Tissue-Specific Influence of Lamin A Mutations on Notch Signaling and Osteogenic Phenotype of Primary Human Mesenchymal Cells


Abstract
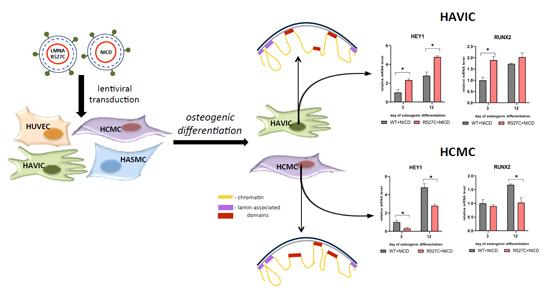
:
1. Introduction
2. Materials and Methods
2.1. Human Samples
2.2. Cell Culture
2.3. Lentiviral Constructs and Transduction
2.4. Lentiviral Production and Transduction
2.5. Immunocytochemical Staining
2.6. Induction of Osteogenic Differentiation
2.7. qPCR
2.8. Promoter Activity Assay
2.9. Statistics
3. Results
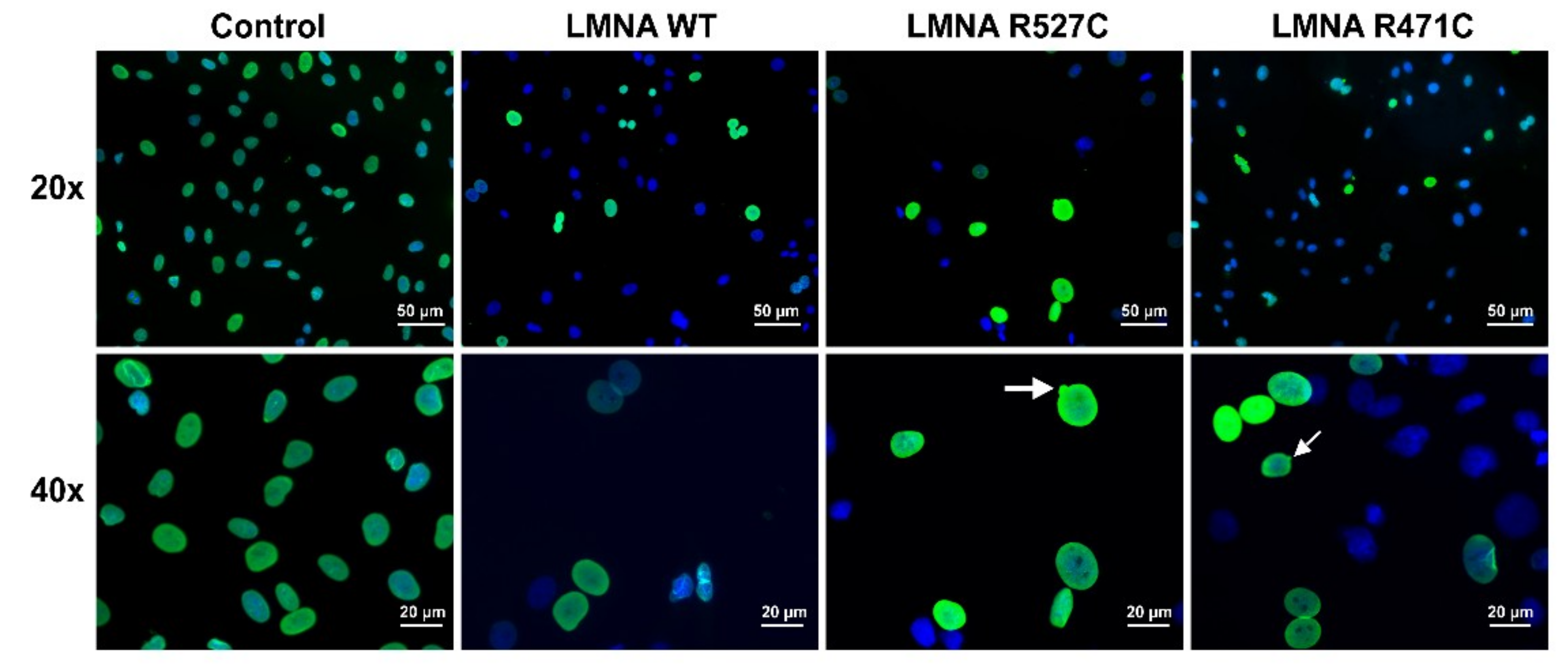
3.1. LMNA R527C and LMNA R471C Causes Disruption of Lamin Organization in CMC
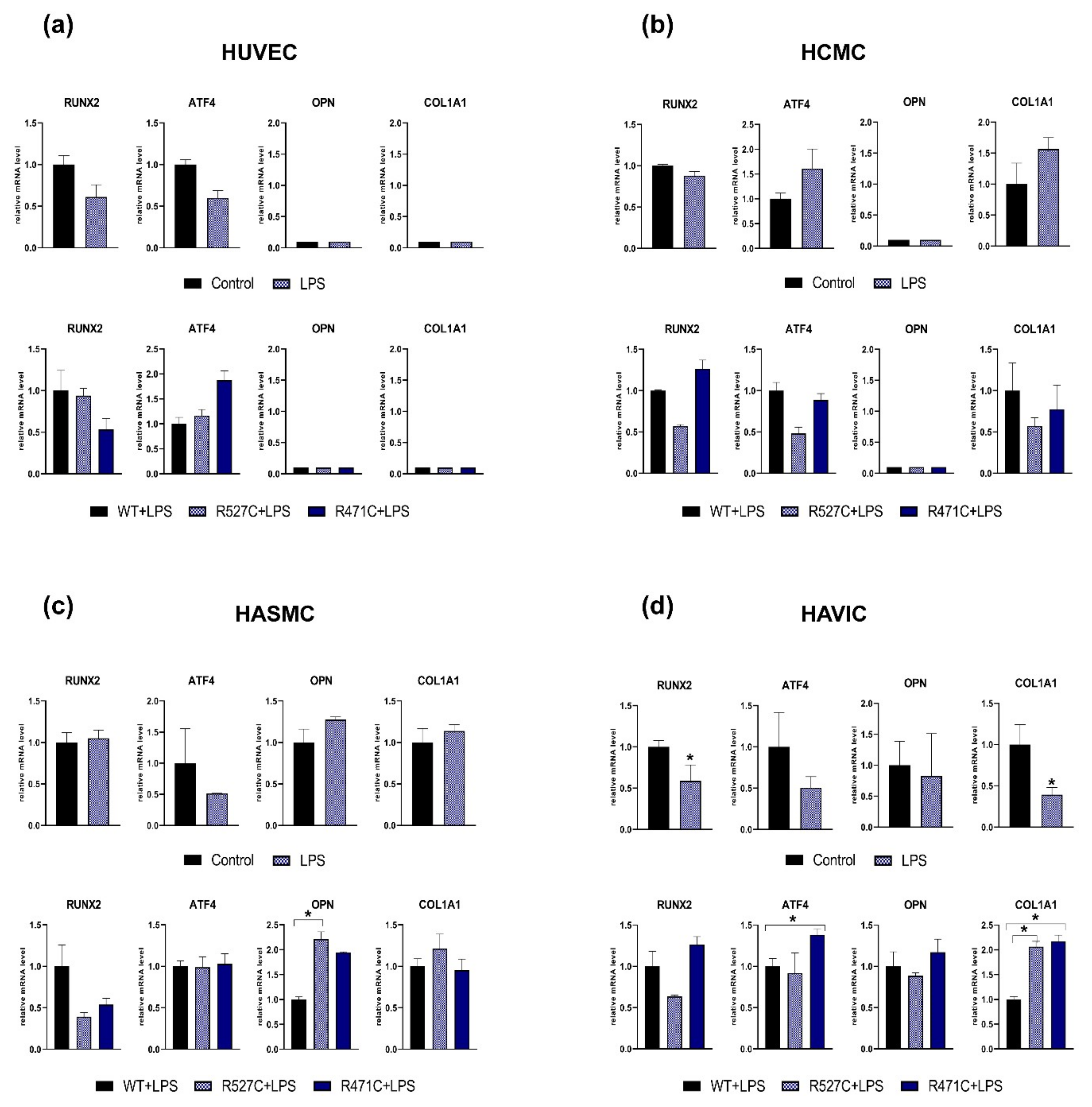
3.2. The Impact of LMNA R527C and LMNA R471C on Osteogenic Markers in Human Mesenchymal Cells in the Presence of LPS
3.3. The Impact of LMNA R527C and LMNA R471C on Osteogenic Differentiation in Human Mesenchymal Cells
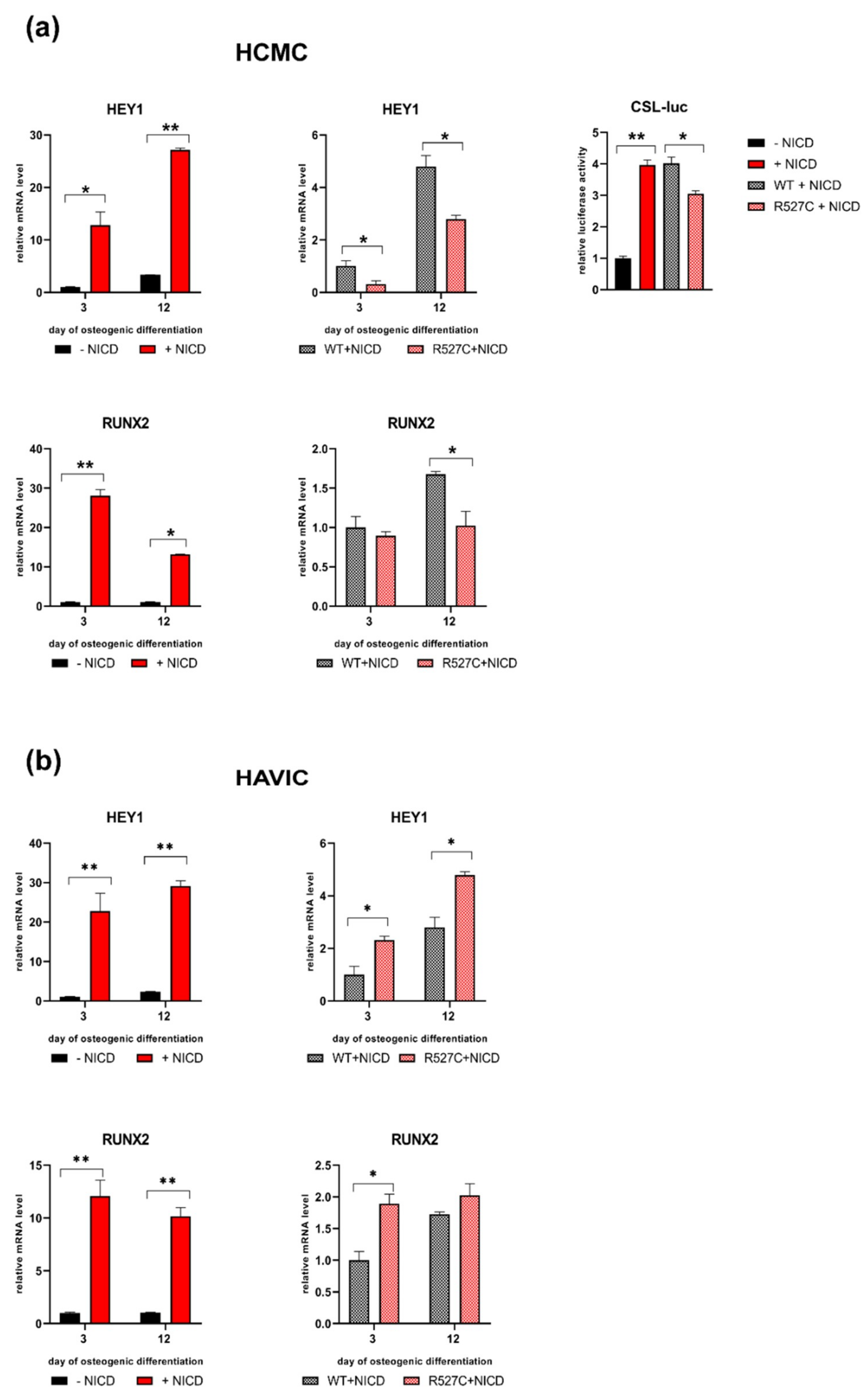
3.4. The Impact of LMNA R527C and LMNA R471C on Expression of Notch Target Genes in Human Mesenchymal Cells
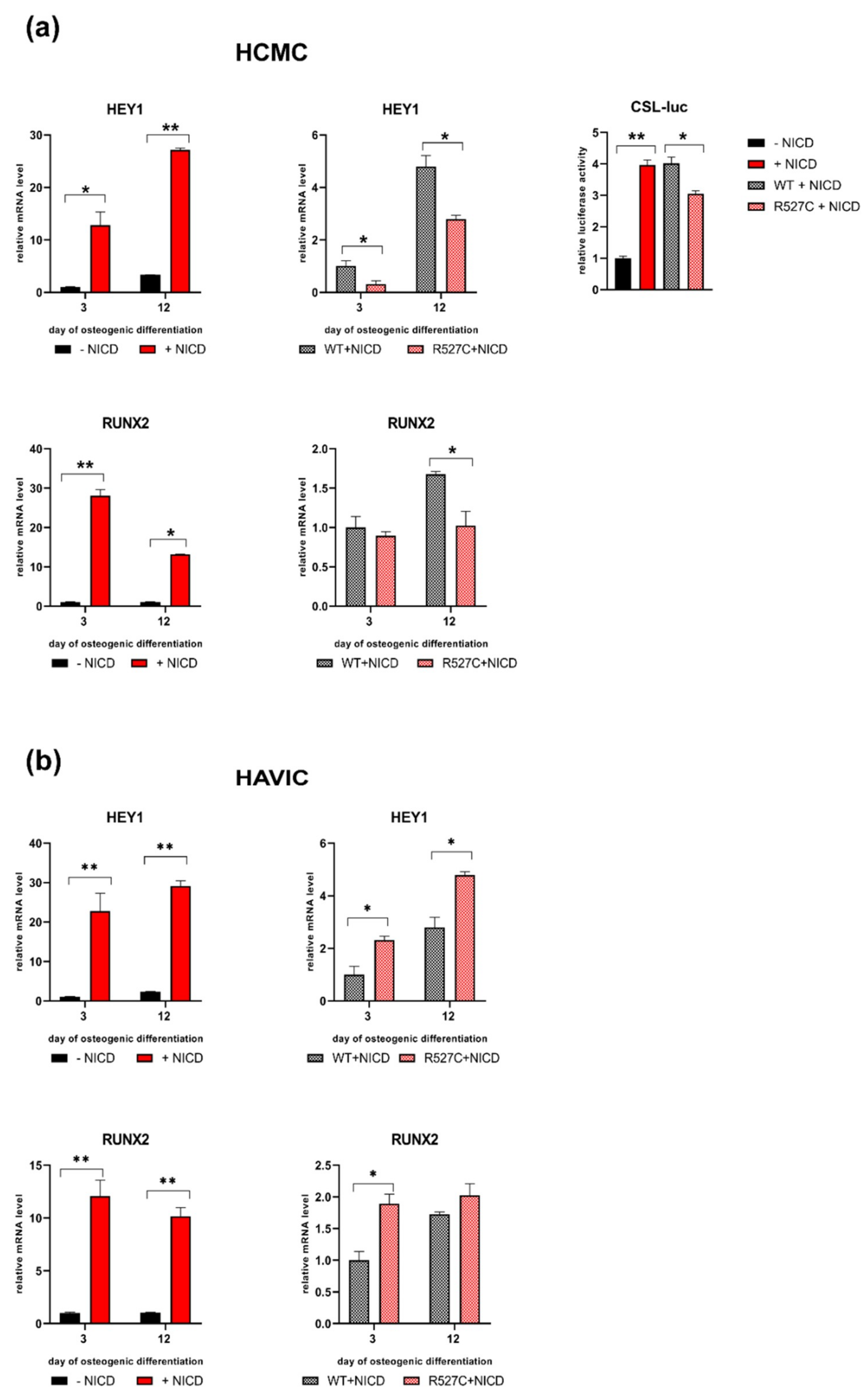
3.5. LMNA R527C Mutation Has the Opposite Effect on HCMC and HAVIC in the Presence of Notch Activation
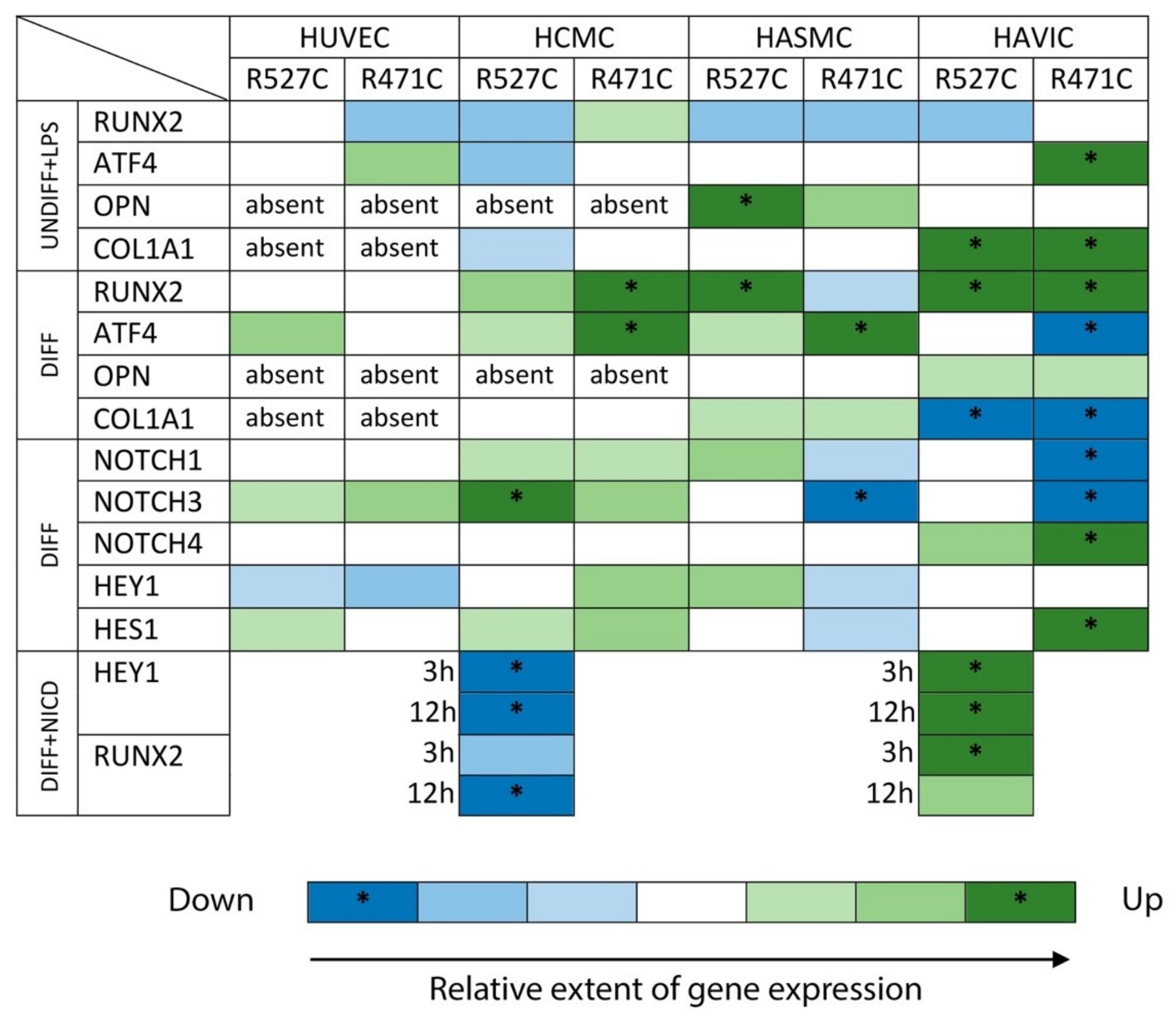
3.6. Analysis of Osteogenic Genes and Notch-Responsive Genes Expression in Different Lines of Undifferentiated Mesenchymal Cells
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brayson, D.; Shanahan, C.M. Current insights into lmna cardiomyopathies: Existing models and missing lincs. Nucleus 2017, 8, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Caux, F.; Dubosclard, E.; Lascols, O.; Buendia, B.; Chazouilleres, O.; Cohen, A.; Courvalin, J.-C.; Laroche, L.; Capeau, J.; Vigouroux, C. A new clinical condition linked to a novel mutation in lamins a and c with generalized lipoatrophy, insulin-resistant diabetes, disseminated leukomelanodermic papules, liver steatosis, and cardiomyopathy. J. Clin. Endocrinol. Metab. 2003, 88, 1006–1013. [Google Scholar] [CrossRef]
