Overexpression of GRP78/BiP in P-Glycoprotein-Positive L1210 Cells is Responsible for Altered Response of Cells to Tunicamycin as a Stressor of the Endoplasmic Reticulum

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Culture Conditions, and Transfections
2.2. Effect of Culturing in Media Containing Tunicamycin on the Viability of S, R and T Cells
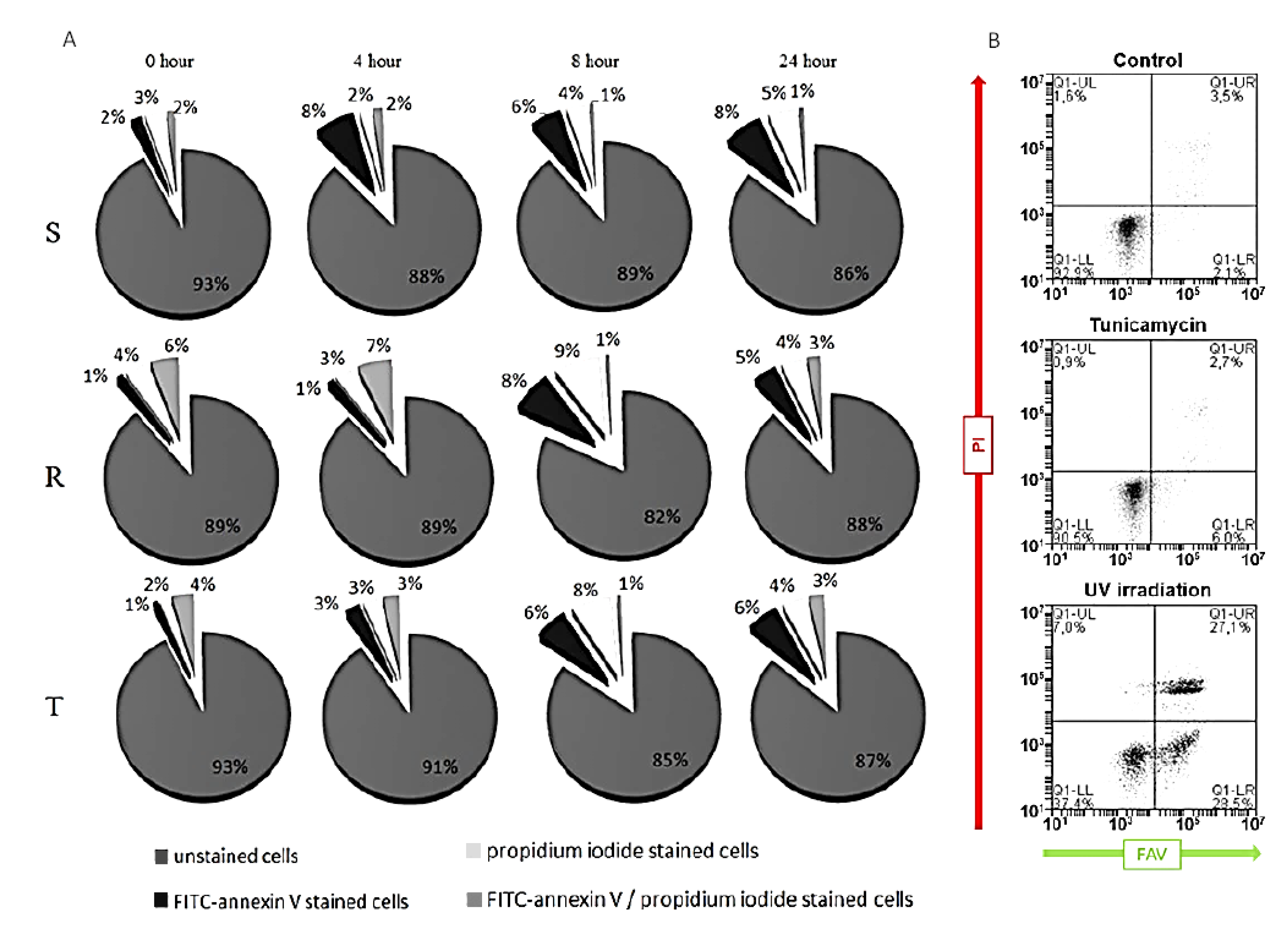
2.3. Detection of Tunicamycin-Induced Apoptosis and Necrosis in S, R, and T Cells
2.4. Monitoring cell cycle progression
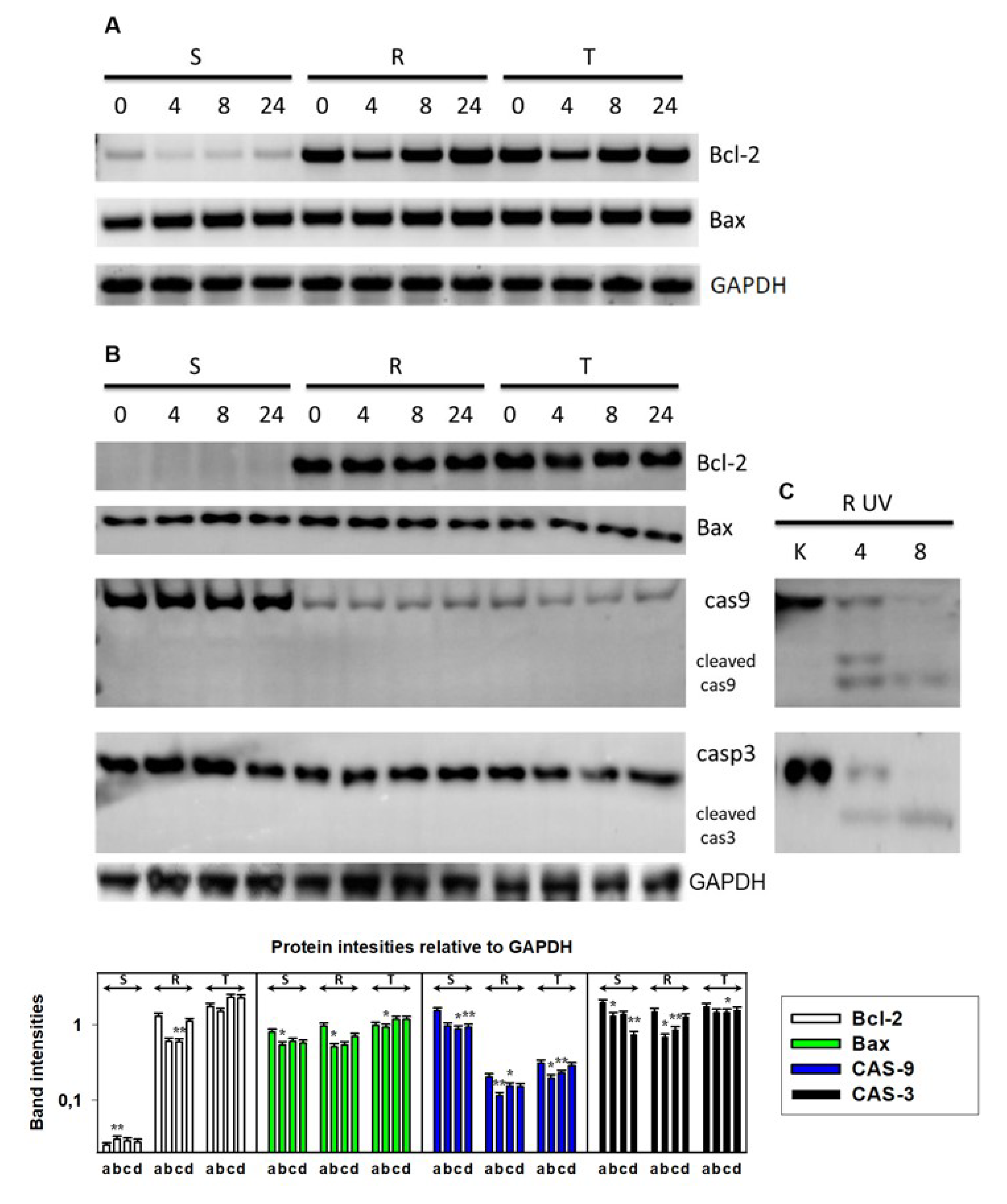
2.5. Western Blotting
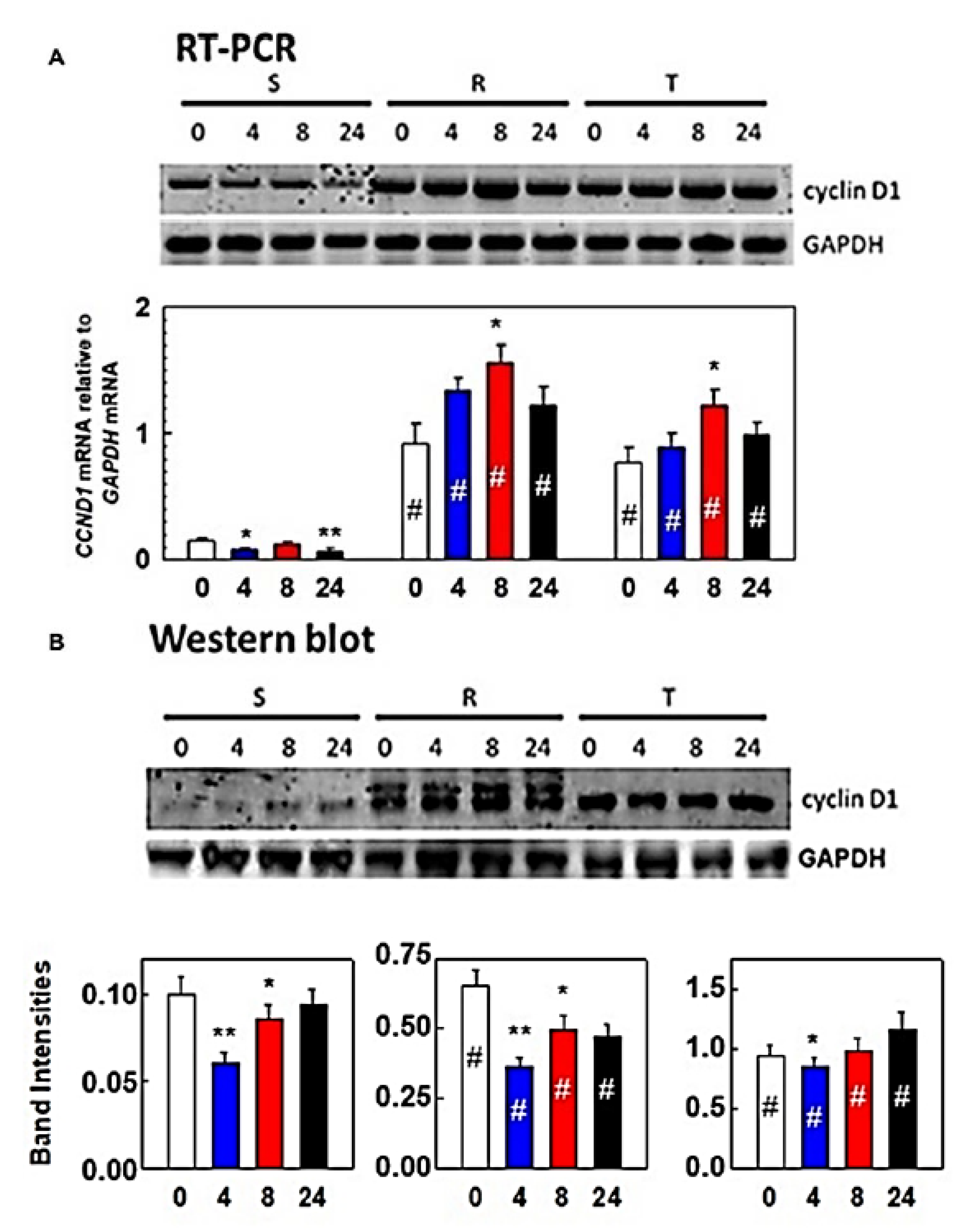
2.6. Determination of ER Stress-Induced Factors and Chaperones by RT-PCR
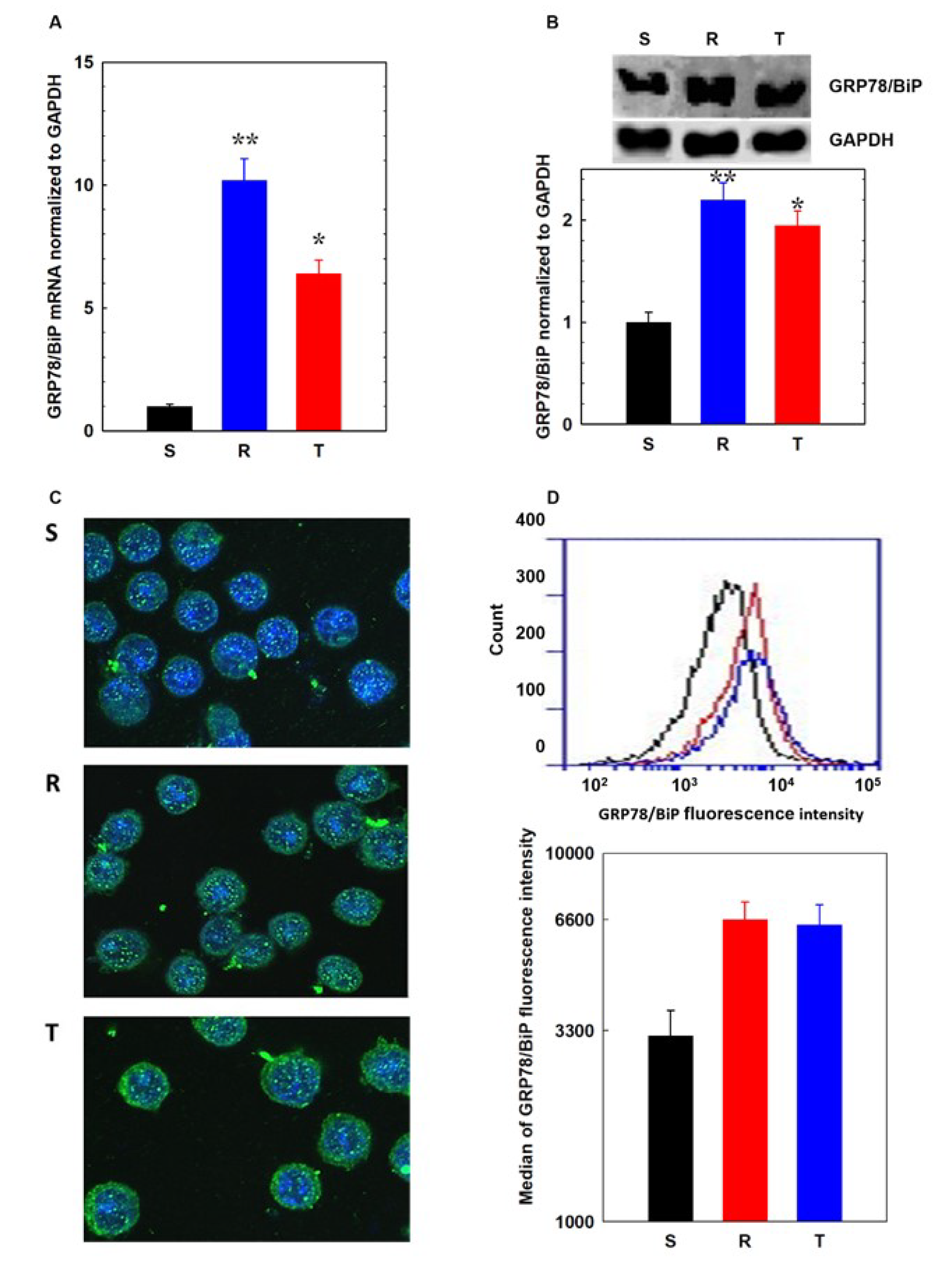
2.7. Determination of Stress Response Proteins by qPCR
2.8. Visualization of GRP78/BiP in S, R, and T Cells Using Immunofluorescence Confocal Microscopy
2.9. Detection of GRP78/Bip on Cell Surface of L1210 cells by Flow Cytometry
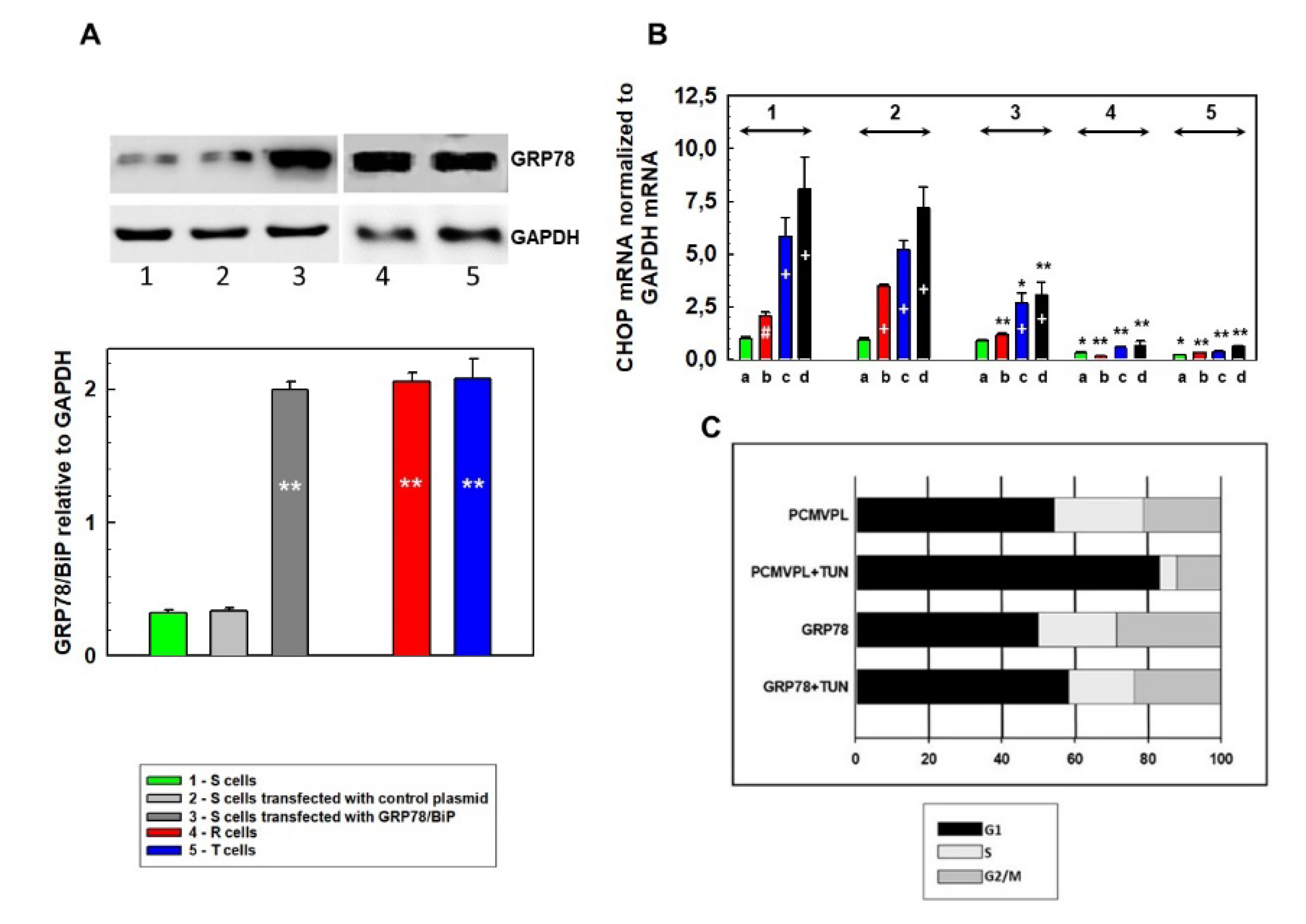
3. Results
3.1. Effect of Tunicamycin on S, R, and T Cell Cycle Progression
3.2. Endoplasmic Stress Receptors and Activation of the UPR
3.3. Downregulation of CHOP Expression After Transfection of S Cells With GRP78/BiP
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette |
| ALL | acute lymphoblasticleukemia |
| calcein AM | calcein acetoxymethyl |
| AML | Acute myeloid leukemia |
| ATF4 | activatingtranscription factor 4 |
| ATF6 | activating transcription factor 6 |
| ATF6/f | 50-kDa proteolytic fragment of ATF6 |
| Bax | Bcl-2-associated X protein |
| Bcl-2 | B-cell lymphoma 2 protein |
| BSA | Bovine serum albumin |
| cAMP | Cyclic adenosine monophosphate |
| CDK4 | Cyclindependentkinase 4 |
| CHOP | C/EBP homologous protein |
| CNX | Calnexin |
| CRT | calreticulin |
| Ct value | cycle threshold value |
| DAPI | 4′-6-diamidino-2-phenylindole |
| DMBA | dimethylbenzanthracene |
| DSMZ | German Collection of Microorganisms and Cell Cultures |
| eIF2 | eukaryotic translation-initiation factor 2 |
| ER | endoplasmic reticulum |
| ERAD | Endoplasmic-reticulum-associated protein degradation |
| FAV | Fluorescein linked annexin V |
| FP | folded protein |
| FITC | Fluorescein isothiocyanate |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GRP78/BiP | glucose-regulated protein 78/Binding immunoglobulin protein |
| GRP94 | glucose-regulated protein 94 |
| SIRE1 | inositol-requiring enzyme 1 |
| MDR | multidrug resistance |
| MTT | 3-(4,5-dimethyl-2-thiazolyl)-2, 5-diphenyl-2H-tetrazolium bromide |
| PBS | Phosphate-buffered saline |
| PCR | polymerasechain reaction |
| PERK | pancreatic ER kinase-like ER kinase |
| P-gp | P-glycoprotein |
| PI | propidium iodide |
| SDS-PAGE | sodium dodecyl sulfate–polyacrylamide gel electrophoresis |
