical network at the nuclear inner membrane. PLoS Biol. 2004, 2, E231. [Google Scholar] [CrossRef] [PubMed]
- Holaska, J.M.; Rais-Bahrami, S.; Wilson, K.L. Lmo7 is an emerin-binding protein that regulates the transcription of emerin and many other muscle-relevant genes. Hum. Mol. Genet. 2006, 15, 3459–3472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, M.D.; Guan, T.; Gerace, L. Overlapping functions of nuclear envelope proteins NET25 (Lem2) and emerin in regulation of extracellular signal-regulated kinase signaling in myoblast differentiation. Mol. Cell Biol. 2009, 29, 5718–5728. [Google Scholar] [CrossRef] [PubMed]
- Markiewicz, E.; Tilgner, K.; Barker, N.; van de Wetering, M.; Clevers, H.; Dorobek, M.; Hausmanowa-Petrusewicz, I.; Ramaekers, F.C.S.; Broers, J.L.V.; Blankesteijn, W.M.; et al. The inner nuclear membrane protein Emerin regulates beta-catenin activity by restricting its accumulation in the nucleus. EMBO J. 2006, 25, 3275–3285. [Google Scholar] [CrossRef]
- Margalit, A.; Segura-Totten, M.; Gruenbaum, Y.; Wilson, K.L. Barrier-to-autointegration factor is required to segregate and enclose chromosomes within the nuclear envelope and assemble the nuclear lamina. Proc. Natl. Acad. Sci. USA 2005, 102, 3290–3295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barkan, R.; Zahand, A.J.; Sharabi, K.; Lamm, A.T.; Feinstein, N.; Haithcock, E.; Wilson, K.L.; Liu, J.; Gruenbaum, Y. Ce-emerin and LEM-2: Essential roles in Caenorhabditis elegans development, muscle function, and mitosis. Mol. Biol. Cell 2012, 23, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.N.; Wriston, A.; Fan, Q.; Shabanowitz, J.; Florwick, A.; Dharmaraj, T.; Peterson, S.B.; Gruenbaum, Y.; Carlson, C.R.; Gronning-Wang, L.M.; et al. OGT (O-GlcNAc Transferase) Selectively Modifies Multiple Residues Unique to Lamin A. Cells 2018, 7, 44. [Google Scholar] [CrossRef] [PubMed]
- Salpingidou, G.; Rzepecki, R.; Kiseleva, E.; Lyon, C.; Lane, B.; Fusiek, K.; Golebiewska, A.; Drummond, S.; Allen, T.; Ellis, J.A.; et al. NEP-A and NEP-B both contribute to nuclear pore formation in Xenopus eggs and oocytes. J. Cell Sci. 2008, 121, 706–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Guan, T.; Gerace, L. Lamin-binding fragment of LAP2 inhibits increase in nuclear volume during the cell cycle and progression into S phase. J. Cell Biol. 1997, 139, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Gant, T.M.; Harris, C.A.; Wilson, K.L. Roles of LAP2 proteins in nuclear assembly and DNA replication: Truncated LAP2beta proteins alter lamina assembly, envelope formation, nuclear size, and DNA replication efficiency in Xenopus laevis extracts. J. Cell Biol. 1999, 144, 1083–1096. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.; Paulin-Levasseur, M.; Gajewski, A.; Alsheimer, M.; Benavente, R.; Krohne, G. Molecular characterization and developmentally regulated expression of Xenopus lamina-associated polypeptide 2 (XLAP2). J. Cell Sci. 1999, 112, 749–759. [Google Scholar]
- Isaji, M.; Iwata, H.; Harayama, H.; Miyake, M. The localization of LAP2 beta during pronuclear formation in bovine oocytes after fertilization or activation. Zygote 2006, 14, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Chmielewska, M.; Dubinska-Magiera, M.; Sopel, M.; Rzepecka, D.; Hutchison, C.J.; Goldberg, M.W.; Rzepecki, R. Embryonic and adult isoforms of XLAP2 form microdomains associated with chromatin and the nuclear envelope. Cell Tissue Res. 2011, 344, 97–110. [Google Scholar] [CrossRef] [Green Version]