- Capell, B.C.; Collins, F.S.; Nabel, E.G. Mechanisms of cardiovascular disease in accelerated aging syndromes. Circul. Res. 2007, 101, 13–26. [Google Scholar] [CrossRef]
- Olive, M.; Harten, I.; Mitchell, R.; Beers, J.K.; Djabali, K.; Cao, K.; Erdos, M.R.; Blair, C.; Funke, B.; Smoot, L. Cardiovascular pathology in hutchinson-gilford progeria: Correlation with the vascular pathology of aging. Atertio. Thromb. Vasc. Biol. 2010, 30, 2301–2309. [Google Scholar] [CrossRef] [PubMed]
- Bidault, G.; Vatier, C.; Capeau, J.; Vigouroux, C.; Béréziat, V. Lmna-linked lipodystrophies: From altered fat distribution to cellular alterations. Biochem. Soc. Trans. 2011, 39, 1752–1757. [Google Scholar] [CrossRef]
- Ragnauth, C.D.; Warren, D.T.; Liu, Y.; McNair, R.; Tajsic, T.; Figg, N.; Shroff, R.; Skepper, J.; Shanahan, C.M. Prelamin a acts to accelerate smooth muscle cell senescence and is a novel biomarker of human vascular aging. Circulation 2010, 121, 2200–2210. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Drozdov, I.; Shroff, R.; Beltran, L.E.; Shanahan, C.M. Prelamin a accelerates vascular calcification via activation of the DNA damage response and senescence-associated secretory phenotype in vascular smooth muscle cells. Circul. Res. 2013, 112, e99–e109. [Google Scholar] [CrossRef]
- Zhang, J.; Lian, Q.; Zhu, G.; Zhou, F.; Sui, L.; Tan, C.; Mutalif, R.A.; Navasankari, R.; Zhang, Y.; Tse, H.-F. A human ipsc model of hutchinson gilford progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 2011, 8, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Wang, L.; Yao, Q.-P.; Zhang, P.; Liu, B.; Wang, G.-L.; Shen, B.-R.; Cheng, B.; Wang, Y.; Jiang, Z.-L. Nuclear envelope proteins nesprin2 and lamina regulate proliferation and apoptosis of vascular endothelial cells in response to shear stress. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 1165–1173. [Google Scholar] [CrossRef] [PubMed]
