Improving Cancer Immunotherapy by Targeting the Hypoxic Tumor Microenvironment: New Opportunities and Challenges

 ,
,  and
and 
{kind=link}
Abstract
:1. Introduction
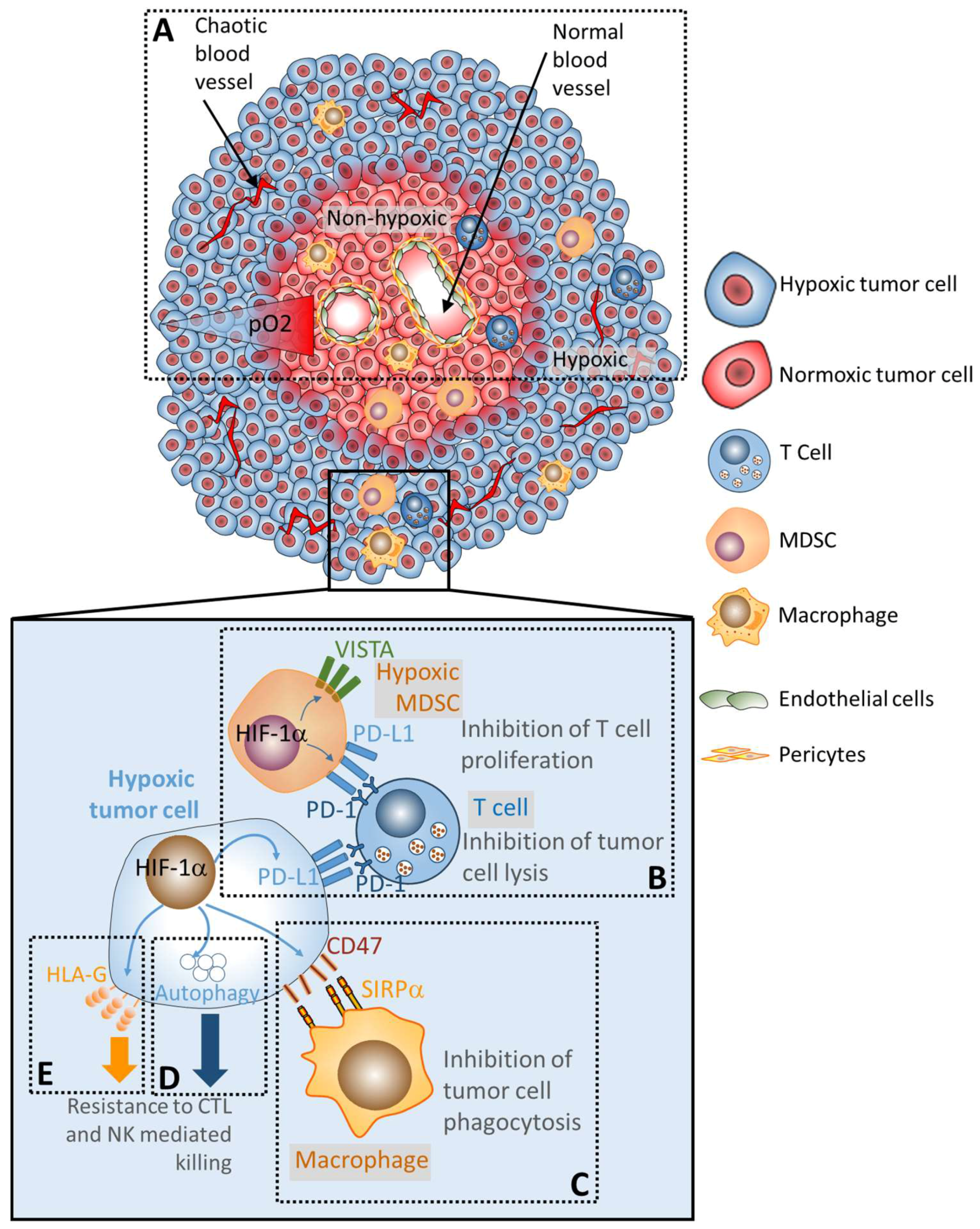
2. The Mechanisms Underlying the Establishment of Hypoxic Tumor Microenvironment and the Impact of Hypoxia on Damping the Anti-Tumor Immune Response
3. Hypoxia Upregulates the Expression of PD-L1 and Promotes the Establishment of the Immunosuppressive Tumor Microenvironment
4. Hypoxia Induces the Expression of the Immune Checkpoint V-Domain Ig Suppressor of T Cell Activation (VISTA) and Promotes the Immunosuppressive Function of Tumoral MDSC
5. Hypoxia Upregulates the Macrophage Immune Checkpoint CD47 “Don’t Eat Me Signal” and Induces Tumor Cell Escape from Phagocytosis
6. Hypoxia-Induced Autophagy Impairs Tumor Cell Susceptibility to Immune Cell Attack
7. Hypoxia Upregulates the Expression of the Non-Classical and Immunosuppressive MHC Class I (HLA-G)
8. The Challenges and Opportunities of Targeting Hypoxia
9. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Skrombolas, D.; Frelinger, J.G. Challenges and developing solutions for increasing the benefits of IL-2 treatment in tumor therapy. Expert Rev. Clin. Immunol. 2014, 10, 207–217. [Google Scholar] [CrossRef] [Green Version]
- June, C.H. Adoptive T cell therapy for cancer in the clinic. J. Clin. Investig. 2007, 117, 1466–1476. [Google Scholar] [CrossRef] [Green Version]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef]
- Smyth, M.J.; Ngiow, S.F.; Ribas, A.; Teng, M.W. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat. Rev. Clin. Oncol. 2016, 13, 143–158. [Google Scholar] [CrossRef]
- McArthur, H.L. Checkpoint inhibitors in breast cancer: hype or promise? Clin. Adv. Hematol. Oncol. 2016, 14, 392–395. [Google Scholar]
- Khan, K.A.; Kerbel, R.S. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat. Rev. Clin. Oncol. 2018, 15, 310–324. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Cooper, Z.A.; Frederick, D.T.; Ahmed, Z.; Wargo, J.A. Combining checkpoint inhibitors and BRAF-targeted agents against metastatic melanoma. Oncoimmunology 2013, 2, e24320. [Google Scholar] [CrossRef] [Green Version]
- Buchbinder, E.I.; Hodi, F.S. Melanoma in 2015: Immune-checkpoint blockade—Durable cancer control. Nat. Rev. Clin. Oncol. 2016, 13, 77–78. [Google Scholar] [CrossRef]
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer immunotherapy: The beginning of the end of cancer? BMC Med. 2016, 14, 73. [Google Scholar] [CrossRef]
- Tredan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef]
- Jain, R.K. Normalizing tumor vasculature with anti-angiogenic therapy: A new paradigm for combination therapy. Nat. Med. 2001, 7, 987–989. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Jewell, U.R.; Kvietikova, I.; Scheid, A.; Bauer, C.; Wenger, R.H.; Gassmann, M. Induction of HIF-1alpha in response to hypoxia is instantaneous. FASEB J. 2001, 15, 1312–1314. [Google Scholar] [CrossRef]