- Dubinska-Magiera, M.; Chmielewska, M.; Koziol, K.; Machowska, M.; Hutchison, C.J.; Goldberg, M.W.; Rzepecki, R. Xenopus LAP2beta protein knockdown affects location of lamin B and nucleoporins and has effect on assembly of cell nucleus and cell viability. Protoplasma 2016, 253, 943–956. [Google Scholar] [CrossRef]
- Dabauvalle, M.C.; Muller, E.; Ewald, A.; Kress, W.; Krohne, G.; Muller, C.R. Distribution of emerin during the cell cycle. Eur. J. Cell Biol. 1999, 78, 749–756. [Google Scholar] [CrossRef]
- Haraguchi, T.; Koujin, T.; Hayakawa, T.; Kaneda, T.; Tsutsumi, C.; Imamoto, N.; Akazawa, C.; Sukegawa, J.; Yoneda, Y.; Hiraoka, Y. Live fluorescence imaging reveals early recruitment of emerin, LBR, RanBP2, and Nup153 to reforming functional nuclear envelopes. J. Cell Sci. 2000, 113, 779–794. [Google Scholar]
- Haraguchi, T.; Kojidani, T.; Koujin, T.; Shimi, T.; Osakada, H.; Mori, C.; Yamamoto, A.; Hiraoka, Y. Live cell imaging and electron microscopy reveal dynamic processes of BAF-directed nuclear envelope assembly. J. Cell Sci. 2008, 121, 2540–2554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salpingidou, G.; Smertenko, A.; Hausmanowa-Petrucewicz, I.; Hussey, P.J.; Hutchison, C.J. A novel role for the nuclear membrane protein emerin in association of the centrosome to the outer nuclear membrane. J. Cell Biol. 2007, 178, 897–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, J.A.; Craxton, M.; Yates, J.R.; Kendrick-Jones, J. Aberrant intracellular targeting and cell cycle-dependent phosphorylation of emerin contribute to the Emery-Dreifuss muscular dystrophy phenotype. J. Cell Sci. 1998, 111, 781–792. [Google Scholar]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, K.; Sugiyama, S.; Osouda, S.; Goto, H.; Inagaki, M.; Horigome, T.; Omata, S.; McConnell, M.; Fisher, P.A.; Nishida, Y. Barrier-to-autointegration factor plays crucial roles in cell cycle progression and nuclear organization in Drosophila. J. Cell Sci. 2003, 116, 3811–3823. [Google Scholar] [CrossRef] [PubMed]
- Dyer, J.A.; Lane, B.E.; Hutchison, C.J. Investigations of the pathway of incorporation and function of lamin A in the nuclear lamina. Microsc. Res. Tech. 1999, 45, 1–12. [Google Scholar] [CrossRef]
- Dechat, T.; Gajewski, A.; Korbei, B.; Gerlich, D.; Daigle, N.; Haraguchi, T.; Furukawa, K.; Ellenberg, J.; Foisner, R. LAP2alpha and BAF transiently localize to telomeres and specific regions on chromatin during nuclear assembly. J. Cell Sci. 2004, 117, 6117–6128. [Google Scholar] [CrossRef] [PubMed]
- Dorner, D.; Vlcek, S.; Foeger, N.; Gajewski, A.; Makolm, C.; Gotzmann, J.; Hutchison, C.J.; Foisner, R. Lamina-associated polypeptide 2alpha regulates cell cycle progression and differentiation via the retinoblastoma-E2F pathway. J. Cell Biol. 2006, 173, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Zaremba-Czogalla, M.; Piekarowicz, K.; Wachowicz, K.; Koziol, K.; Dubinska-Magiera, M.; Rzepecki, R. The Different Function of Single Phosphorylation Sites of Drosophila melanogaster Lamin Dm and Lamin C. PLoS ONE 2012, 7, e32649. [Google Scholar] [CrossRef] [PubMed]