| UPR | unfolded proteinresponse |
| UP | unfolded protein |
| UV | ultraviolet |
| s/us/t XBP1 | spliced/unspliced/total X-boxbinding protein 1 |
| qRT-PCR | quantitative RT-PCR |
| RPMI 1640 | Roswell Park MemorialInstitute 1640 medium |
| HRP | horseradish peroxidase |
| VCR | vincristine |
References
- Pahl, H.L. Signal transduction from the endoplasmic reticulum to the cell nucleus. Physiol. Rev. 1999, 79, 683–701. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Ng, D.T. Glycosylation-directed quality control of protein folding. Nat. Rev. Mol. Cell Biol. 2015, 16, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Kaufman, R.J. Unfolded protein response. Curr. Biol. 2012, 22, R622–R626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutkowski, D.T.; Kaufman, R.J. A trip to the er: Coping with stress. Trends Cell Biol. 2004, 14, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Schroder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator grp78/bip in development, cancer, and neurological disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef]
- Shi, Y.; Vattem, K.M.; Sood, R.; An, J.; Liang, J.; Stramm, L.; Wek, R.C. Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, pek, involved in translational control. Mol. Cell Biol. 1998, 18, 7499–7509. [Google Scholar] [CrossRef] [Green Version]
- Nishitoh, H. Chop is a multifunctional transcription factor in the er stress response. J. Biochem. 2012, 151, 217–219. [Google Scholar] [CrossRef] [Green Version]
- Klymenko, O.; Henneke, I.; Ruppert, C.; Mahavadi, P.; Seeger, W.; Guenther, A.; Korfei, M. Regulation and role of the pro-apoptotic transcription factor c/ebp homologous protein (chop) in type ii cells apoptosis and idiopathic pulmonary fibrosis (ipf). Am. J. Respir. Crit. Care Med. 2015, 191, A3439. [Google Scholar]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Dave, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. Er stress induces cleavage of membrane-bound atf6 by the same proteases that process srebps. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Park, H.R.; Oh, R.; Wagner, P.; Panganiban, R.; Lu, Q. New insights into cellular stress responses to environmental metal toxicants. Int. Rev. Cell Mol. Biol. 2017, 331, 55–82. [Google Scholar] [PubMed]
- Harrington, P.E.; Biswas, K.; Malwitz, D.; Tasker, A.S.; Mohr, C.; Andrews, K.L.; Dellamaggiore, K.; Kendall, R.; Beckmann, H.; Jaeckel, P.; et al. Unfolded protein response in cancer: Ire1alpha inhibition by selective kinase ligands does not impair tumor cell viability. ACS Med. Chem. Lett. 2014, 6, 68–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirota, M.; Kitagaki, M.; Itagaki, H.; Aiba, S. Quantitative measurement of spliced xbp1 mrna as an indicator of endoplasmic reticulum stress. J. Toxicol. Sci. 2006, 31, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Wiersma, V.R.; Michalak, M.; Abdullah, T.M.; Bremer, E.; Eggleton, P. Mechanisms of translocation of er chaperones to the cell surface and immunomodulatory roles in cancer and autoimmunity. Front. Oncol. 