Systematic Identification of Housekeeping Genes Possibly Used as References in Caenorhabditis elegans by Large-Scale Data Integration


Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection and Filtration
2.2. Methodology
2.2.1. Normalization of Microarray Data
2.2.2. First-Round Ranking of Gene Expression Stability
2.2.3. Second-Round Ranking of Gene Expression Stability
2.2.4. Identification of HKG Candidates Used as Reference Genes
2.3. Functional Annotation and Enrichment Analysis
2.4. Evaluation of HKG Candidates
2.4.1. Evaluation of HKG Candidates Using Newly Published Datasets
2.4.2. Comparison of Newly Identified HKG Candidates and Commonly Used Reference Genes
3. Results
3.1. Large-Scale Microarray Datasets Collection
3.2. Data Normalization and Gene Probe Matching
3.3. First-Round Ranking Based on Gene Expression Stability
3.4. Second-Round Ranking Using RankAggreg
3.5. Overlapping for Determining the HKG Candidates Used as Reference Genes
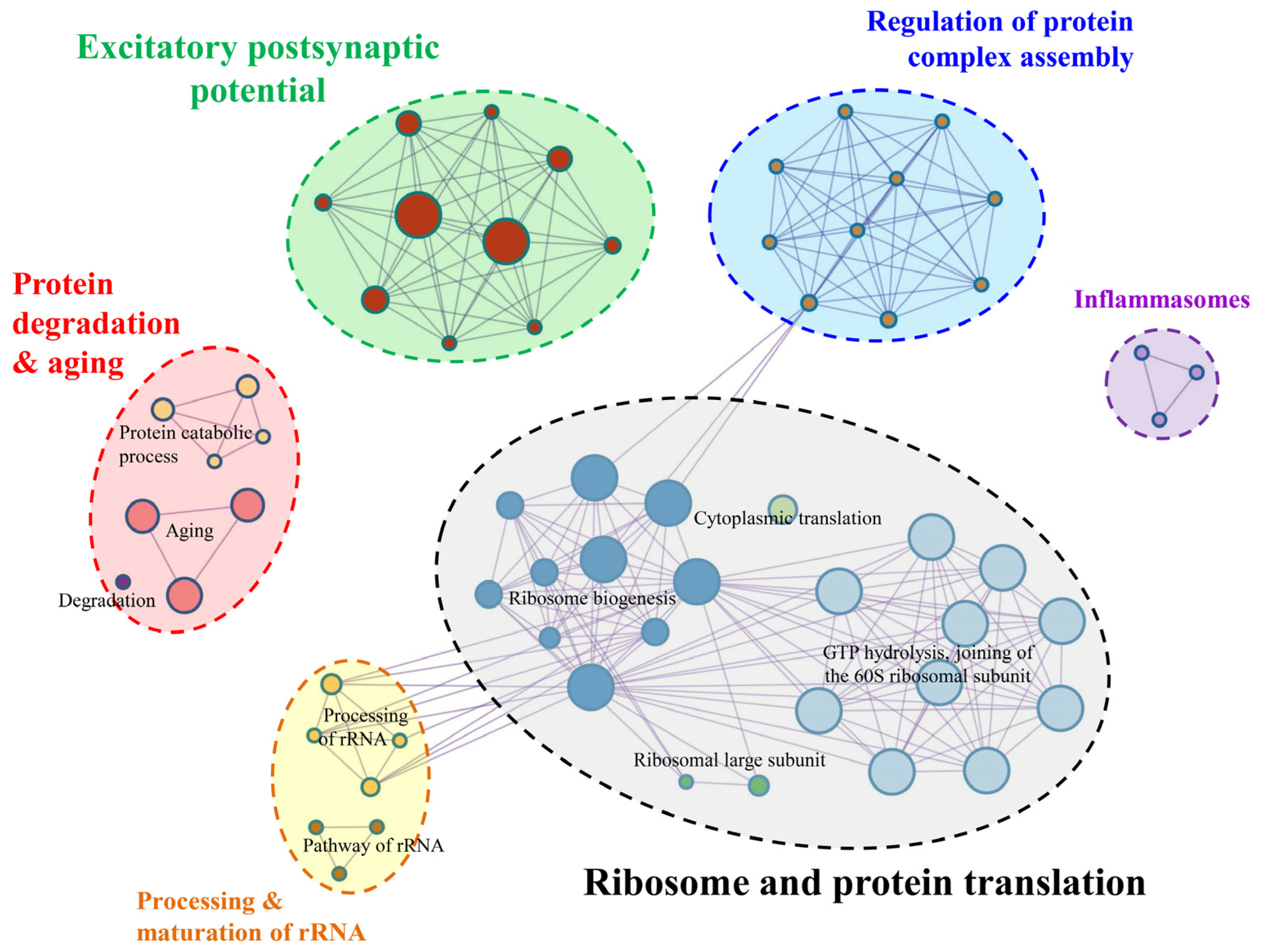
3.6. Gene Annotation and Enrichment
3.7. Validation of HKG Candidates Used as Reference Genes
3.8. Comparison between HKG Candidates Identified in This Study and Commonly Used Reference Gene
4. Discussion
4.1. Data Integration and Its Advantages
4.2. Why Microarray Datasets Were Adopted?
4.3. Importance of Normalization Methods
4.4. Reliability of Newly Identified HKG Candidates Possibly Used as Reference Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Data Availability
References
- Jameson, P.E. Selection of reference genes for flowering pathway analysis in the masting plants, Celmisia lyallii and Chionochloa pallens, under variable environmental conditions. Sci. Rep. 2019, 9, 1–16. [Google Scholar]
- Czechowski, T.; Bari, R.P.; Stitt, M.; Scheible, W.R.; Udvardi, M.K. Real-time RT-PCR profiling of over 1400 Arabidopsis transcription factors: Unprecedented sensitivity reveals novel root-and shoot-specific genes. Plant J. 2004, 38, 366–379. [Google Scholar] [CrossRef]
- Nolan, T.; Hands, R.E.; Bustin, S.A. Quantification of mRNA using real-time RT-PCR. Nat. Protoc. 2006, 1, 1559. [Google Scholar] [CrossRef]
- Derveaux, S.; Vandesompele, J.; Hellemans, J. How to do successful gene expression analysis using real-time PCR. Methods 2010, 50, 227–230. [Google Scholar] [CrossRef]
- Bustin, S.A.; Nolan, T. Pitfalls of quantitative real-time reverse-transcription polymerase chain reaction. J. Biomol. Tech. JBT 2004, 15, 155. [Google Scholar]
- Huggett, J.; Dheda, K.; Bustin, S.; Zumla, A. Real-time RT-PCR normalisation; strategies and considerations. Genes Immun. 2005, 6, 279. [Google Scholar] [CrossRef] [Green Version]