- Semenza, G.L. A Compendium of Proteins that Interact with HIF-1alpha. Exp. Cell Res. 2017. [Google Scholar] [CrossRef]
- Harris, A.L. Hypoxia--a key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef]
- Shannon, A.M.; Bouchier-Hayes, D.J.; Condron, C.M.; Toomey, D. Tumour hypoxia, chemotherapeutic resistance and hypoxia-related therapies. Cancer Treat Rev. 2003, 29, 297–307. [Google Scholar] [CrossRef]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef]
- Lequeux, A.; Noman, M.Z.; Xiao, M.; Sauvage, D.; Van Moer, K.; Viry, E.; Bocci, I.; Hasmim, M.; Bosseler, M.; Berchem, G.; et al. Impact of hypoxic tumor microenvironment and tumor cell plasticity on the expression of immune checkpoints. Cancer Lett. 2019, 458, 13–20. [Google Scholar] [CrossRef]
- Daniel, S.K.; Sullivan, K.M.; Labadie, K.P.; Pillarisetty, V.G. Hypoxia as a barrier to immunotherapy in pancreatic adenocarcinoma. Clin. Transl. Med. 2019, 8, 10. [Google Scholar] [CrossRef]
- Chouaib, S.; Messai, Y.; Couve, S.; Escudier, B.; Hasmim, M.; Noman, M.Z. Hypoxia promotes tumor growth in linking angiogenesis to immune escape. Front. Immunol. 2012, 3, 21. [Google Scholar] [CrossRef]
- Chouaib, S.; Janji, B.; Tittarelli, A.; Eggermont, A.; Thiery, J.P. Tumor plasticity interferes with anti-tumor immunity. Crit. Rev. Immunol. 2014, 34, 91–102. [Google Scholar] [CrossRef]
- Hasmim, M.; Messai, Y.; Ziani, L.; Thiery, J.; Bouhris, J.H.; Noman, M.Z.; Chouaib, S. Critical Role of Tumor Microenvironment in Shaping NK Cell Functions: Implication of Hypoxic Stress. Front. Immunol. 2015, 6, 482. [Google Scholar] [CrossRef]
- Noman, M.Z.; Hasmim, M.; Messai, Y.; Terry, S.; Kieda, C.; Janji, B.; Chouaib, S. Hypoxia: A key player in antitumor immune response. A Review in the Theme: Cellular Responses to Hypoxia. Am. J. Physiol. Cell Physiol. 2015, 309, C569–C579. [Google Scholar] [CrossRef]
- Fridman, W.H.; Pages, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [Green Version]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Barsoum, I.B.; Smallwood, C.A.; Siemens, D.R.; Graham, C.H. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014, 74, 665–674. [Google Scholar] [CrossRef]
- Messai, Y.; Gad, S.; Noman, M.Z.; Le Teuff, G.; Couve, S.; Janji, B.; Kammerer, S.F.; Rioux-Leclerc, N.; Hasmim, M.; Ferlicot, S.; et al. Renal Cell Carcinoma Programmed Death-ligand 1, a New Direct Target of Hypoxia-inducible Factor-2 Alpha, is Regulated by von Hippel-Lindau Gene Mutation Status. Eur. Urol. 2015. [Google Scholar] [CrossRef]
- Noman, M.Z.; Chouaib, S. Targeting hypoxia at the forefront of anticancer immune responses. Oncoimmunology 2014, 3, e954463. [Google Scholar] [CrossRef] [Green Version]