2015, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.K.; Dubeau, L.; Kleiner, H.; Parr, T.; Nichols, P.; Ko, B.; Dong, D.; Ko, H.; Mao, C.; DiGiovanni, J.; et al. Cancer-inducible transgene expression by the grp94 promoter: Spontaneous activation in tumors of various origins and cancer-associated macrophages. Cancer Res. 2002, 62, 7207–7212. [Google Scholar]
- Wang, Q.; Groenendyk, J.; Michalak, M. Glycoprotein quality control and endoplasmic reticulum stress. Molecules 2015, 20, 13689–13704. [Google Scholar] [CrossRef] [Green Version]
- Heifetz, A.; Keenan, R.W.; Elbein, A.D. Mechanism of action of tunicamycin on the udp-glcnac:Dolichyl-phosphate glc-nac-1-phosphate transferase. Biochemistry 1979, 18, 2186–2192. [Google Scholar] [CrossRef]
- Morin, M.J.; Bernacki, R.J. Biochemical effects and therapeutic potential of tunicamycin in murine l1210 leukemia. Cancer Res. 1983, 43, 1669–1674. [Google Scholar]
- Hemming, F. Glycosyl phosphopolyprenols. In Glycolipids; Wiegandt, H., Ed.; Elsevier Biomedical Press: Amsterdam, The Netherlands, 1985; pp. 261–305. [Google Scholar]
- You, S.; Li, W.; Guan, Y. Tunicamycin inhibits colon carcinoma growth and aggressiveness via modulation of the erk-jnk-mediated akt/mtor signaling pathway. Mol. Med. Rep. 2018, 17, 4203–4212. [Google Scholar] [CrossRef]
- Han, C.; Jin, L.; Mei, Y.; Wu, M. Endoplasmic reticulum stress inhibits cell cycle progression via induction of p27 in melanoma cells. Cell Signal. 2013, 25, 144–149. [Google Scholar] [CrossRef]
- Wang, X.; Xiong, W.; Tang, Y. Tunicamycin suppresses breast cancer cell growth and metastasis via regulation of the protein kinase b/nuclear factor-kappab signaling pathway. Oncol. Lett. 2018, 15, 4137–4142. [Google Scholar] [PubMed] [Green Version]
- Qi, W.; Zeng, S.; Liu, M.; Yang, S.; Tan, X.; Yu, B. Tunicamycin induces apoptosis in non-small cell lung cancer cells through c/ebp homologous protein activation-mediated endoplasmic reticulum stress. Int. J. Clin. Exp. Med. 2018, 11, 5310–5322. [Google Scholar]
- da Silva, D.C.; Valentao, P.; Andrade, P.B.; Pereira, D.M. Endoplasmic reticulum stress signaling in cancer and neurodegenerative disorders: Tools and strategies to understand its complexity. Pharmacol. Res. 2020, 155, 104702. [Google Scholar] [CrossRef] [PubMed]
- Kelly, G.L.; Strasser, A. The essential role of evasion from cell death in cancer. Adv. Cancer Res. 2011, 111, 39–96. [Google Scholar]