- Su, L.; Li, H.; Huang, C.; Zhao, T.; Zhang, Y.; Ba, X.; Li, Z.; Zhang, Y.; Huang, B.; Lu, J. Muscle-specific histone H3K36 dimethyltransferase SET-18 shortens lifespan of Caenorhabditis elegans by repressing daf-16a expression. Cell Rep. 2018, 22, 2716–2729. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Schroeder, E.A.; Silva-García, C.G.; Hebestreit, K.; Mair, W.B.; Brunet, A. Mono-unsaturated fatty acids link H3K4me3 modifiers to C. elegans lifespan. Nature 2017, 544, 185. [Google Scholar] [CrossRef] [PubMed]
- Rajan, M.; Anderson, C.P.; Rindler, P.M.; Romney, S.J.; dos Santos, M.C.F.; Gertz, J.; Leibold, E.A. NHR-14 loss of function couples intestinal iron uptake with innate immunity in C. elegans through PQM-1 signaling. eLife 2019, 8. [Google Scholar] [CrossRef]
- Almeida, M.V.; Dietz, S.; Redl, S.; Karaulanov, E.; Hildebrandt, A.; Renz, C.; Ulrich, H.D.; König, J.; Butter, F.; Ketting, R.F. GTSF-1 is required for formation of a functional RNA-dependent RNA Polymerase complex in Caenorhabditis elegans. EMBO J. 2018, 37, e99325. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Perentis, R.J.; Caldwell, G.A.; Caldwell, K.A. Gene-by-environment interactions that disrupt mitochondrial homeostasis cause neurodegeneration in C. elegans Parkinson’s models. Cell Death Dis. 2018, 9, 555. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, J.; Pinter, K.; Samuel, T.; Beardsley, S.; Yuan, X.; Zhang, J.; Meng, K.; Yun, S.; Krause, M.; Hamza, I. Inter-organ signalling by HRG-7 promotes systemic haem homeostasis. Nat. Cell Biol. 2017, 19, 799. [Google Scholar] [CrossRef] [PubMed]
- Bilal, B.; Azim, M.K. Nematicidal activity of ‘major royal jelly protein’-containing glycoproteins from Acacia honey. Exp. Parasitol. 2018, 192, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Kurshan, P.T.; Merrill, S.A.; Dong, Y.; Ding, C.; Hammarlund, M.; Bai, J.; Jorgensen, E.M.; Shen, K. γ-neurexin and frizzled mediate parallel synapse assembly pathways antagonized by receptor endocytosis. Neuron 2018, 100, 150–166.e4. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; Janssens, G.E.; McIntyre, R.L.; Molenaars, M.; Kamble, R.; Gao, A.W.; Jongejan, A.; van Weeghel, M.; MacInnes, A.W.; Houtkooper, R.H. Glycine promotes longevity in Caenorhabditis elegans in a methionine cycle-dependent fashion. PLoS Genet. 2019, 15, e1007633. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Wang, Y.; Fu, J.; Zhou, Y.; Luo, Y.; Zhang, Y.; Li, B.; Yang, Q.; Xue, W.; Lou, Y. A critical assessment of the feature selection methods used for biomarker discovery in current metaproteomics studies. Brief. Bioinform. 2019, bbz061. [Google Scholar] [CrossRef]
- Köhnlein, K.; Urban, N.; Guerrero-Gómez, D.; Steinbrenner, H.; Urbánek, P.; Priebs, J.; Koch, P.; Kaether, C.; Miranda-Vizuete, A.; Klotz, L.-O. A Caenorhabditis elegans ortholog of human selenium-binding protein 1 is a pro-aging factor protecting against selenite toxicity. Redox Biol. 2020, 28, 101323. [Google Scholar] [CrossRef]