- Bonello-Palot, N.; Simoncini, S.; Robert, S.; Bourgeois, P.; Sabatier, F.; Levy, N.; Dignat-George, F.; Badens, C. Prelamin a accumulation in endothelial cells induces premature senescence and functional impairment. Atherosclerosis 2014, 237, 45–52. [Google Scholar] [CrossRef]
- Afonso, P.; Auclair, M.; Boccara, F.; Vantyghem, M.-C.; Katlama, C.; Capeau, J.; Vigouroux, C.; Caron-Debarle, M. Lmna mutations resulting in lipodystrophy and hiv protease inhibitors trigger vascular smooth muscle cell senescence and calcification: Role of zmpste24 downregulation. Atherosclerosis 2016, 245, 200–211. [Google Scholar] [CrossRef]
- Naetar, N.; Ferraioli, S.; Foisner, R. Lamins in the nuclear interior− life outside the lamina. J. Cell Sci. 2017, 130, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Lund, E.; Oldenburg, A.R.; Delbarre, E.; Freberg, C.T.; Duband-Goulet, I.; Eskeland, R.; Buendia, B.; Collas, P. Lamin a/c-promoter interactions specify chromatin state–dependent transcription outcomes. Genome Res. 2013, 23, 1580–1589. [Google Scholar] [CrossRef]
- Amendola, M.; van Steensel, B. Mechanisms and dynamics of nuclear lamina–genome interactions. Curr. Opin. Cell Biol. 2014, 28, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Braun, S.; Barrales, R.R. Beyond tethering and the lem domain: Mscellaneous functions of the inner nuclear membrane lem2. Nucleus 2016, 7, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Perepelina, K.; Dmitrieva, R.; Ignatieva, E.; Borodkina, A.; Kostareva, A.; Malashicheva, A. Lamin a/c mutation associated with lipodystrophy influences adipogenic differentiation of stem cells through interaction with notch signaling. Biochem. Cell Biol. 2017, 96, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Clements, L.; Manilal, S.; Love, D.; Morris, G. Direct interaction between emerin and lamin a. Biochem. Biophys. Res. Commun. 