GPS-PBS: A Deep Learning Framework to Predict Phosphorylation Sites that Specifically Interact with Phosphoprotein-Binding Domains

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection and Preparation
2.2. Performance Evaluation
2.3. An Improved GPS Algorithm
- (i)
- The basic scoring strategy. Initially, the average similarity score (S) between a PBP(10, 10) item A and the whole positive data set was defined as:where K is the length of the PBP(10, 10) peptide and equal to 21, and N is the number of positive PBP(10, 10) entries. Pij is the amino acid residue at position j of a positive PBP(10, 10) Pi (i = 1, 2, 3, …, N). Wj is the weight value of position j, and M denotes an amino acid substitution matrix BLOSUM62 used in this study.
- (ii)
- PWD. In this part, the weight value of each position in the PBP(10, 10) item was initialized as 1. Then, we adopted the original PLR algorithm with the LASSO regularization to optimize the weight values of different positions. The 10-fold cross-validation was conducted, and the corresponding AUC value was calculated. To further enhance accuracy and avoid overfitting, we added two methods including random mutation and random zeroing. In the step of random mutation, we randomly chose a weight value for +1 or −1 per time, and re-calculated the AUC value. The manipulation was accepted if the AUC value was increased. In the step of random zeroing, a weight value was randomly selected and set to 0, and the manipulation was adopted if the AUC value was increased. The two steps were iteratively repeated, and the optimal Wj vectors were determined if the AUC value was not increased any longer, with a numeric criterion of 1*10-5 after 50 iterations. The PLR algorithm was implemented in Python 3.6 with Scikit-learn 0.21 [34].
- iii)
- PVT. Given the final Wj vectors, the average similarity score (Sab) of residue a in the given PBP(10, 10) item A and the amino acid b in the positive data set was defined as below:where Dj is the number of ab amino acid pairs at position j. For the 21 types of pseudo amino acids listed in an alphabetical order (A, C, D, …, Y, *), there were a number of [21*(21+1)]/2 = 231 unique Sab scores (Sab = Sba). These scores reflect the position-weighted similarity of amino acids between the given PBP(10, 10) item and all positive PBP(10, 10) entries. Thus, PVT represents a PBP(10, 10) item into a 231-dimensional vector, as below:
2.4. The Deep Learning Framework
2.5. A Permutation Test to Detect Significant Associations of PPBDs and PKs
2.6. Implementation of the Web Service
3. Results
3.1. A Deep Learning Plus Transfer Learning Strategy for Predicting PBSs
3.2. The Data Statistics and Analysis of Known PBSs
3.3. Development of GPS-PBS to Predict PBSs Recognized by Various PPBDs
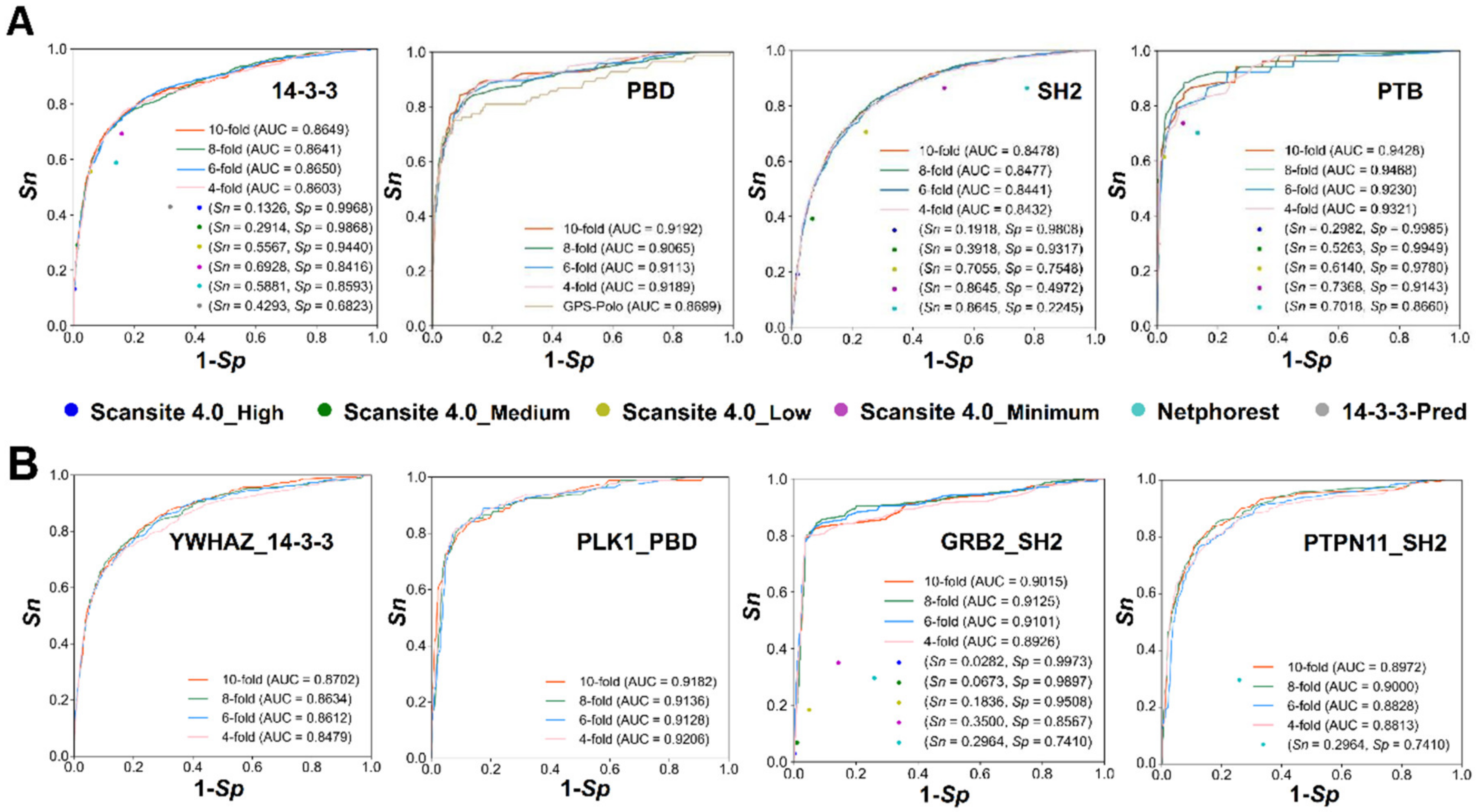
3.4. Comparison of GPS-PBS to Other Existing Tools
3.5. A Large-Scale Prediction of Potential PBSs from the Phosphoproteomic Data