- Kumsta, C.; Chang, J.T.; Schmalz, J.; Hansen, M. Hormetic heat stress and HSF-1 induce autophagy to improve survival and proteostasis in C. elegans. Nat. Commun. 2017, 8, 14337. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Huang, X.-B.; Wan, Q.-L.; Ding, A.-J.; Yang, Z.-L.; Qiu, M.-H.; Sun, H.-Y.; Qi, S.-H.; Luo, H.-R. Otophylloside B protects against Aβ toxicity in Caenorhabditis elegans models of Alzheimer’s Disease. Nat. Prod. Bioprospect. 2017, 7, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Klosin, A.; Casas, E.; Hidalgo-Carcedo, C.; Vavouri, T.; Lehner, B. Transgenerational transmission of environmental information in C. elegans. Science 2017, 356, 320–323. [Google Scholar] [CrossRef] [Green Version]
- Osman, G.A.; Fasseas, M.K.; Koneru, S.L.; Essmann, C.L.; Kyrou, K.; Srinivasan, M.A.; Zhang, G.; Sarkies, P.; Félix, M.-A.; Barkoulas, M. Natural infection of C. elegans by an oomycete reveals a new pathogen-specific immune response. Curr. Biol. 2018, 28, 640–648.e5. [Google Scholar] [CrossRef] [Green Version]
- Thellin, O.; Zorzi, W.; Lakaye, B.; De Borman, B.; Coumans, B.; Hennen, G.; Grisar, T.; Igout, A.; Heinen, E. Housekeeping genes as internal standards: Use and limits. J. Biotechnol. 1999, 75, 291–295. [Google Scholar] [CrossRef] [Green Version]
- Hoogewijs, D.; Houthoofd, K.; Matthijssens, F.; Vandesompele, J.; Vanfleteren, J.R. Selection and validation of a set of reliable reference genes for quantitative sod gene expression analysis in C. elegans. BMC Mol. Biol. 2008, 9, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacRae, T.; Sargeant, T.; Lemieux, S.; Hébert, J.; Deneault, É.; Sauvageau, G. RNA-Seq reveals spliceosome and proteasome genes as most consistent transcripts in human cancer cells. PLoS ONE 2013, 8, e72884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, P.K.; Mafra, V.; De Souza, W.R.; Ribeiro, A.P.; Vinecky, F.; Basso, M.F.; Da Cunha, B.A.D.B.; Kobayashi, A.K.; Molinari, H.B.C. Selection of reliable reference genes for RT-qPCR analysis during developmental stages and abiotic stress in Setaria viridis. Sci. Rep. 2016, 6, 28348. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Cong, P.; Tian, Y.; Zhu, Y. Using RNA-seq data to select reference genes for normalizing gene expression in apple roots. PLoS ONE 2017, 12, e0185288. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, L.; Li, R.; Zhang, M.; Li, Y.; Wang, H.; Wang, S.; Bao, Z. Systematic identification and validation of the reference genes from 60 RNA-Seq libraries in the scallop Mizuhopecten yessoensis. BMC Genom. 2019, 20, 288. [Google Scholar] [CrossRef]
- Smitha, P.; Vishnupriyan, K.; Kar, A.S.; Kumar, M.A.; Bathula, C.; Chandrashekara, K.; Dhar, S.K.; Das, M. Genome wide search to identify reference genes candidates for gene expression analysis in Gossypium hirsutum. BMC Plant Biol. 2019, 19, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zhan, A.; Huang, X.; Li, S. Genome-wide identification and evaluation of new reference genes for gene expression analysis under temperature and salinity stresses in Ciona savignyi. Front. Genet. 2019, 10, 71. [Google Scholar]