- Bublik, D.R.; Bursac, S.; Sheffer, M.; Orsolic, I.; Shalit, T.; Tarcic, O.; Kotler, E.; Mouhadeb, O.; Hoffman, Y.; Fuchs, G.; et al. Regulatory module involving fgf13, mir-504, and p53 regulates ribosomal biogenesis and supports cancer cell survival. Proc. Natl. Acad. Sci. USA 2017, 114, E496–E505. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.H. Regulation of protein degradation by proteasomes in cancer. J. Cancer Prev. 2018, 23, 153–161. [Google Scholar] [CrossRef]
- Sonneveld, P. Multidrug resistance in haematological malignancies. J. Intern. Med. 2000, 247, 521–534. [Google Scholar] [CrossRef]
- Breier, A.; Barancik, M.; Sulova, Z.; Uhrik, B. P-glycoprotein--implications of metabolism of neoplastic cells and cancer therapy. Curr. Cancer Drug Targets 2005, 5, 457–468. [Google Scholar] [CrossRef]
- Breier, A.; Gibalova, L.; Seres, M.; Barancik, M.; Sulova, Z. New insight into p-glycoprotein as a drug target. Anticancer Agents Med. Chem. 2013, 13, 159–170. [Google Scholar] [CrossRef]
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in chinese hamster ovary cell mutants. Biochim. Biophys. Acta 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Greer, D.A.; Ivey, S. Distinct n-glycan glycosylation of p-glycoprotein isolated from the human uterine sarcoma cell line mes-sa/dx5. Biochim. Biophys. Acta 2007, 1770, 1275–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seres, M.; Cholujova, D.; Bubencikova, T.; Breier, A.; Sulova, Z. Tunicamycin depresses p-glycoprotein glycosylation without an effect on its membrane localization and drug efflux activity in l1210 cells. Int. J. Mol. Sci. 2011, 12, 7772–7784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breier, A.; Stetka, J.; Bohacova, V.; Macejova, D.; Brtko, J.; Sulova, Z. Effect of 9-cis retinoic acid and all-trans retinoic acid in combination with verapamil on p-glycoprotein expression in l1210 cells. Neoplasma 2014, 61, 553–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlikova, L.; Seres, M.; Imrichova, D.; Hano, M.; Rusnak, A.; Zamorova, M.; Katrlik, J.; Breier, A.; Sulova, Z. The expression of p-gp in leukemia cells is associated with cross-resistance to protein n-glycosylation inhibitor tunicamycin. Gen. Physiol. Biophys. 2016, 35, 497–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hano, M.; Tomasova, L.; Seres, M.; Pavlikova, L.; Breier, A.; Sulova, Z. Interplay between p-glycoprotein expression and resistance to endoplasmic reticulum stressors. Molecules 2018, 23, 337. [Google Scholar] [CrossRef] [Green Version]
- Polekova, L.; Barancik, M.; Mrazova, T.; Pirker, R.; Wallner, J.; Sulova, Z.; Breier, A. Adaptation of mouse leukemia cells l1210 to vincristine. Evidence for expression of p-glycoprotein. Neoplasma 1992, 39, 73–77. [Google Scholar]