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Reinhardt, H.C.; Yaffe, M.B. Phospho-ser/thr-binding domains: Navigating the cell cycle and DNA damage response. Nat. Rev. Mol. Cell Biol. 2013, 14, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.K. The 14-3-3 proteins: Integrators of diverse signaling cues that impact cell fate and cancer development. Trends Cell Biol. 2009, 19, 16–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, W.A.; Pawson, T. Phosphotyrosine signaling: Evolving a new cellular communication system. Cell 2010, 142, 661–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaffe, M.B. Phosphotyrosine-binding domains in signal transduction. Nat. Rev. Mol. Cell Biol. 2002, 3, 177–186. [Google Scholar] [CrossRef]
- Pawson, T.; Scott, J.D. Signaling through scaffold, anchoring, and adaptor proteins. Science 1997, 278, 2075–2080. [Google Scholar] [CrossRef] [Green Version]
- Yaffe, M.B.; Elia, A.E. Phosphoserine/threonine-binding domains. Curr. Opin. Cell Biol. 2001, 13, 131–138. [Google Scholar] [CrossRef]
- Pawson, T. Specificity in signal transduction: From phosphotyrosine-sh2 domain interactions to complex cellular systems. Cell 2004, 116, 191–203. [Google Scholar] [CrossRef] [Green Version]
- Hermeking, H. The 14-3-3 cancer connection. Nat. Rev. Cancer 2003, 3, 931–943. [Google Scholar] [CrossRef]
- Garnett, M.J.; Rana, S.; Paterson, H.; Barford, D.; Marais, R. Wild-type and mutant b-raf activate c-raf through distinct mechanisms involving heterodimerization. Mol. Cell 2005, 20, 963–969. [Google Scholar] [CrossRef]
- Yuan, Z.; Becker, E.B.; Merlo, P.; Yamada, T.; DiBacco, S.; Konishi, Y.; Schaefer, E.M.; Bonni, A. Activation of foxo1 by cdk1 in cycling cells and postmitotic neurons. Science 2008, 319, 1665–1668. [Google Scholar] [CrossRef]
- DeClue, J.E.; Sadowski, I.; Martin, G.S.; Pawson, T. A conserved domain regulates interactions of the v-fps protein-tyrosine kinase with the host cell. Proc. Natl. Acad. Sci. USA 1987, 84, 9064–9068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, M.; Mayer, B.J.; Fukui, Y.; Hanafusa, H. Binding of transforming protein, p47gag-crk, to a broad range of phosphotyrosine-containing proteins. Science 1990, 248, 1537–1539. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, M.B.; Cantley, L.C. Mapping specificity determinants for protein-protein association using protein fusions and random peptide libraries. Methods Enzymol. 2000, 328, 157–170. [Google Scholar] [PubMed]
- Keilhack, H.; Tenev, T.; Nyakatura, E.; Godovac-Zimmermann, J.; Nielsen, L.; Seedorf, K.; Bohmer, F.D. Phosphotyrosine 1173 mediates binding of the protein-tyrosine phosphatase shp-1 to the epidermal growth factor receptor and attenuation of receptor signaling. J. Biol. Chem. 1998, 273, 24839–24846. [Google Scholar] [CrossRef] [Green Version]
- Elia, A.E.; Cantley, L.C.; Yaffe, M.B. Proteomic screen finds pser/pthr-binding domain localizing plk1 to mitotic substrates. Science 2003, 299, 1228–1231. [Google Scholar] [CrossRef]
- Lowery, D.M.; Clauser, K.R.; Hjerrild, M.; Lim, D.; Alexander, J.; Kishi, K.; Ong, S.E.; Gammeltoft, S.; Carr, S.A.; Yaffe, M.B. Proteomic screen defines the polo-box domain interactome and identifies rock2 as a plk1 substrate. Embo J. 2007, 26, 2262–2273. [Google Scholar] [CrossRef]
- Gong, W.; Zhou, D.; Ren, Y.; Wang, Y.; Zuo, Z.; Shen, Y.; Xiao, F.; Zhu, Q.; Hong, A.; Zhou, X.; et al. Pepcyber:P~pep: A database of human protein protein interactions mediated by phosphoprotein-binding domains. Nucleic Acids Res. 2008, 36, D679–D683. [Google Scholar] [CrossRef] [Green Version]