2000, 267, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Sakaki, M.; Koike, H.; Takahashi, N.; Sasagawa, N.; Tomioka, S.; Arahata, K.; Ishiura, S. Interaction between emerin and nuclear lamins. J. Biochem. 2001, 129, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Lee, T.-H.; Shim, J. Emerin suppresses notch signaling by restricting the notch intracellular domain to the nuclear membrane. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, T.; Escalante-Alcalde, D.; Bhatt, H.; Anver, M.; Bhat, N.; Nagashima, K.; Stewart, C.L.; Burke, B. Loss of a-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol. 1999, 147, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Lamin a-dependent misregulation of adult stem cells associated with accelerated ageing. Nat. Cell Biol. 2008, 10, 452–459. [Google Scholar] [CrossRef]
- Canalis, E. Notch in skeletal physiology and disease. Osteoporos. Int. 2018, 29, 2611–2621. [Google Scholar] [CrossRef]
- Luo, D.-Q.; Wang, X.-Z.; Meng, Y.; He, D.-Y.; Chen, Y.-M.; Ke, Z.-Y.; Yan, M.; Huang, Y.; Chen, D.-F. Mandibuloacral dysplasia type a-associated progeria caused by homozygous lmna mutation in a family from southern china. BMC Pediatr. 2014, 14, 256. [Google Scholar] [CrossRef] [PubMed]
- Garavelli, L.; D’Apice, M.R.; Rivieri, F.; Bertoli, M.; Wischmeijer, A.; Gelmini, C.; De Nigris, V.; Albertini, E.; Rosato, S.; Virdis, R.; et al. Mandibuloacral dysplasia type a in childhood. Am. J. Med. Genet. A 2009, 149, 2258–2264. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Cogulu, O.; Ozkinay, F.; Onay, H.; Agarwal, A.K. A novel homozygous ala529val lmna mutation in turkish patients with mandibuloacral dysplasia. J. Clin. Endocrinol. Metab. 2005, 90, 5259–5264. [Google Scholar] [CrossRef] [PubMed]