- Nowak, E.C.; Lines, J.L.; Varn, F.S.; Deng, J.; Sarde, A.; Mabaera, R.; Kuta, A.; Le Mercier, I.; Cheng, C.; Noelle, R.J. Immunoregulatory functions of VISTA. Immunol. Rev. 2017, 276, 66–79. [Google Scholar] [CrossRef]
- Zou, W.; Chen, L. Inhibitory B7-family molecules in the tumour microenvironment. Nat. Rev. Immunol. 2008, 8, 467–477. [Google Scholar] [CrossRef]
- Deng, J.; Li, J.; Sarde, A.; Lines, J.L.; Lee, Y.C.; Qian, D.C.; Pechenick, D.A.; Manivanh, R.; Le Mercier, I.; Lowrey, C.H.; et al. Hypoxia-Induced VISTA Promotes the Suppressive Function of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Cancer Immunol. Res. 2019, 7, 1079–1090. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef]
- Willingham, S.B.; Volkmer, J.P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [Green Version]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D., Jr.; van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gutgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Lu, H.; Xiang, L.; Bullen, J.W.; Zhang, C.; Samanta, D.; Gilkes, D.M.; He, J.; Semenza, G.L. HIF-1 regulates CD47 expression in breast cancer cells to promote evasion of phagocytosis and maintenance of cancer stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, E6215–E6223. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Wang, Y.; Li, Y.-Z. MicroRNA-133a suppresses the proliferation, migration, and invasion of laryngeal carcinoma cells by targeting CD47. Tumor Biol. 2016, 37, 16103–16113. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Liu, X.; Pu, Y.; Cron, K.; Deng, L.; Kline, J.; Frazier, W.A.; Xu, H.; Peng, H.; Fu, Y.X.; Xu, M.M. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat. Med. 2015, 21, 1209–1215. [Google Scholar] [CrossRef]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Jan, M.; Weissman-Tsukamoto, R.; Zhao, F.; Park, C.Y.; Weissman, I.L.; Majeti, R. Therapeutic antibody targeting of CD47 eliminates human acute lymphoblastic leukemia. Cancer Res. 2011, 71, 1374–1384. [Google Scholar] [CrossRef]
- Weiskopf, K.; Jahchan, N.S.; Schnorr, P.J.; Cristea, S.; Ring, A.M.; Maute, R.L.; Volkmer, A.K.; Volkmer, J.P.; Liu, J.; Lim, J.S.; et al. CD47-blocking immunotherapies stimulate macrophage-mediated destruction of small-cell lung cancer. J. Clin. Investig. 2016, 126, 2610–2620. [Google Scholar] [CrossRef]
- Matlung, H.L.; Szilagyi, K.; Barclay, N.A.; van den Berg, T.K. The CD47-SIRPalpha signaling axis as an innate immune checkpoint in cancer. Immunol. Rev. 2017, 276, 145–164. [Google Scholar] [CrossRef]
- Park, S.; Jiang, Z.; Mortenson, E.D.; Deng, L.; Radkevich-Brown, O.; Yang, X.; Sattar, H.; Wang, Y.; Brown, N.K.; Greene, M.; et al. The therapeutic effect of anti-HER2/neu antibody depends on both innate and adaptive immunity. Cancer Cell 2010, 18, 160–170. [Google Scholar] [CrossRef]