- Dinkel, H.; Chica, C.; Via, A.; Gould, C.M.; Jensen, L.J.; Gibson, T.J.; Diella, F. Phospho.Elm: A database of phosphorylation sites--update 2011. Nucleic Acids Res. 2011, 39, D261–D267. [Google Scholar] [CrossRef] [Green Version]
- Tinti, M.; Madeira, F.; Murugesan, G.; Hoxhaj, G.; Toth, R.; Mackintosh, C. Ania: Annotation and integrated analysis of the 14-3-3 interactome. Database J. Biol. Databases Curation 2014, 2014, bat085. [Google Scholar] [CrossRef]
- Miller, M.L.; Jensen, L.J.; Diella, F.; Jorgensen, C.; Tinti, M.; Li, L.; Hsiung, M.; Parker, S.A.; Bordeaux, J.; Sicheritz-Ponten, T.; et al. Linear motif atlas for phosphorylation-dependent signaling. Sci. Signal. 2008, 1, ra2. [Google Scholar] [CrossRef]
- Goel, R.; Harsha, H.C.; Pandey, A.; Prasad, T.S. Human protein reference database and human proteinpedia as resources for phosphoproteome analysis. Mol. Biosyst. 2012, 8, 453–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaffe, M.B.; Leparc, G.G.; Lai, J.; Obata, T.; Volinia, S.; Cantley, L.C. A motif-based profile scanning approach for genome-wide prediction of signaling pathways. Nat. Biotechnol. 2001, 19, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Obenauer, J.C.; Cantley, L.C.; Yaffe, M.B. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003, 31, 3635–3641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Wu, C.; Huang, H.; Zhang, K.; Gan, J.; Li, S.S. Prediction of phosphotyrosine signaling networks using a scoring matrix-assisted ligand identification approach. Nucleic Acids Res. 2008, 36, 3263–3273. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Ren, J.; Cao, J.; He, J.; Yao, X.; Jin, C.; Xue, Y. Systematic analysis of the plk-mediated phosphoregulation in eukaryotes. Brief. Bioinform. 2013, 14, 344–360. [Google Scholar] [CrossRef] [Green Version]
- Tinti, M.; Kiemer, L.; Costa, S.; Miller, M.L.; Sacco, F.; Olsen, J.V.; Carducci, M.; Paoluzi, S.; Langone, F.; Workman, C.T. , et al. The sh2 domain interaction landscape. Cell Rep. 2013, 3, 1293–1305. [Google Scholar] [CrossRef] [Green Version]
- Madeira, F.; Tinti, M.; Murugesan, G.; Berrett, E.; Stafford, M.; Toth, R.; Cole, C.; MacKintosh, C.; Barton, G.J. 14-3-3-pred: Improved methods to predict 14-3-3-binding phosphopeptides. Bioinformatics 2015, 31, 2276–2283. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Peng, D.; Zhou, J.; Lin, S.; Wang, C.; Ning, W.; Xu, H.; Deng, W.; Xue, Y. Iekpd 2.0: An update with rich annotations for eukaryotic protein kinases, protein phosphatases and proteins containing phosphoprotein-binding domains. Nucleic Acids Res. 2019, 47, D344–D350. [Google Scholar] [CrossRef]
- Xue, Y.; Ren, J.; Gao, X.; Jin, C.; Wen, L.; Yao, X. Gps 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell. Proteom. MCP 2008, 7, 1598–1608. [Google Scholar] [CrossRef] [Green Version]
- Ning, W.; Lin, S.; Zhou, J.; Guo, Y.; Zhang, Y.; Peng, D.; Deng, W.; Xue, Y. Wocea: The visualization of functional enrichment results in word clouds. J. Genet. Genom. Yi Chuan Xue Bao 2018, 45, 415–417. [Google Scholar] [CrossRef]
- GPB-PBS. Prediction of PPBD-specific binding p-sites. Available online: http://pbs.biocuckoo.cn/ (accessed on 22 April 2020).