- Malashicheva, A.; Bogdanova, M.; Zabirnyk, A.; Smolina, N.; Ignatieva, E.; Freilikhman, O.; Fedorov, A.; Dmitrieva, R.; Sjöberg, G.; Sejersen, T. Various lamin a/c mutations alter expression profile of mesenchymal stem cells in mutation specific manner. Mol. Genet. Metab. 2015, 115, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Li, W.; Vidal, C.; Yeo, L.S.; Fatkin, D.; Duque, G. Lamin a/c deficiency is associated with fat infiltration of muscle and bone. Mech. Ageing Dev. 2011, 132, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Kosho, T.; Takahashi, J.; Momose, T.; Nakamura, A.; Sakurai, A.; Wada, T.; Yoshida, K.; Wakui, K.; Suzuki, T.; Kasuga, K.; et al. Mandibuloacral dysplasia and a novel lmna mutation in a woman with severe progressive skeletal changes. Am. J. Med. Genet. A 2007, 143a, 2598–2603. [Google Scholar] [CrossRef]
- Smits, A.M.; Van Vliet, P.; Metz, C.H.; Korfage, T.; Sluijter, J.P.; Doevendans, P.A.; Goumans, M.-J. Human cardiomyocyte progenitor cells differentiate into functional mature cardiomyocytes: An in vitro model for studying human cardiac physiology and pathophysiology. Nat. Protoc. 2009, 4, 232–243. [Google Scholar] [CrossRef]
- Malashicheva, A.; Kostina, D.; Kostina, A.; Irtyuga, O.; Voronkina, I.; Smagina, L.; Ignatieva, E.; Gavriliuk, N.; Uspensky, V.; Moiseeva, O. Phenotypic and functional changes of endothelial and smooth muscle cells in thoracic aortic aneurysms. Int. J. Vasc. Med. 2016, 2016, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kostina, A.; Shishkova, A.; Ignatieva, E.; Irtyuga, O.; Bogdanova, M.; Levchuk, K.; Golovkin, A.; Zhiduleva, E.; Uspenskiy, V.; Moiseeva, O. Different notch signaling in cells from calcified bicuspid and tricuspid aortic valves. J. Mol. Cell. Cardiol. 2018, 114, 211–219. [Google Scholar] [CrossRef]
- Baudin, B.; Bruneel, A.; Bosselut, N.; Vaubourdolle, M. A protocol for isolation and culture of human umbilical vein endothelial cells. Nat. Protoc. 2007, 2, 481–485. [Google Scholar] [CrossRef]