- Sulova, Z.; Ditte, P.; Kurucova, T.; Polakova, E.; Rogozanova, K.; Gibalova, L.; Seres, M.; Skvarkova, L.; Sedlak, J.; Pastorek, J.; et al. The presence of p-glycoprotein in l1210 cells directly induces down-regulation of cell surface saccharide targets of concanavalin a. Anticancer Res. 2010, 30, 3661–3668. [Google Scholar]
- Pastan, I.; Gottesman, M.M.; Ueda, K.; Lovelace, E.; Rutherford, A.V.; Willingham, M.C. A retrovirus carrying an mdr1 cdna confers multidrug resistance and polarized expression of p-glycoprotein in mdck cells. Proc. Natl. Acad. Sci. USA 1988, 85, 4486–4490. [Google Scholar] [CrossRef] [Green Version]
- Werstuck, G.H.; Lentz, S.R.; Dayal, S.; Hossain, G.S.; Sood, S.K.; Shi, Y.Y.; Zhou, J.; Maeda, N.; Krisans, S.K.; Malinow, M.R.; et al. Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J. Clin. Investig. 2001, 107, 1263–1273. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Y.; Wagner, E.J.; Cullen, B.R. Both natural and designed micro rnas can inhibit the expression of cognate mrnas when expressed in human cells. Mol. Cell 2002, 9, 1327–1333. [Google Scholar] [CrossRef]
- Samali, A.; Fitzgerald, U.; Deegan, S.; Gupta, S. Methods for monitoring endoplasmic reticulum stress and the unfolded protein response. Int. J. Cell Biol. 2010, 2010, 830307. [Google Scholar] [CrossRef] [PubMed]
- Oslowski, C.M.; Urano, F. Measuring er stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 2011, 490, 71–92. [Google Scholar] [PubMed] [Green Version]
- Rastogi, S.; Boylan, M.; Wright, E.G.; Coates, P.J. Interactions of apoptotic cells with macrophages in radiation-induced bystander signaling. Radiat. Res. 2013, 179, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Prischi, F.; Nowak, P.R.; Carrara, M.; Ali, M.M. Phosphoregulation of ire1 rnase splicing activity. Nat. Commun. 2014, 5, 3554. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.A.; Fang, S.U.; Su, C.L.; Hsiao, C.J.; Chang, C.C.; Lin, Y.F.; Cheng, C.W. Silencing glucose-regulated protein 78 induced renal cell carcinoma cell line g1 cell-cycle arrest and resistance to conventional chemotherapy. Urol. Oncol. 2014, 32, 29.e1–29.e11. [Google Scholar] [CrossRef]
- Guha, P.; Kaptan, E.; Gade, P.; Kalvakolanu, D.V.; Ahmed, H. Tunicamycin induced endoplasmic reticulum stress promotes apoptosis of prostate cancer cells by activating mtorc1. Oncotarget 2017, 8, 68191–68207. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Luo, X.; Yang, Y.; Dai, Z.; Wu, G.; Wu, Z. Endoplasmic reticulum stress-induced apoptosis in intestinal epithelial cells: A feed-back regulation by mechanistic target of rapamycin complex 1 (mtorc1). J. Anim. Sci. Biotechnol. 2018, 9, 38. [Google Scholar] [CrossRef]