- Li, S.; Wang, W.; Li, X.; Fan, K.; Yang, K. Genome-wide identification and characterization of reference genes with different transcript abundances for Streptomyces coelicolor. Sci. Rep. 2015, 5, 15840. [Google Scholar] [CrossRef] [Green Version]
- Czechowski, T.; Stitt, M.; Altmann, T.; Udvardi, M.K.; Scheible, W.-R. Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol. 2005, 139, 5–17. [Google Scholar] [CrossRef] [Green Version]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034.1. [Google Scholar]
- Andersen, C.L.; Jensen, J.L.; Ørntoft, T.F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C T method. Nat. Protoc. 2008, 3, 1101. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, B.S.; Irizarry, R.A. A framework for oligonucleotide microarray preprocessing. Bioinformatics 2010, 26, 2363–2367. [Google Scholar] [CrossRef] [PubMed]
- Pepper, S.D.; Saunders, E.K.; Edwards, L.E.; Wilson, C.L.; Miller, C.J. The utility of MAS5 expression summary and detection call algorithms. BMC Bioinform. 2007, 8, 273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Wong, W.H. Model-based analysis of oligonucleotide arrays: Expression index computation and outlier detection. Proc. Natl. Acad. Sci. USA 2001, 98, 31–36. [Google Scholar] [CrossRef]
- Wu, Z.; Irizarry, R.A. Preprocessing of oligonucleotide array data. Nat. Biotechnol. 2004, 22, 656. [Google Scholar] [CrossRef]
- Shippy, R.; Fulmer-Smentek, S.; Jensen, R.V.; Jones, W.D.; Wolber, P.K.; Johnson, C.D.; Pine, P.S.; Boysen, C.; Guo, X.; Chudin, E. Using RNA sample titrations to assess microarray platform performance and normalization techniques. Nat. Biotechnol. 2006, 24, 1123. [Google Scholar] [CrossRef] [Green Version]
- Huber, W.; Von Heydebreck, A.; Sültmann, H.; Poustka, A.; Vingron, M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 2002, 18, S96–S104. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [Green Version]
- Pihur, V.; Datta, S.; Datta, S. RankAggreg, an R package for weighted rank aggregation. BMC Bioinform. 2009, 10, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Cline, M.S.; Smoot, M.; Cerami, E.; Kuchinsky, A.; Landys, N.; Workman, C.; Christmas, R.; Avila-Campilo, I.; Creech, M.; Gross, B. Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc. 2007, 2, 2366. [Google Scholar] [CrossRef] [Green Version]
- Kryuchkova-Mostacci, N.; Robinson-Rechavi, M. A benchmark of gene expression tissue-specificity metrics. Brief. Bioinform. 2017, 18, 205–214. [Google Scholar] [CrossRef]
- O’Hagan, S.; Muelas, M.W.; Day, P.J.; Lundberg, E.; Kell, D.B. GeneGini: Assessment via the Gini coefficient of reference “housekeeping” genes and diverse human transporter expression profiles. Cell Syst. 2018, 6, 230–244.e1. [Google Scholar] [CrossRef] [Green Version]