- Kostina, A.S.; Uspensky, V.E.; Irtyuga, O.B.; Ignatieva, E.V.; Freylikhman, O.; Gavriliuk, N.D.; Moiseeva, O.M.; Zhuk, S.; Tomilin, A.; Kostareva, A.A.; et al. Notch-dependent emt is attenuated in patients with aortic aneurysm and bicuspid aortic valve. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1862, 733–740. [Google Scholar] [CrossRef]
- Herzmann, N.; Salamon, A.; Fiedler, T.; Peters, K. Lipopolysaccharide induces proliferation and osteogenic differentiation of adipose-derived mesenchymal stromal cells in vitro via tlr4 activation. Exp. Cell Res. 2017, 350, 115–122. [Google Scholar] [CrossRef]
- Zanotti, S.; Canalis, E. Notch signaling and the skeleton. Endocr. Rev. 2016, 37, 223–253. [Google Scholar] [CrossRef] [PubMed]
- Boguslavsky, R.L.; Stewart, C.L.; Worman, H.J. Nuclear lamin a inhibits adipocyte differentiation: Implications for dunnigan-type familial partial lipodystrophy. Hum. Mol. Genet. 2006, 15, 653–663. [Google Scholar] [CrossRef]
- Favreau, C.; Higuet, D.; Courvalin, J.C.; Buendia, B. Expression of a mutant lamin a that causes emery-dreifuss muscular dystrophy inhibits in vitro differentiation of c2c12 myoblasts. Mol. Cell. Biol. 2004, 24, 1481–1492. [Google Scholar] [CrossRef]
- Hakelien, A.M.; Delbarre, E.; Gaustad, K.G.; Buendia, B.; Collas, P. Expression of the myodystrophic r453w mutation of lamin a in c2c12 myoblasts causes promoter-specific and global epigenetic defects. Exp. Cell Res. 2008, 314, 1869–1880. [Google Scholar] [CrossRef]
- Poleshko, A.; Shah, P.P.; Gupta, M.; Babu, A.; Morley, M.P.; Manderfield, L.J.; Ifkovits, J.L.; Calderon, D.; Aghajanian, H.; Sierra-Pagán, J.E. Genome-nuclear lamina interactions regulate cardiac stem cell lineage restriction. Cell 2017, 171, 573–587. [Google Scholar] [CrossRef] [PubMed]
- Perovanovic, J.; Dell’Orso, S.; Gnochi, V.F.; Jaiswal, J.K.; Sartorelli, V.; Vigouroux, C.; Mamchaoui, K.; Mouly, V.; Bonne, G.; Hoffman, E.P. Laminopathies disrupt epigenomic developmental programs and cell fate. Sci. Transl. Med. 2016, 8, 335–358. [Google Scholar] [CrossRef] [PubMed]