- UniProt Consortium. Uniprot: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [CrossRef] [PubMed] [Green Version]
- Ning, W.; Jiang, P.; Guo, Y.; Wang, C.; Tan, X.; Zhang, W.; Peng, D.; Xue, Y. Gps-palm: A deep learning-based graphic presentation system for the prediction of s-palmitoylation sites in proteins. Brief. Bioinform. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.T.; Chen, Y.F.; Hastie, T.; Sobel, E.; Lange, K. Genome-wide association analysis by lasso penalized logistic regression. Bioinformatics 2009, 25, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xu, H.; Lin, S.; Deng, W.; Zhou, J.; Zhang, Y.; Shi, Y.; Peng, D.; Xue, Y. Gps 5.0: An update on the prediction of kinase-specific phosphorylation sites in proteins. Genom. Proteom. Bioinform. 2020. [Google Scholar] [CrossRef]
- O’Shea, J.P.; Chou, M.F.; Quader, S.A.; Ryan, J.K.; Church, G.M.; Schwartz, D. Plogo: A probabilistic approach to visualizing sequence motifs. Nat. Methods 2013, 10, 1211–1212. [Google Scholar] [CrossRef]
- Muslin, A.J.; Tanner, J.W.; Allen, P.M.; Shaw, A.S. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell 1996, 84, 889–897. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, T.; Huang, H.; Zhao, B.; Li, L.; Liu, H.; Voss, C.K.; Wu, C.; Schiller, M.R.; Li, S.S. Loops govern sh2 domain specificity by controlling access to binding pockets. Sci. Signal. 2010, 3, ra34. [Google Scholar] [CrossRef]
- Kumar, M.; Gouw, M.; Michael, S.; Samano-Sanchez, H.; Pancsa, R.; Glavina, J.; Diakogianni, A.; Valverde, J.A.; Bukirova, D.; Calyseva, J.; et al. Elm-the eukaryotic linear motif resource in 2020. Nucleic Acids Res. 2019, 48, D296–D306. [Google Scholar] [CrossRef] [Green Version]
- Engels, N.; Konig, L.M.; Schulze, W.; Radtke, D.; Vanshylla, K.; Lutz, J.; Winkler, T.H.; Nitschke, L.; Wienands, J. The immunoglobulin tail tyrosine motif upgrades memory-type bcrs by incorporating a grb2-btk signalling module. Nat. Commun. 2014, 5, 5456. [Google Scholar] [CrossRef] [Green Version]
- The Gene Ontology Consortium. Expansion of the gene ontology knowledgebase and resources. Nucleic Acids Res. 2017, 45, D331–D338. [Google Scholar] [CrossRef] [Green Version]
- Woods, N.T.; Mesquita, R.D.; Sweet, M.; Carvalho, M.A.; Li, X.; Liu, Y.; Nguyen, H.; Thomas, C.E.; Iversen, E.S., Jr.; Marsillac, S.; et al. Charting the landscape of tandem brct domain-mediated protein interactions. Sci. Signal. 2012, 5, rs6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petegrosso, R.; Park, S.; Hwang, T.H.; Kuang, R. Transfer learning across ontologies for phenome-genome association prediction. Bioinformatics 2017, 33, 529–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Wang, C.; Zhou, J.; Shi, Y.; Ruan, C.; Tu, Y.; Yao, L.; Peng, D.; Xue, Y. Epsd: A well-annotated data resource of protein phosphorylation sites in eukaryotes. Brief. Bioinform. 2020, bbz169. [Google Scholar] [CrossRef] [PubMed]