- Büttner, M.; Miao, Z.; Wolf, F.A.; Teichmann, S.A.; Theis, F.J. A test metric for assessing single-cell RNA-seq batch correction. Nat. Methods 2019, 16, 43. [Google Scholar] [CrossRef] [Green Version]
- Hartman, J.H.; Richie, C.T.; Gordon, K.L.; Mello, D.F.; Castillo, P.; Zhu, A.; Wang, Y.; Hoffer, B.J.; Sherwood, D.R.; Meyer, J.N.; et al. MANF deletion abrogates early larval Caenorhabditis elegans stress response to tunicamycin and Pseudomonas aeruginosa. Eur. J. Cell Biol. 2019, 98. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Lautrup, S.; Jensen, M.B.; Yang, B.; SenGupta, T.; Caponio, D.; Khezri, R.; Demarest, T.G.; Aman, Y.; et al. NAD(+) augmentation restores mitophagy and limits accelerated aging in Werner syndrome. Nat. Commun. 2019, 10, 5284. [Google Scholar] [CrossRef] [Green Version]
- Fang, E.F.; Kassahun, H.; Croteau, D.L.; Scheibye-Knudsen, M.; Marosi, K.; Lu, H.; Shamanna, R.A.; Kalyanasundaram, S.; Bollineni, R.C.; Wilson, M.A.; et al. NAD(+) Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab. 2016, 24, 566–581. [Google Scholar] [CrossRef] [Green Version]
- Rangaraju, S.; Solis, G.M.; Thompson, R.C.; Gomez-Amaro, R.L.; Kurian, L.; Encalada, S.E.; Niculescu, A.B., 3rd; Salomon, D.R.; Petrascheck, M. Suppression of transcriptional drift extends C. elegans lifespan by postponing the onset of mortality. eLife 2015, 4, e08833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, M.; Anavy, L.; Cole, A.G.; Winter, E.; Mostov, N.; Khair, S.; Senderovich, N.; Kovalev, E.; Silver, D.H.; Feder, M.; et al. The mid-developmental transition and the evolution of animal body plans. Nature 2016, 531, 637–641. [Google Scholar] [CrossRef]
- Harvald, E.B.; Sprenger, R.R.; Dall, K.B.; Ejsing, C.S.; Nielsen, R.; Mandrup, S.; Murillo, A.B.; Larance, M.; Gartner, A.; Lamond, A.I. Multi-omics Analyses of Starvation Responses Reveal a Central Role for Lipoprotein Metabolism in Acute Starvation Survival in C. elegans. Cell Syst. 2017, 5, 38–52.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Snyder, M.P. Integrative omics for health and disease. Nat. Rev. Genet. 2018, 19, 299. [Google Scholar] [CrossRef] [PubMed]
- Haghverdi, L.; Lun, A.T.; Morgan, M.D.; Marioni, J.C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 2018, 36, 421. [Google Scholar] [CrossRef] [PubMed]
- Goh, W.W.B.; Wang, W.; Wong, L. Why batch effects matter in omics data, and how to avoid them. Trends Biotechnol. 2017, 35, 498–507. [Google Scholar] [CrossRef]
- Panagiotou, O.A.; Willer, C.J.; Hirschhorn, J.N.; Ioannidis, J.P. The power of meta-analysis in genome-wide association studies. Ann. Rev. Genom. Hum. Genet. 2013, 14, 441–465. [Google Scholar] [CrossRef] [Green Version]