- Kumar, V.; Gabrilovich, D.I. Hypoxia-inducible factors in regulation of immune responses in tumour microenvironment. Immunology 2014, 143, 512–519. [Google Scholar] [CrossRef] [Green Version]
- Zhu, E.F.; Gai, S.A.; Opel, C.F.; Kwan, B.H.; Surana, R.; Mihm, M.C.; Kauke, M.J.; Moynihan, K.D.; Angelini, A.; Williams, R.T.; et al. Synergistic innate and adaptive immune response to combination immunotherapy with anti-tumor antigen antibodies and extended serum half-life IL-2. Cancer Cell 2015, 27, 489–501. [Google Scholar] [CrossRef]
- Michaels, A.D.; Newhook, T.E.; Adair, S.J.; Morioka, S.; Goudreau, B.J.; Nagdas, S.; Mullen, M.G.; Persily, J.B.; Bullock, T.N.J.; Slingluff, C.L., Jr.; et al. CD47 Blockade as an Adjuvant Immunotherapy for Resectable Pancreatic Cancer. Clin. Cancer Res. 2018, 24, 1415–1425. [Google Scholar] [CrossRef]
- Soto-Pantoja, D.R.; Terabe, M.; Ghosh, A.; Ridnour, L.A.; DeGraff, W.G.; Wink, D.A.; Berzofsky, J.A.; Roberts, D.D. CD47 in the tumor microenvironment limits cooperation between antitumor T-cell immunity and radiotherapy. Cancer Res. 2014, 74, 6771–6783. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef]
- Mazure, N.M.; Pouyssegur, J. Hypoxia-induced autophagy: Cell death or cell survival? Curr. Opin. Cell Biol. 2010, 22, 177–180. [Google Scholar] [CrossRef]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef]
- Noman, M.Z.; Janji, B.; Kaminska, B.; Van Moer, K.; Pierson, S.; Przanowski, P.; Buart, S.; Berchem, G.; Romero, P.; Mami-Chouaib, F.; et al. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res. 2011, 71, 5976–5986. [Google Scholar] [CrossRef]
- Noman, M.Z.; Janji, B.; Berchem, G.; Mami-Chouaib, F.; Chouaib, S. Hypoxia-induced autophagy: A new player in cancer immunotherapy? Autophagy 2012, 8, 704–706. [Google Scholar] [CrossRef]
- Hasmim, M.; Janji, B.; Khaled, M.; Noman, M.Z.; Louache, F.; Bordereaux, D.; Abderamane, A.; Baud, V.; Mami-Chouaib, F.; Chouaib, S. Cutting Edge: NANOG Activates Autophagy under Hypoxic Stress by Binding to BNIP3L Promoter. J. Immunol. 2017, 198, 1423–1428. [Google Scholar] [CrossRef]
- Hasmim, M.; Noman, M.Z.; Lauriol, J.; Benlalam, H.; Mallavialle, A.; Rosselli, F.; Mami-Chouaib, F.; Alcaide-Loridan, C.; Chouaib, S. Hypoxia-dependent inhibition of tumor cell susceptibility to CTL-mediated lysis involves NANOG induction in target cells. J. Immunol. 2011, 187, 4031–4039. [Google Scholar] [CrossRef]
- Hasmim, M.; Noman, M.Z.; Messai, Y.; Bordereaux, D.; Gros, G.; Baud, V.; Chouaib, S. Cutting edge: Hypoxia-induced Nanog favors the intratumoral infiltration of regulatory T cells and macrophages via direct regulation of TGF-beta1. J. Immunol. 2013, 191, 5802–5806. [Google Scholar] [CrossRef]