- Dubois, F.; Vandermoere, F.; Gernez, A.; Murphy, J.; Toth, R.; Chen, S.; Geraghty, K.M.; Morrice, N.A.; MacKintosh, C. Differential 14-3-3 affinity capture reveals new downstream targets of phosphatidylinositol 3-kinase signaling. Mol. Cell. Proteom. MCP 2009, 8, 2487–2499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yip, M.F.; Ramm, G.; Larance, M.; Hoehn, K.L.; Wagner, M.C.; Guilhaus, M.; James, D.E. Camkii-mediated phosphorylation of the myosin motor myo1c is required for insulin-stimulated glut4 translocation in adipocytes. Cell Metab. 2008, 8, 384–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. Kegg: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Cao, M.; Chen, G.; Yu, J.; Shi, S. Computational prediction and analysis of species-specific fungi phosphorylation via feature optimization strategy. Brief. Bioinform. 2020, 21, 595–608. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Hoxhaj, G.; Manning, B.D. The pi3k-akt network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Shoelson, S.E.; Chatterjee, S.; Chaudhuri, M.; White, M.F. Ymxm motifs of irs-1 define substrate specificity of the insulin receptor kinase. Proc. Natl. Acad. Sci. USA 1992, 89, 2027–2031. [Google Scholar] [CrossRef] [Green Version]
- Felder, S.; Zhou, M.; Hu, P.; Urena, J.; Ullrich, A.; Chaudhuri, M.; White, M.; Shoelson, S.E.; Schlessinger, J. Sh2 domains exhibit high-affinity binding to tyrosine-phosphorylated peptides yet also exhibit rapid dissociation and exchange. Mol. Cell. Biol. 1993, 13, 1449–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holgado-Madruga, M.; Emlet, D.R.; Moscatello, D.K.; Godwin, A.K.; Wong, A.J. A grb2-associated docking protein in egf- and insulin-receptor signalling. Nature 1996, 379, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zhang, C.; Croucher, D.R.; Soliman, M.A.; St-Denis, N.; Pasculescu, A.; Taylor, L.; Tate, S.A.; Hardy, W.R.; Colwill, K.; et al. Temporal regulation of egf signalling networks by the scaffold protein shc1. Nature 2013, 499, 166–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the pten tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, E.B.; Snel, M.; Guigas, B.; van der Zon, G.C.; Kriek, J.; Maassen, J.A.; Jazet, I.M.; Diamant, M.; Ouwens, D.M. Phosphorylation of pras40 on thr246 by pkb/akt facilitates efficient phosphorylation of ser183 by mtorc1. Cell. Signal. 2010, 22, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lu, H. 14-3-3gamma inhibition of mdmx-mediated p21 turnover independent of p53. J. Biol. Chem. 2011, 286, 5136–5142. [Google Scholar] [CrossRef] [Green Version]
- Koumanov, F.; Richardson, J.D.; Murrow, B.A.; Holman, G.D. As160 phosphotyrosine-binding domain constructs inhibit insulin-stimulated glut4 vesicle fusion with the plasma membrane. J. Biol. Chem. 2011, 286, 16574–16582. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Ning, W.; Jiang, P.; Lin, S.; Wang, C.; Tan, X.; Yao, L.; Peng, D.; Xue, Y. GPS-PBS: A Deep Learning Framework to Predict Phosphorylation Sites that Specifically Interact with Phosphoprotein-Binding Domains. Cells 2020, 9, 1266. https://doi.org/10.3390/cells9051266
Guo Y, Ning W, Jiang P, Lin S, Wang C, Tan X, Yao L, Peng D, Xue Y. GPS-PBS: A Deep Learning Framework to Predict Phosphorylation Sites that Specifically Interact with Phosphoprotein-Binding Domains. Cells. 2020; 9(5):1266. https://doi.org/10.3390/cells9051266
Chicago/Turabian StyleGuo, Yaping, Wanshan Ning, Peiran Jiang, Shaofeng Lin, Chenwei Wang, Xiaodan Tan, Lan Yao, Di Peng, and Yu Xue. 2020. "GPS-PBS: A Deep Learning Framework to Predict Phosphorylation Sites that Specifically Interact with Phosphoprotein-Binding Domains" Cells 9, no. 5: 1266. https://doi.org/10.3390/cells9051266