- Dapas, M.; Kandpal, M.; Bi, Y.; Davuluri, R.V. Comparative evaluation of isoform-level gene expression estimation algorithms for RNA-seq and exon-array platforms. Brief. Bioinform. 2016, 18, 260–269. [Google Scholar] [CrossRef] [Green Version]
- Romero, J.P.; Ortiz-Estévez, M.; Muniategui, A.; Carrancio, S.; de Miguel, F.J.; Carazo, F.; Montuenga, L.M.; Loos, R.; Pío, R.; Trotter, M.W. Comparison of RNA-seq and microarray platforms for splice event detection using a cross-platform algorithm. BMC Genom. 2018, 19, 703. [Google Scholar] [CrossRef] [Green Version]
- Mantione, K.J.; Kream, R.M.; Kuzelova, H.; Ptacek, R.; Raboch, J.; Samuel, J.M.; Stefano, G.B. Comparing bioinformatic gene expression profiling methods: Microarray and RNA-Seq. Med. Sci. Monit. Basic Res. 2014, 20, 138. [Google Scholar] [PubMed] [Green Version]
- Meng, Q.; Catchpoole, D.; Skillicorn, D.; Kennedy, P.J. DBNorm: Normalizing high-density oligonucleotide microarray data based on distributions. BMC Bioinform. 2017, 18, 527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Tang, J.; Yang, Q.; Li, S.; Cui, X.; Li, Y.; Chen, Y.; Xue, W.; Li, X.; Zhu, F. NOREVA: Normalization and evaluation of MS-based metabolomics data. Nucleic Acids Res. 2017, 45, W162–W170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Tang, J.; Yang, Q.; Cui, X.; Li, S.; Chen, S.; Cao, Q.; Xue, W.; Chen, N.; Zhu, F. Performance evaluation and online realization of data-driven normalization methods used in LC/MS based untargeted metabolomics analysis. Sci. Rep. 2016, 6, 38881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hicks, S.C.; Irizarry, R.A. quantro: A data-driven approach to guide the choice of an appropriate normalization method. Genome Biol. 2015, 16, 117. [Google Scholar] [CrossRef] [Green Version]
- Casadei, R.; Pelleri, M.C.; Vitale, L.; Facchin, F.; Lenzi, L.; Canaider, S.; Strippoli, P.; Frabetti, F. Identification of housekeeping genes suitable for gene expression analysis in the zebrafish. Gene Expr. Patterns 2011, 11, 271–276. [Google Scholar] [CrossRef]
- Simsek, D.; Barna, M. An emerging role for the ribosome as a nexus for post-translational modifications. Curr. Opin. Cell Biol. 2017, 45, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, A.; Kenmochi, N. Ribosomes and Ribosomal Proteins: More Than Just ‘Housekeeping’. In e LS; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2001. [Google Scholar] [CrossRef]
- Smagin, D.A.; Kovalenko, I.L.; Galyamina, A.G.; Orlov, Y.L.; Babenko, V.N.; Kudryavtseva, N.N. Heterogeneity of brain ribosomal genes expression following positive fighting experience in male mice as revealed by RNA-Seq. Mol. Neurobiol. 2018, 55, 390–401. [Google Scholar] [CrossRef]
- Eisenberg, E.; Levanon, E.Y. Human housekeeping genes, revisited. Trends Genet. 2013, 29, 569–574. [Google Scholar] [CrossRef]