- Briand, N.; Collas, P. Laminopathy-causing lamin a mutations reconfigure lamina-associated domains and local spatial chromatin conformation. Nucleus 2018, 9, 216–226. [Google Scholar] [CrossRef]
- Andersson, E.R.; Sandberg, R.; Lendahl, U. Notch signaling: Simplicity in design, versatility in function. Development 2011, 138, 3593–3612. [Google Scholar] [CrossRef] [PubMed]
- Tsukune, N.; Naito, M.; Kubota, T.; Ozawa, Y.; Nagao, M.; Ohashi, A.; Sato, S.; Takahashi, T. Lamin a overexpression promotes osteoblast differentiation and calcification in the mc3t3-e1 preosteoblastic cell line. Biochem. Biophys. Res. Commun. 2017, 488, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Blondel, S.; Jaskowiak, A.L.; Egesipe, A.L.; Le Corf, A.; Navarro, C.; Cordette, V.; Martinat, C.; Laabi, Y.; Djabali, K.; de Sandre-Giovannoli, A.; et al. Induced pluripotent stem cells reveal functional differences between drugs currently investigated in patients with hutchinson-gilford progeria syndrome. Stem Cells Transl. Med. 2014, 3, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, C.; Bernasconi, P.; Cavalcante, P.; Cappelletti, C.; D’Apice, M.R.; Sbraccia, P.; Novelli, G.; Prencipe, S.; Lemma, S.; Baldini, N.; et al. Modulation of tgfbeta 2 levels by lamin a in u2-os osteoblast-like cells: Understanding the osteolytic process triggered by altered lamins. Oncotarget 2015, 6, 7424–7437. [Google Scholar] [CrossRef] [PubMed]
- Piekarowicz, K.; Machowska, M.; Dratkiewicz, E.; Lorek, D.; Madej-Pilarczyk, A.; Rzepecki, R. The effect of the lamin a and its mutants on nuclear structure, cell proliferation, protein stability, and mobility in embryonic cells. Chromosoma 2016, 126, 501–517. [Google Scholar] [CrossRef] [PubMed]
- Rønningen, T.; Shah, A.; Oldenburg, A.R.; Vekterud, K.; Delbarre, E.; Moskaug, J.Ø.; Collas, P. Prepatterning of differentiation-driven nuclear lamin a/c-associated chromatin domains by glcnacylated histone h2b. Genome Res. 2015, 25, 1825–1835. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer (5′→3′) |