- Oda, T.; Kosuge, Y.; Arakawa, M.; Ishige, K.; Ito, Y. Distinct mechanism of cell death is responsible for tunicamycin-induced er stress in sk-n-sh and sh-sy5y cells. Neurosci. Res. 2008, 60, 29–39. [Google Scholar] [CrossRef]
- Pavlikova, L.; Seres, M.; Hano, M.; Bohacova, V.; Sevcikova, I.; Kyca, T.; Breier, A.; Sulova, Z. L1210 cells overexpressing abcb1 drug transporters are resistant to inhibitors of the n- and o-glycosylation of proteins. Molecules 2017, 22, 1104. [Google Scholar] [CrossRef] [Green Version]
- Seres, M.; Pavliková, L.; Sulova, Z.; Breier, A. Lectin detection of cell surface saccharides remodeling induced by development of p-glycoprotein mediated multidrug resistance phenotype in l1210 leukemia cells. Acta Chim. Slovaca 2014, 7, 52–56. [Google Scholar] [CrossRef] [Green Version]
- Del Principe, M.I.; Del Poeta, G.; Maurillo, L.; Buccisano, F.; Venditti, A.; Tamburini, A.; Bruno, A.; Cox, M.C.; Suppo, G.; Tendas, A.; et al. P-glycoprotein and bcl-2 levels predict outcome in adult acute lymphoblastic leukaemia. Br. J. Haematol. 2003, 121, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Kasimir-Bauer, S.; Beelen, D.; Flasshove, M.; Noppeney, R.; Seeber, S.; Scheulen, M.E. Impact of the expression of p glycoprotein, the multidrug resistance-related protein, bcl-2, mutant p53, and heat shock protein 27 on response to induction therapy and long-term survival in patients with de novo acute myeloid leukemia. Exp. Hematol. 2002, 30, 1302–1308. [Google Scholar] [CrossRef]
- Lee, J.S.; Jung, W.K.; Jeong, M.H.; Yoon, T.R.; Kim, H.K. Sanguinarine induces apoptosis of ht-29 human colon cancer cells via the regulation of bax/bcl-2 ratio and caspase-9-dependent pathway. Int. J. Toxicol. 2012, 31, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Karpel-Massler, G.; Ishida, C.T.; Zhang, Y.; Halatsch, M.E.; Westhoff, M.A.; Siegelin, M.D. Targeting intrinsic apoptosis and other forms of cell death by bh3-mimetics in glioblastoma. Expert Opin. Drug Discov. 2017, 12, 1031–1040. [Google Scholar] [CrossRef]
- Hsu, J.L.; Chiang, P.C.; Guh, J.H. Tunicamycin induces resistance to camptothecin and etoposide in human hepatocellular carcinoma cells: Role of cell-cycle arrest and grp78. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2009, 380, 373–382. [Google Scholar] [CrossRef]
- Du, Z.; Tong, X.; Ye, X. Cyclin d1 promotes cell cycle progression through enhancing ndr1/2 kinase activity independent of cyclin-dependent kinase 4. J. Biol. Chem. 2013, 288, 26678–26687. [Google Scholar] [CrossRef] [Green Version]
- Cooley, A.; Zelivianski, S.; Jeruss, J.S. Impact of cyclin e overexpression on smad3 activity in breast cancer cell lines. Cell Cycle 2010, 9, 4900–4907. [Google Scholar] [CrossRef] [Green Version]