- Choudhary, R.; Kumar, S.; Singh, S.V.; Sharma, A.K.; Goud, T.S.; Srivastava, A.K.; Kumar, A.; Mohanty, A.K.; Upadhyay, R.C. Validation of putative reference genes for gene expression studies in heat stressed and α-MSH treated melanocyte cells of Bos indicus using real-time quantitative PCR. Mol. Cell. Probes 2016, 30, 161–167. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Y.Q.; Jin, G.; Wu, S.; Cui, J.; Wang, R.F. Selection of reference genes for gene expression studies in human bladder cancer using SYBR-Green quantitative polymerase chain reaction. Oncol. Lett. 2017, 14, 6001–6011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.; Han, S.; Han, H.; Zhao, F.; He, Y. Selection of reference genes for Harmonia axyridis (Coleoptera: Coccinellidae) feeding on different diets. J. Asia-Pac. Entomol. 2019, 22, 1115–1122. [Google Scholar] [CrossRef]
- Mhatre, K.N.; Wakula, P.; Klein, O.; Bisping, E.; Völkl, J.; Pieske, B.; Heinzel, F.R. Crosstalk between FGF23-and angiotensin II-mediated Ca 2+ signaling in pathological cardiac hypertrophy. Cell. Mol. Life Sci. 2018, 75, 4403–4416. [Google Scholar] [CrossRef] [PubMed]
- Kfir, S.; Basavaraja, R.; Wigoda, N.; Ben-Dor, S.; Orr, I.; Meidan, R. Genomic profiling of bovine corpus luteum maturation. PLoS ONE 2018, 13, e0194456. [Google Scholar] [CrossRef] [PubMed]
- Gruzdev, E.V.; Mardanov, A.V.; Beletsky, A.V.; Kochieva, E.Z.; Ravin, N.V.; Skryabin, K.G. The complete chloroplast genome of parasitic flowering plant Monotropa hypopitys: Extensive gene losses and size reduction. Mitochondrial DNA Part B 2016, 1, 212–213. [Google Scholar] [CrossRef] [Green Version]
- Mallet, S.; Weiss, S.; Jacques, N.; Leh-Louis, V.; Sacerdot, C.; Casaregola, S. Insights into the life cycle of yeasts from the CTG clade revealed by the analysis of the Millerozyma (Pichia) farinosa species complex. PLoS ONE 2012, 7, e35842. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Entrez Gene ID | Description | Chromosome Location | Size (bp) |
|---|---|---|---|---|
| rps-23 | 178188 | 40S ribosomal protein S23 | Chr. IV: 12390264–12391396 | 550 |
| rps-27 | 178538 | 40S ribosomal protein S27 | Chr. V: 103394–104064 | 356 |
| rps-16 | 179998 | 40S ribosomal protein S16 | Chr. V: 15000011–15000594 | 533 |
| rps-26 | 173342 | 40S ribosomal protein S26 | Chr. I: 14759918–14760654 | 440 |
| rps-4 | 177481 | 40S ribosomal protein S4 | Chr. IV: 7083694–7084682 | 849 |
| rps-2 | 177583 | 40S ribosomal protein S2 | Chr. IV: 7925298–7926391 | 998 |
| rps-17 | 172313 | 40S ribosomal protein S17 | Chr. I: 6220090–6220766 | 465 |
| rpl-24.1 | 172062 | 60S ribosomal protein L24 | Chr. I: 4585115–4586177 | 552 |
| rpl-15 | 176891 | 60S ribosomal protein L15 | Chr. IV: 653436–654576 | 732 |
| rpl-35 | 176097 | 60S ribosomal protein L35 | Chr. III: 7855118–7855680 | 460 |
| rpl-36 | 176007 | 60S ribosomal protein L36 | Chr. III: 7180249–7180677 | 355 |