|---|---|
| GAPDH | F: AATGAAGGGGTCATTGATGG |
| R: AAGGTGAAGGTCGGAGTCAA | |
| NOTCH1 | F: GTCAACGCCGTAGATGACC |
| R: TTGTTAGCCCCGTTCTTCAG | |
| NOTCH2 | F: ATGGTGGCAGAACTGATCAAC |
| R: TTGGCAAAATGGTCTAACAGG | |
| NOTCH3 | F: GGAGCCAATAAGGACATGCAGGAT |
| R: GGCAAAGTGGTCCAACAGCAGC | |
| NOTCH4 | F: GTTGTGACAGGGTTGGGACT |
| R: CAGCCCAGTGGGTATCTCTG | |
| DLL4 | F: AGGCCTGTTTTGTGACCAAG |
| R: CTCCAGCTCACAGTCCACAC | |
| JAG1 | F: TGCCAAGTGCCAGGAAGT |
| R: GCCCCATCTGGTATCACACT | |
| HEY1 | F: TGGATCACCTGAAAATGCTG |
| R: CGAAATCCCAAACTCCGATA | |
| HES1 | F: AGCACAGAAAGTCATCAAAG |
| R: AGGTGCTTCACTGTCATTTC | |
| RUNX2 | F: TGGATCACCTGAAAATGCTG |
| R: CGAAATCCCAACTCCGATA | |
| BMP4 | F: AGCACTGGTCTTGAGTATCCTG |
| R: GCAGAGTTTTCACTGGTCCC | |
| COL1A1 | F: GACCTAAAGGTGCTGCTGGAG |
| R: CTTGTTCACCTCTCTCGCCA | |
| OPN | F: TCACCTGTGCCATACCAGTTAAA |
| R: TGGGTATTTGTTGTAAAGCTGCTT | |
| OGN | F: GGCAATAACACCATTACCTCCC |
| R: AGGGTGGTACAGCATCAATGT | |
| ATF4 (TaqMan) | HS00909569_g1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perepelina, K.; Klauzen, P.; Kostareva, A.; Malashicheva, A. Tissue-Specific Influence of Lamin A Mutations on Notch Signaling and Osteogenic Phenotype of Primary Human Mesenchymal Cells. Cells 2019, 8, 266. https://doi.org/10.3390/cells8030266
Perepelina K, Klauzen P, Kostareva A, Malashicheva A. Tissue-Specific Influence of Lamin A Mutations on Notch Signaling and Osteogenic Phenotype of Primary Human Mesenchymal Cells. Cells. 2019; 8(3):266. https://doi.org/10.3390/cells8030266
Chicago/Turabian StylePerepelina, Kseniya, Polina Klauzen, Anna Kostareva, and Anna Malashicheva. 2019. "Tissue-Specific Influence of Lamin A Mutations on Notch Signaling and Osteogenic Phenotype of Primary Human Mesenchymal Cells" Cells 8, no. 3: 266. https://doi.org/10.3390/cells8030266