- Dechat, T.; Gotzmann, J.; Stockinger, A.; Harris, C.A.; Talle, M.A.; Siekierka, J.J.; Foisner, R. Detergent-salt resistance of LAP2alpha in interphase nuclei and phosphorylation-dependent association with chromosomes early in nuclear assembly implies functions in nuclear structure dynamics. EMBO J. 1998, 17, 4887–4902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyrpasopoulou, A.; Meier, J.; Maison, C.; Simos, G.; Georgatos, S.D. The lamin B receptor (LBR) provides essential chromatin docking sites at the nuclear envelope. EMBO J. 1996, 15, 7108–7119. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.A.; Callebaut, I.; Pezhman, A.; Courvalin, J.C.; Worman, H.J. Domain-specific interactions of human HP1-type chromodomain proteins and inner nuclear membrane protein LBR. J. Biol. Chem. 1997, 272, 14983–14989. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, T.; Koujin, T.; Segura, M.; Wilson, K.L.; Hiraoka, Y. Dynamic behavior of emerin and BAF at early stages of nuclear assembly in living HeLa cells. Mol. Biol. Cell 2000, 11, 21a. [Google Scholar]
- Shumaker, D.K.; Lee, K.K.; Tanhehco, Y.C.; Craigie, R.; Wilson, K.L. LAP2 binds to BAF.DNA complexes: Requirement for the LEM domain and modulation by variable regions. EMBO J. 2001, 20, 1754–1764. [Google Scholar] [CrossRef] [PubMed]
- Mansharamani, M.; Wilson, K.L. Direct binding of nuclear membrane protein MAN1 to emerin in vitro and two modes of binding to barrier-to-autointegration factor. J. Biol. Chem. 2005, 280, 13863–13870. [Google Scholar] [CrossRef] [PubMed]
- Solovei, I.; Wang, A.S.; Thanisch, K.; Schmidt, C.S.; Krebs, S.; Zwerger, M.; Cohen, T.V.; Devys, D.; Foisner, R.; Peichl, L.; et al. LBR and Lamin A/C Sequentially Tether Peripheral Heterochromatin and Inversely Regulate Differentiation. Cell 2013, 152, 584–598. [Google Scholar] [CrossRef] [Green Version]
- Giannios, I.; Chatzantonaki, E.; Georgatos, S. Dynamics and Structure-Function Relationships of the Lamin B Receptor (LBR). PLoS ONE 2017, 12, e0169626. [Google Scholar] [CrossRef]
- Hirano, Y.; Segawa, M.; Ouchi, F.S.; Yamakawa, Y.; Furukawa, K.; Takeyasu, K.; Horigome, T. Dissociation of emerin from barrier-to-autointegration factor is regulated through mitotic phosphorylation of emerin in a Xenopus egg cell-free system. J. Biol. Chem. 2005, 280, 39925–39933. [Google Scholar] [CrossRef]
- Wilson, K.L.; Holaska, J.M.; Montes de Oca, R.; Tifft, K.; Zastrow, M.; Segura-Totten, M.; Mansharamani, M.; Bengtsson, L. Nuclear membrane protein emerin: Roles in gene regulation, actin dynamics and human disease. Novartis Found. Symp. 2005, 264, 51–58, discussion 58–62, 227–230. [Google Scholar]
- Tunnah, D.; Sewry, C.A.; Vaux, D.; Schirmer, E.C.; Morris, G.E. The apparent absence of lamin B1 and emerin in many tissue nuclei is due to epitope masking. J. Mol. Histol. 2005, 36, 337–344. [Google Scholar] [CrossRef] [Green Version]
- Hozak, P.; Sasseville, A.M.; Raymond, Y.; Cook, P.R. Lamin proteins form an internal nucleoskeleton as well as a peripheral lamina in human cells. J. Cell Sci. 1995, 108, 635–644. [Google Scholar]
- Furukawa, K. LAP2 binding protein 1 (L2BP1/BAF) is a candidate mediator of LAP2-chromatin interaction. J. Cell Sci. 1999, 112, 2485–2492. [Google Scholar] [PubMed]
- Cai, M.; Huang, Y.; Ghirlando, R.; Wilson, K.L.; Craigie, R.; Clore, G.M. Solution structure of the constant region of nuclear envelope protein LAP2 reveals two LEM-domain structures: One binds BAF and the other binds DNA. EMBO J. 2001, 20, 4399–4407. [Google Scholar] [CrossRef] [PubMed]