| rpl-27 | 171750 | 60S ribosomal protein L27 | Chr. I: 1834881–1835439 | 466 |
| rpl-33 | 174166 | 60S ribosomal protein L35 | Chr. II: 7105556–7106462 | 440 |
| Accession Number | Journal, Year | Technique | Sample Size | Data Source | Normalization Method | Top 10 Genes Sorted by SD 1 | Top 10 Genes Sorted by GC 2 |
|---|---|---|---|---|---|---|---|
| GSE118294 | Eur. J. Cell Biol., 2019 | DNA Microarray | 12 | GPL200 Affymetrix C. elegans Genome Array | RMA | rps-26, rpl-36, rpl-33, rpl-15, rps-27, rpl-35, rpl-27, rps-4, rps-23, rpl-24.1 | rps-26, rpl-33, rpl-36, rps-27, rpl-15, rpl-35, rpl-27, rps-4, rps-23, rpl-24.1 |
| GSE108968 | Nat. Commun., 2019 | DNA Microarray | 32 | GPL10094 Agilent-020186 C. elegans (V2) Microarray | Quantile, log | rpl-24.1, rpl-27, rps-16, rpl-35, cyc-1, rps-23, rpl-33, atp-3, rps-2, rpl-36 | rpl-33, rpl-24.1, rps-16, rpl-35, rpl-27, rps-23, rpl-36, atp-3, rps-2, cyc-1 |
| GSE76380 | Cell Metab., 2016 | DNA Microarray | 48 | GPL10094 Agilent-020186 C. elegans (V2) Microarray | Quantile, log | rpl-33, rps-2, rps-16, rpl-24.1, rpl-35, rps-23, rps-4, rpl-36, rpl-27, cyc-1 | rpl-33, rps-23, rps-2, rpl-35, rps-4, rpl-36, rps-16, rpl-15, rpl-24.1, cyc-1 |
| GSE63528 | eLife, 2015 | RNA-sequencing | 36 | GPL13657 Illumina HiSeq 2000 (C. elegans) | log2(CPM+1) | rps-2, rps-4, pmp-3, atp-3, rbd-1, rpl-27, rps-17, rpl-35, rps-26, act-2 | rps-4, rps-2, atp-3, rps-26, rps-17, rpl-27, rpl-35, rps-16, pmp-3, rpl-24.1 |
| GSE60755 | Nature, 2016 | RNA-sequencing | 139 | GPL13657 Illumina HiSeq 2000 (C. elegans) | log2(CPM+1) | rps-2, atp-3, mdh-1, rpl-36, rps-17, rpl-33, rpl-35, rps-4, cyc-1, rpl-15 | rps-2, atp-3, rpl-36, cyc-1, rpl-35, rpl-33, mdh-1, rps-27, rps-17, rpl-24.1 |
| GSE98919 | Cell Syst., 2017 | RNA-sequencing | 42 | GPL18730 Illumina HiSeq 1500 (C. elegans) | log2(CPM+1) | tba-1, rps-4, rpl-27, mdh-1, rpl-15, rps-26, rps-2, rps-23, cyc-1, rpl-24.1 | rpl-4, rpl-15, rps-23, rpl-27, rpl-24.1, rps-2, rps-26, rps-27, rps-17, rps-16 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tao, J.; Hao, Y.; Li, X.; Yin, H.; Nie, X.; Zhang, J.; Xu, B.; Chen, Q.; Li, B. Systematic Identification of Housekeeping Genes Possibly Used as References in Caenorhabditis elegans by Large-Scale Data Integration. Cells 2020, 9, 786. https://doi.org/10.3390/cells9030786
Tao J, Hao Y, Li X, Yin H, Nie X, Zhang J, Xu B, Chen Q, Li B. Systematic Identification of Housekeeping Genes Possibly Used as References in Caenorhabditis elegans by Large-Scale Data Integration. Cells. 2020; 9(3):786. https://doi.org/10.3390/cells9030786
Chicago/Turabian StyleTao, Jingxin, Youjin Hao, Xudong Li, Huachun Yin, Xiner Nie, Jie Zhang, Boying Xu, Qiao Chen, and Bo Li. 2020. "Systematic Identification of Housekeeping Genes Possibly Used as References in Caenorhabditis elegans by Large-Scale Data Integration" Cells 9, no. 3: 786. https://doi.org/10.3390/cells9030786