- Noman, M.Z.; Buart, S.; Romero, P.; Ketari, S.; Janji, B.; Mari, B.; Mami-Chouaib, F.; Chouaib, S. Hypoxia-inducible miR-210 regulates the susceptibility of tumor cells to lysis by cytotoxic T cells. Cancer Res. 2012, 72, 4629–4641. [Google Scholar] [CrossRef]
- Barsoum, I.B.; Hamilton, T.K.; Li, X.; Cotechini, T.; Miles, E.A.; Siemens, D.R.; Graham, C.H. Hypoxia induces escape from innate immunity in cancer cells via increased expression of ADAM10: Role of nitric oxide. Cancer Res. 2011, 71, 7433–7441. [Google Scholar] [CrossRef]
- Siemens, D.R.; Hu, N.; Sheikhi, A.K.; Chung, E.; Frederiksen, L.J.; Pross, H.; Graham, C.H. Hypoxia increases tumor cell shedding of MHC class I chain-related molecule: Role of nitric oxide. Cancer Res. 2008, 68, 4746–4753. [Google Scholar] [CrossRef]
- Yamada, N.; Yamanegi, K.; Ohyama, H.; Hata, M.; Nakasho, K.; Futani, H.; Okamura, H.; Terada, N. Hypoxia downregulates the expression of cell surface MICA without increasing soluble MICA in osteosarcoma cells in a HIF-1alpha-dependent manner. Int. J. Oncol. 2012, 41, 2005–2012. [Google Scholar] [CrossRef]
- Baginska, J.; Viry, E.; Berchem, G.; Poli, A.; Noman, M.Z.; van Moer, K.; Medves, S.; Zimmer, J.; Oudin, A.; Niclou, S.P.; et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc. Natl. Acad. Sci. USA 2013, 110, 17450–17455. [Google Scholar] [CrossRef] [Green Version]
- Viry, E.; Baginska, J.; Berchem, G.; Noman, M.Z.; Medves, S.; Chouaib, S.; Janji, B. Autophagic degradation of GZMB/granzyme B: A new mechanism of hypoxic tumor cell escape from natural killer cell-mediated lysis. Autophagy 2014, 10, 173–175. [Google Scholar] [CrossRef]
- Andersson, E.; Poschke, I.; Villabona, L.; Carlson, J.W.; Lundqvist, A.; Kiessling, R.; Seliger, B.; Masucci, G.V. Non-classical HLA-class I expression in serous ovarian carcinoma: Correlation with the HLA-genotype, tumor infiltrating immune cells and prognosis. Oncoimmunology 2016, 5, e1052213. [Google Scholar] [CrossRef]
- Mouillot, G.; Marcou, C.; Zidi, I.; Guillard, C.; Sangrouber, D.; Carosella, E.D.; Moreau, P. Hypoxia modulates HLA-G gene expression in tumor cells. Hum. Immunol. 2007, 68, 277–285. [Google Scholar] [CrossRef]
- Kren, L.; Slaby, O.; Muckova, K.; Lzicarova, E.; Sova, M.; Vybihal, V.; Svoboda, T.; Fadrus, P.; Lakomy, R.; Vanhara, P.; et al. Expression of immune-modulatory molecules HLA-G and HLA-E by tumor cells in glioblastomas: An unexpected prognostic significance? Neuropathology 2011, 31, 129–134. [Google Scholar] [CrossRef]
- Yan, W.H. HLA-G expression in cancers: Potential role in diagnosis, prognosis and therapy. Endocr. Metab. Immune Disord. Drug Targets 2011, 11, 76–89. [Google Scholar] [CrossRef]
- Carosella, E.D.; Favier, B.; Rouas-Freiss, N.; Moreau, P.; Lemaoult, J. Beyond the increasing complexity of the immunomodulatory HLA-G molecule. Blood 2008, 111, 4862–4870. [Google Scholar] [CrossRef]