- Darzynkiewicz, Z.; Zhao, H.; Zhang, S.; Lee, M.Y.; Lee, E.Y.; Zhang, Z. Initiation and termination of DNA replication during s phase in relation to cyclins d1, e and a, p21waf1, cdt1 and the p12 subunit of DNA polymerase delta revealed in individual cells by cytometry. Oncotarget 2015, 6, 11735–11750. [Google Scholar] [CrossRef]
- Xie, X.; Lin, W.; Zheng, W.; Chen, T.; Yang, H.; Sun, L.; Huang, F.; Wang, Z.; Lin, H.; Chen, L.; et al. Downregulation of g2/mitotic-specific cyclinb1 triggers autophagy via ampk-ulk1-dependent signal pathway in nasopharyngeal carcinoma cells. Cell Death Dis. 2019, 10, 94. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, R.; Ni, M.; Gill, P.; Lee, A.S. Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator grp78/bip. J. Biol. Chem. 2010, 285, 15065–15075. [Google Scholar] [CrossRef] [Green Version]
- Sulova, Z.; Seres, M.; Barancik, M.; Gibalova, L.; Uhrik, B.; Polekova, L.; Breier, A. Does any relationship exist between p-glycoprotein-mediated multidrug resistance and intracellular calcium homeostasis. Gen. Physiol. Biophys. 2009, 28, F89–F95. [Google Scholar] [PubMed]
- Seres, M.; Polakova, E.; Krizanova, O.; Hudecova, S.; Klymenko, S.V.; Breier, A.; Sulova, Z. Overexpression of p-glycoprotein in l1210/vcr cells is associated with changes in several endoplasmic reticulum proteins that may be partially responsible for the lack of thapsigargin sensitivity. Gen. Physiol. Biophys. 2008, 27, 211–221. [Google Scholar] [PubMed]
- Bubencikova, T.; Cholujova, D.; Messingerova, L.; Mislovicova, D.; Seres, M.; Breier, A.; Sulova, Z. Detection of glycomic alterations induced by overexpression of p-glycoprotein on the surfaces of l1210 cells using sialic acid binding lectins. Int. J. Mol. Sci. 2012, 13, 15177–15192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulova, Z.; Mislovicova, D.; Gibalova, L.; Vajcnerova, Z.; Polakova, E.; Uhrik, B.; Tylkova, L.; Kovarova, A.; Sedlak, J.; Breier, A. Vincristine-induced overexpression of p-glycoprotein in l1210 cells is associated with remodeling of cell surface saccharides. J. Proteome Res. 2009, 8, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Xia, Y.; Li, J.; Zhang, C.; Zhang, H.; Ma, T.; Yang, L.; Kong, L. Grp78 inhibition enhances atf4-induced cell death by the deubiquitination and stabilization of chop in human osteosarcoma. Cancer Lett. 2017, 410, 112–123. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Revers Primer | bp |
|---|---|---|---|
| GAPDH b | 5′-TAT GTC GTG GAG TCT ACT GGT GTC-3′ | 5′-GTC ATC ATA CTT GGC AGG TTT CTC-3′ | 492 |
| IRE1α b | 5′-AAC ACA CCG ACC ACC GTA TC-3′ | 5′-AGG GTC CTG GGT AAG GTC TC-3′ | 282 |
| PERK b | 5′-GCC GAC GAT CAA ATG GAA GC-3′ | 5′-GTG GGG CTG AGG ATG GAA AA-3′ | 370 |