- Molitor, T.P.; Traktman, P. Depletion of the protein kinase VRK1 disrupts nuclear envelope morphology and leads to BAF retention on mitotic chromosomes. Mol. Biol. Cell 2014, 25, 891–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asencio, C.; Davidson, I.F.; Santarella-Mellwig, R.; Thi, B.N.L.H.; Mall, M.; Wallenfang, M.R.; Mattaj, I.W.; Gorjanacz, M. Coordination of Kinase and Phosphatase Activities by Lem4 Enables Nuclear Envelope Reassembly during Mitosis. Cell 2012, 150, 122–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyers, L.; Erhart, R.; Laffer, S.; Pusch, O.; Weipoltshammer, K.; Schofer, C. LEM4/ANKLE-2 deficiency impairs post-mitotic re-localization of BAF, LAP2 alpha and LaminA to the nucleus, causes nuclear envelope instability in telophase and leads to hyperploidy in HeLa cells. Eur. J. Cell Biol. 2018, 97, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Piekarowicz, K.; Machowska, M.; Dratkiewicz, E.; Lorek, D.; Madej-Pilarczyk, A.; Rzepecki, R. The effect of the lamin A and its mutants on nuclear structure, cell proliferation, protein stability, and mobility in embryonic cells. Chromosoma 2017, 126, 501–517. [Google Scholar] [CrossRef] [PubMed]
- Gudise, S.; Figueroa, R.A.; Lindberg, R.; Larsson, V.; Hallberg, E. Samp1 is functionally associated with the LINC complex and A-type lamina networks. J. Cell Sci. 2011, 124, 2077–2085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jafferali, M.H.; Figueroa, R.A.; Hasan, M.; Hallberg, E. Spindle associated membrane protein 1 (Samp1) is required for the differentiation of muscle cells (vol 7, pg 16655, 2017). Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Buendia, B.; Courvalin, J.C. Domain-specific disassembly and reassembly of nuclear membranes during mitosis. Exp. Cell Res. 1997, 230, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Guan, T.; Gerace, L. Integral membrane proteins of the nuclear envelope are dispersed throughout the endoplasmic reticulum during mitosis. J. Cell Biol. 1997, 137, 1199–1210. [Google Scholar] [CrossRef]
- Vijayaraghavan, B.; Figueroa, R.A.; Bergqvist, C.; Gupta, A.J.; Sousa, P.; Hallberg, E. RanGTPase regulates the interaction between the inner nuclear membrane proteins, Samp1 and Emerin. BBA Biomembr. 2018, 1860, 1326–1334. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, R.; Ranade, D.; Sengupta, K. Emerin modulates spatial organization of chromosome territories in cells on softer matrices. Nucleic Acids Res. 2018, 46, 5561–5586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, S.; Gilchrist, S.; Bridger, J.M.; Mahy, N.L.; Ellis, J.A.; Bickmore, W.A. The spatial organization of human chromosomes within the nuclei of normal and emerin-mutant cells. Hum. Mol. Genet. 2001, 10, 211–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchiya, Y.; Hase, A.; Ogawa, M.; Yorifuji, H.; Arahata, K. Distinct regions specify the nuclear membrane targeting of emerin, the responsible protein for Emery-Dreifuss muscular dystrophy. Eur. J. Biochem. 1999, 259, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Vigers, G.P.; Lohka, M.J. A distinct vesicle population targets membranes and pore complexes to the nuclear envelope in Xenopus eggs. J. Cell Biol. 1991, 112, 545–556. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.Y.; Wang, S.; Heidinger, J.M.; Shumaker, D.K.; Adam, S.A.; Goldman, R.D.; Zheng, Y. A mitotic lamin B matrix induced by RanGTP required for spindle assembly. Science 2006, 311, 1887–1893. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Tsai, M.Y. The mitotic spindle matrix: A fibro-membranous lamin connection. Cell Cycle 2006, 5, 2345–2347. [Google Scholar] [CrossRef]