- Amodio, G.; Sales de Albuquerque, R.; Gregori, S. New insights into HLA-G mediated tolerance. Tissue Antigens 2014, 84, 255–263. [Google Scholar] [CrossRef]
- Carosella, E.D.; Rouas-Freiss, N.; Tronik-Le Roux, D.; Moreau, P.; LeMaoult, J. HLA-G: An Immune Checkpoint Molecule. Adv. Immunol. 2015, 127, 33–144. [Google Scholar] [CrossRef]
- Curigliano, G.; Criscitiello, C.; Gelao, L.; Goldhirsch, A. Molecular pathways: Human leukocyte antigen G (HLA-G). Clin. Cancer Res. 2013, 19, 5564–5571. [Google Scholar] [CrossRef]
- Yaghi, L.; Poras, I.; Simoes, R.T.; Donadi, E.A.; Tost, J.; Daunay, A.; de Almeida, B.S.; Carosella, E.D.; Moreau, P. Hypoxia inducible factor-1 mediates the expression of the immune checkpoint HLA-G in glioma cells through hypoxia response element located in exon 2. Oncotarget 2016, 7, 63690–63707. [Google Scholar] [CrossRef]
- Garziera, M.; Scarabel, L.; Toffoli, G. Hypoxic Modulation of HLA-G Expression through the Metabolic Sensor HIF-1 in Human Cancer Cells. J. Immunol. Res. 2017, 2017, 4587520. [Google Scholar] [CrossRef]
- Baran, N.; Konopleva, M. Molecular Pathways: Hypoxia-Activated Prodrugs in Cancer Therapy. Clin. Cancer Res. 2017, 23, 2382–2390. [Google Scholar] [CrossRef]
- Borad, M.J.; Reddy, S.G.; Bahary, N.; Uronis, H.E.; Sigal, D.; Cohn, A.L.; Schelman, W.R.; Stephenson, J., Jr.; Chiorean, E.G.; Rosen, P.J.; et al. Randomized Phase II Trial of Gemcitabine Plus TH-302 Versus Gemcitabine in Patients With Advanced Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 1475–1481. [Google Scholar] [CrossRef] [Green Version]
- Chawla, S.P.; Cranmer, L.D.; Van Tine, B.A.; Reed, D.R.; Okuno, S.H.; Butrynski, J.E.; Adkins, D.R.; Hendifar, A.E.; Kroll, S.; Ganjoo, K.N. Phase II study of the safety and antitumor activity of the hypoxia-activated prodrug TH-302 in combination with doxorubicin in patients with advanced soft tissue sarcoma. J. Clin. Oncol. 2014, 32, 3299–3306. [Google Scholar] [CrossRef]
- Jayaprakash, P.; Ai, M.; Liu, A.; Budhani, P.; Bartkowiak, T.; Sheng, J.; Ager, C.; Nicholas, C.; Jaiswal, A.R.; Sun, Y.; et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J. Clin. Investig. 2018, 128, 5137–5149. [Google Scholar] [CrossRef]
- Onnis, B.; Rapisarda, A.; Melillo, G. Development of HIF-1 inhibitors for cancer therapy. J. Cell. Mol. Med. 2009, 13, 2780–2786. [Google Scholar] [CrossRef]
- Wigerup, C.; Pahlman, S.; Bexell, D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol. Ther. 2016, 164, 152–169. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Patel, S.P.; Roszik, J.; Qin, Y. Hypoxia-Driven Immunosuppressive Metabolites in the Tumor Microenvironment: New Approaches for Combinational Immunotherapy. Front. Immunol. 2018, 9, 1591. [Google Scholar] [CrossRef] [Green Version]
- Greenberger, L.M.; Horak, I.D.; Filpula, D.; Sapra, P.; Westergaard, M.; Frydenlund, H.F.; Albaek, C.; Schroder, H.; Orum, H. A RNA antagonist of hypoxia-inducible factor-1alpha, EZN-2968, inhibits tumor cell growth. Mol. Cancer Ther. 2008, 7, 3598–3608. [Google Scholar] [CrossRef]