| ATF6 b | 5′-TGG AAG TGG GAA GAT CGG GA-3′ | 5′-AGC CAC AGG TCC TCT TTA GG-3′ | 312 |
| XBP1 a | 5′-GAA CCA GGA GTT AAG AAC ACG-3′ | 5′-AGG CAA CAG TGT CAG AGT CC-3′ | us 205 s 179 |
| CHOP b | 5′-GGA ACC TGA GGA GAG GTG TTC-3′ | 5′-TGC AGA TCC TCA TAC CAG G-3′ | 162 |
| GRP94 b | 5′-GGG GAG GTC ACC TTC AAG TC-3′ | 5′-TGA GGG GGA GAT CAT CGG AA-3′ | 199 |
| Cnx b | 5′-AGT GGG AAG TAG ATG AGA TGA AGG-3′ | 5′-ATA CAC CTG TCT TGG GAT TTT TGT-3′ | 333 |
| cyclin D1 b | 5′-TCA CCC TGA GAG TAG GGA GC-3′ | 5′-GGC CTT CAG GCA AAA ACC AG-3′ | 592 |
| cyclin A b | 5′-AGC AGA ACT CAT TCG GCT CT-3′ | 5′-CAA GGG AAA AGG AAG AAG AAG AGA A-3′ | 297 |
| cyclin E b | 5′-ATG TTA CAG ATG GCG CTT GC-3′ | 5′-GAG GAC ACC ATA AGG AAA TCT GA-3′ | 254 |
| cyclin B1 b | 5′-CAG TTG TGT GCC CAA GAA GA-3′ | 5′-CTA CGG AGG AAG TGC AGA GG-3′ | 216 |
| Bcl-2 b | 5′-GGC TGG GGA TGA CTT CTC TC-3′ | 5′-GCA TGC TGG GGC CAT ATA GTT-3‘ | 323 |
| Bax b | 5′-ATC CAA GAC CAG GGT GGC T-3′ | 5′-CTT CCC CCA TTC ATC CCA GG-3′ | 197 |
| Gene | Forward Primer | Revers Primer |
|---|---|---|
| sXBP1 c,s | 5′-GTC CAT GGG AAG ATG TTC TGG-3′ | 5′-CTG AGT CCG AAT CAG GTG CAG-3′ |
| usXBP1 c, us | 5′-GTC CAT GGG AAG ATG TTC TGG-3′ | 5′-CAG CAC TCA GAC TAT GTG CA-3′ |
| totalXBP1 c,t | 5′-GTC CAT GGG AAG ATG TTC TGG-3′ | 5′-TGG CCG GGT CTG CTG AGT CCG-3′ |
| CHOP c | 5′-AGG TGA AAG GCA GGG ACT CA-3′ | 5′-CCA CCA CAC CTG AAA GCA GAA-3′ |
| GRP78/BiP c | 5′-TTT TCT GAT GTA TCC TCT TCA CCA GT-3′ | 5′-TTC AGC CAA TTA TCA GCA AAC TCT-3′ |
| GRP94 c | 5′-CAA ATG GAG AAG ATT CCG CC-3′ | 5′-AAG AAT GAA GGA AAA ACA GGA CAA AA-3′ |
| β-actin c | 5′-TGT CCA CCT TCC AGC AGA T-3′ | 5′-AGC TCA GTA ACA GTC CGC C-3′ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šereš, M.; Pavlíková, L.; Boháčová, V.; Kyca, T.; Borovská, I.; Lakatoš, B.; Breier, A.; Sulová, Z. Overexpression of GRP78/BiP in P-Glycoprotein-Positive L1210 Cells is Responsible for Altered Response of Cells to Tunicamycin as a Stressor of the Endoplasmic Reticulum. Cells 2020, 9, 890. https://doi.org/10.3390/cells9040890
Šereš M, Pavlíková L, Boháčová V, Kyca T, Borovská I, Lakatoš B, Breier A, Sulová Z. Overexpression of GRP78/BiP in P-Glycoprotein-Positive L1210 Cells is Responsible for Altered Response of Cells to Tunicamycin as a Stressor of the Endoplasmic Reticulum. Cells. 2020; 9(4):890. https://doi.org/10.3390/cells9040890
Chicago/Turabian StyleŠereš, Mário, Lucia Pavlíková, Viera Boháčová, Tomáš Kyca, Ivana Borovská, Boris Lakatoš, Albert Breier, and Zdena Sulová. 2020. "Overexpression of GRP78/BiP in P-Glycoprotein-Positive L1210 Cells is Responsible for Altered Response of Cells to Tunicamycin as a Stressor of the Endoplasmic Reticulum" Cells 9, no. 4: 890. https://doi.org/10.3390/cells9040890