- Hayashi, D.; Tanabe, K.; Katsube, H.; Inoue, Y.H. B-type nuclear lamin and the nuclear pore complex Nup107-160 influences maintenance of the spindle envelope required for cytokinesis in Drosophila male meiosis. Biol. Open 2016, 5, 1011–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smythe, C.; Jenkins, H.E.; Hutchison, C.J. Incorporation of the nuclear pore basket protein nup153 into nuclear pore structures is dependent upon lamina assembly: Evidence from cell-free extracts of Xenopus eggs. EMBO J. 2000, 19, 3918–3931. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.; Krohne, G. Lamina-associated polypeptide 2beta (LAP2beta) is contained in a protein complex together with A- and B-type lamins. Eur. J. Cell Biol. 2003, 82, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Blake, D.L.; Callebaut, I.; Skerjanc, I.S.; Holmer, L.; McBurney, M.W.; Paulin-Levasseur, M.; Worman, H.J. MAN1, an inner nuclear membrane protein that shares the LEM domain with lamina-associated polypeptide 2 and emerin. J. Biol. Chem. 2000, 275, 4840–4847. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.K.; Haraguchi, T.; Lee, R.S.; Koujin, T.; Hiraoka, Y.; Wilson, K.L. Distinct functional domains in emerin bind lamin A and DNA-bridging protein BAF. J. Cell Sci. 2001, 114, 4567–4573. [Google Scholar]
- Bonne, G.; Di Barletta, M.R.; Varnous, S.; Becane, H.M.; Hammouda, E.H.; Merlini, L.; Muntoni, F.; Greenberg, C.R.; Gary, F.; Urtizberea, J.A.; et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet. 1999, 21, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xu, R.; Zhu, B.; Yang, X.; Ding, X.; Duan, S.; Xu, T.; Zhuang, Y.; Han, M. Syne-1 and Syne-2 play crucial roles in myonuclear anchorage and motor neuron innervation. Development 2007, 134, 901–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, W.C.; Mitsuhashi, H.; Keduka, E.; Nonaka, I.; Noguchi, S.; Nishino, I.; Hayashi, Y.K. TMEM43 Mutations in Emery-Dreifuss Muscular Dystrophy-Related Myopathy. Ann. Neurol. 2011, 69, 1005–1013. [Google Scholar] [CrossRef]
- Yuan, J.H.; Ando, M.; Higuchi, I.; Sakiyama, Y.; Matsuura, E.; Michizono, K.; Watanabe, O.; Nagano, S.; Inamori, Y.; Hashiguchi, A.; et al. Partial Deficiency of Emerin Caused by a Splice Site Mutation in EMD. Internal. Med. 2014, 53, 1563–1568. [Google Scholar] [CrossRef] [Green Version]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubińska-Magiera, M.; Kozioł, K.; Machowska, M.; Piekarowicz, K.; Filipczak, D.; Rzepecki, R. Emerin Is Required for Proper Nucleus Reassembly after Mitosis: Implications for New Pathogenetic Mechanisms for Laminopathies Detected in EDMD1 Patients. Cells 2019, 8, 240. https://doi.org/10.3390/cells8030240
Dubińska-Magiera M, Kozioł K, Machowska M, Piekarowicz K, Filipczak D, Rzepecki R. Emerin Is Required for Proper Nucleus Reassembly after Mitosis: Implications for New Pathogenetic Mechanisms for Laminopathies Detected in EDMD1 Patients. Cells. 2019; 8(3):240. https://doi.org/10.3390/cells8030240
Chicago/Turabian StyleDubińska-Magiera, Magda, Katarzyna Kozioł, Magdalena Machowska, Katarzyna Piekarowicz, Daria Filipczak, and Ryszard Rzepecki. 2019. "Emerin Is Required for Proper Nucleus Reassembly after Mitosis: Implications for New Pathogenetic Mechanisms for Laminopathies Detected in EDMD1 Patients" Cells 8, no. 3: 240. https://doi.org/10.3390/cells8030240