- Terzuoli, E.; Puppo, M.; Rapisarda, A.; Uranchimeg, B.; Cao, L.; Burger, A.M.; Ziche, M.; Melillo, G. Aminoflavone, a ligand of the aryl hydrocarbon receptor, inhibits HIF-1alpha expression in an AhR-independent fashion. Cancer Res. 2010, 70, 6837–6848. [Google Scholar] [CrossRef]
- Jones, D.T.; Pugh, C.W.; Wigfield, S.; Stevens, M.F.; Harris, A.L. Novel thioredoxin inhibitors paradoxically increase hypoxia-inducible factor-alpha expression but decrease functional transcriptional activity, DNA binding, and degradation. Clin. Cancer Res. 2006, 12, 5384–5394. [Google Scholar] [CrossRef]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol. Cell. Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef]
- Thomas, G.V.; Tran, C.; Mellinghoff, I.K.; Welsbie, D.S.; Chan, E.; Fueger, B.; Czernin, J.; Sawyers, C.L. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat. Med. 2006, 12, 122–127. [Google Scholar] [CrossRef]
- Majumder, P.K.; Febbo, P.G.; Bikoff, R.; Berger, R.; Xue, Q.; McMahon, L.M.; Manola, J.; Brugarolas, J.; McDonnell, T.J.; Golub, T.R.; et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat. Med. 2004, 10, 594–601. [Google Scholar] [CrossRef]
- Rapisarda, A.; Uranchimeg, B.; Scudiero, D.A.; Selby, M.; Sausville, E.A.; Shoemaker, R.H.; Melillo, G. Identification of small molecule inhibitors of hypoxia-inducible factor 1 transcriptional activation pathway. Cancer Res. 2002, 62, 4316–4324. [Google Scholar]
- Rapisarda, A.; Zalek, J.; Hollingshead, M.; Braunschweig, T.; Uranchimeg, B.; Bonomi, C.A.; Borgel, S.D.; Carter, J.P.; Hewitt, S.M.; Shoemaker, R.H.; et al. Schedule-dependent inhibition of hypoxia-inducible factor-1alpha protein accumulation, angiogenesis, and tumor growth by topotecan in U251-HRE glioblastoma xenografts. Cancer Res. 2004, 64, 6845–6848. [Google Scholar] [CrossRef]
- Mabjeesh, N.J.; Escuin, D.; LaVallee, T.M.; Pribluda, V.S.; Swartz, G.M.; Johnson, M.S.; Willard, M.T.; Zhong, H.; Simons, J.W.; Giannakakou, P. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell 2003, 3, 363–375. [Google Scholar] [CrossRef] [Green Version]
- Bohonowych, J.E.; Peng, S.; Gopal, U.; Hance, M.W.; Wing, S.B.; Argraves, K.M.; Lundgren, K.; Isaacs, J.S. Comparative analysis of novel and conventional Hsp90 inhibitors on HIF activity and angiogenic potential in clear cell renal cell carcinoma: Implications for clinical evaluation. BMC Cancer 2011, 11, 520. [Google Scholar] [CrossRef]
- Hutt, D.M.; Roth, D.M.; Vignaud, H.; Cullin, C.; Bouchecareilh, M. The histone deacetylase inhibitor, Vorinostat, represses hypoxia inducible factor 1 alpha expression through translational inhibition. PLoS ONE 2014, 9, e106224. [Google Scholar] [CrossRef]
- Koh, M.Y.; Spivak-Kroizman, T.; Venturini, S.; Welsh, S.; Williams, R.R.; Kirkpatrick, D.L.; Powis, G. Molecular mechanisms for the activity of PX-478, an antitumor inhibitor of the hypoxia-inducible factor-1alpha. Mol. Cancer Ther. 2008, 7, 90–100. [Google Scholar] [CrossRef]
- Moreno-Manzano, V.; Rodriguez-Jimenez, F.J.; Acena-Bonilla, J.L.; Fustero-Lardies, S.; Erceg, S.; Dopazo, J.; Montaner, D.; Stojkovic, M.; Sanchez-Puelles, J.M. FM19G11, a new hypoxia-inducible factor (HIF) modulator, affects stem cell differentiation status. J. Biol. Chem. 2010, 285, 1333–1342. [Google Scholar] [CrossRef]
- Lee, K.; Zhang, H.; Qian, D.Z.; Rey, S.; Liu, J.O.; Semenza, G.L. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc. Natl. Acad. Sci. USA 2009, 106, 17910–17915. [Google Scholar] [CrossRef] [Green Version]
- Cook, K.M.; Hilton, S.T.; Mecinovic, J.; Motherwell, W.B.; Figg, W.D.; Schofield, C.J. Epidithiodiketopiperazines block the interaction between hypoxia-inducible factor-1alpha (HIF-1alpha) and p300 by a zinc ejection mechanism. J. Biol. Chem. 2009, 284, 26831–26838. [Google Scholar] [CrossRef]
- Chan, M.C.; Holt-Martyn, J.P.; Schofield, C.J.; Ratcliffe, P.J. Pharmacological targeting of the HIF hydroxylases--A new field in medicine development. Mol. Asp. Med. 2016, 47–48, 54–75. [Google Scholar] [CrossRef]
- Haase, V.H. Therapeutic targeting of the HIF oxygen-sensing pathway: Lessons learned from clinical studies. Exp. Cell Res. 2017, 356, 160–165. [Google Scholar] [CrossRef]
- Yu, T.; Tang, B.; Sun, X. Development of Inhibitors Targeting Hypoxia-Inducible Factor 1 and 2 for Cancer Therapy. Yonsei Med. J. 2017, 58, 489–496. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noman, M.Z.; Hasmim, M.; Lequeux, A.; Xiao, M.; Duhem, C.; Chouaib, S.; Berchem, G.; Janji, B. Improving Cancer Immunotherapy by Targeting the Hypoxic Tumor Microenvironment: New Opportunities and Challenges. Cells 2019, 8, 1083. https://doi.org/10.3390/cells8091083
Noman MZ, Hasmim M, Lequeux A, Xiao M, Duhem C, Chouaib S, Berchem G, Janji B. Improving Cancer Immunotherapy by Targeting the Hypoxic Tumor Microenvironment: New Opportunities and Challenges. Cells. 2019; 8(9):1083. https://doi.org/10.3390/cells8091083
Chicago/Turabian StyleNoman, Muhammad Zaeem, Meriem Hasmim, Audrey Lequeux, Malina Xiao, Caroline Duhem, Salem Chouaib, Guy Berchem, and Bassam Janji. 2019. "Improving Cancer Immunotherapy by Targeting the Hypoxic Tumor Microenvironment: New Opportunities and Challenges" Cells 8, no. 9: 1083. https://doi.org/10.3390/cells8091083
APA StyleNoman, M. Z., Hasmim, M., Lequeux, A., Xiao, M., Duhem, C., Chouaib, S., Berchem, G., & Janji, B. (2019). Improving Cancer Immunotherapy by Targeting the Hypoxic Tumor Microenvironment: New Opportunities and Challenges. Cells, 8(9), 1083. https://doi.org/10.3390/cells8091083