The Cellular Protein CAD is Recruited into Ebola Virus Inclusion Bodies by the Nucleoprotein NP to Facilitate Genome Replication and Transcription


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Plasmids and Cloning
2.3. Antibodies
2.4. Viruses
2.5. Chemical Compounds
2.6. siRNA Knockdown with EBOV Minigenomes and Pyrimidine Complementation
2.7. RNA Isolation and RT-qPCR
2.8. Immunofluorescence Analysis
2.9. Infection of Transfected Huh7 Cells
2.10. Co-Immunoprecipitation of Viral Proteins
2.11. Western Blotting
2.12. Statistical Analyses
3. Results
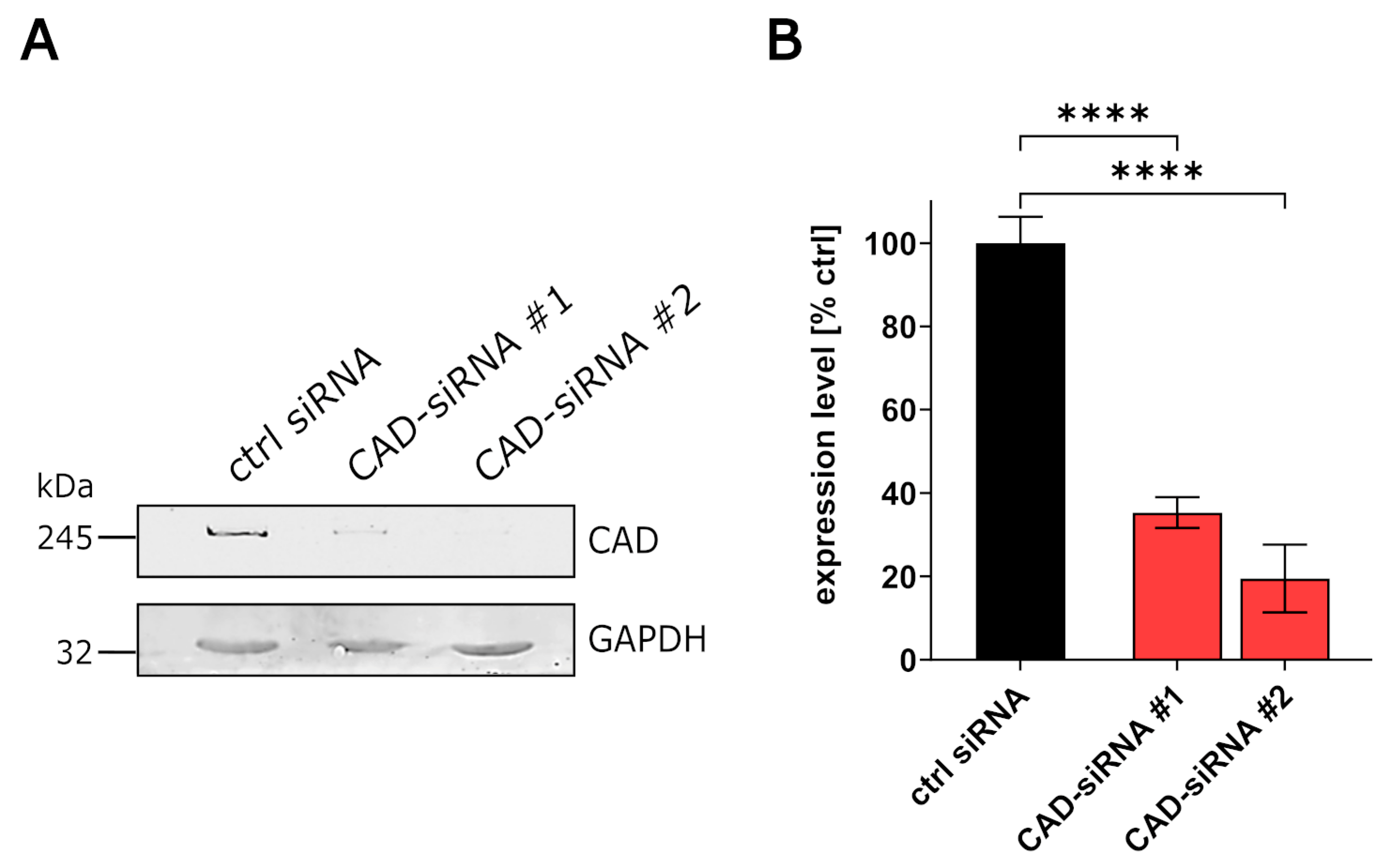
3.1. CAD Knockdown Affects Both EBOV Genome Replication and Transcription
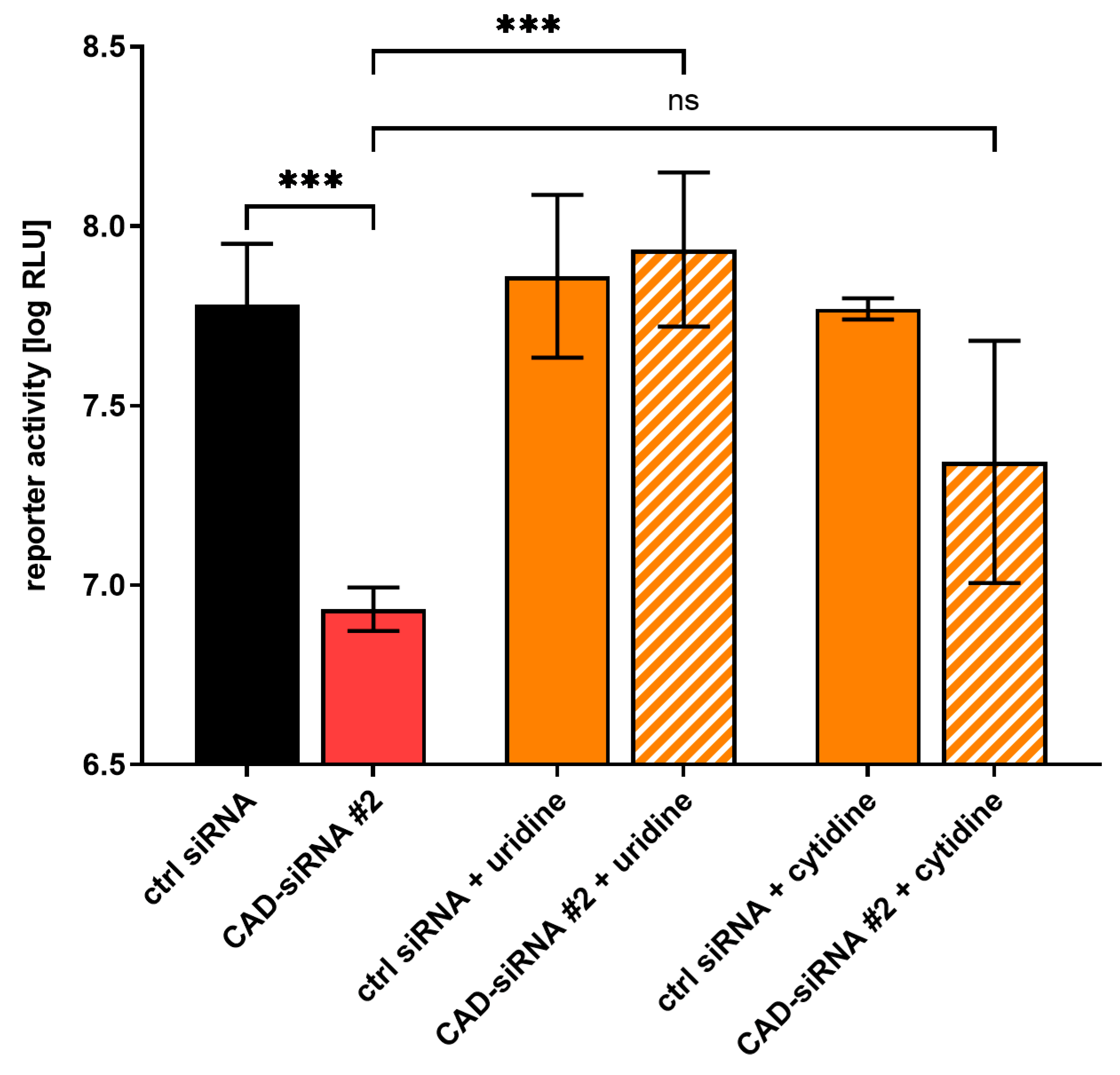
3.2. The Effect of CAD Knockdown Can Be Compensated for by Exogenous Pyrimidines
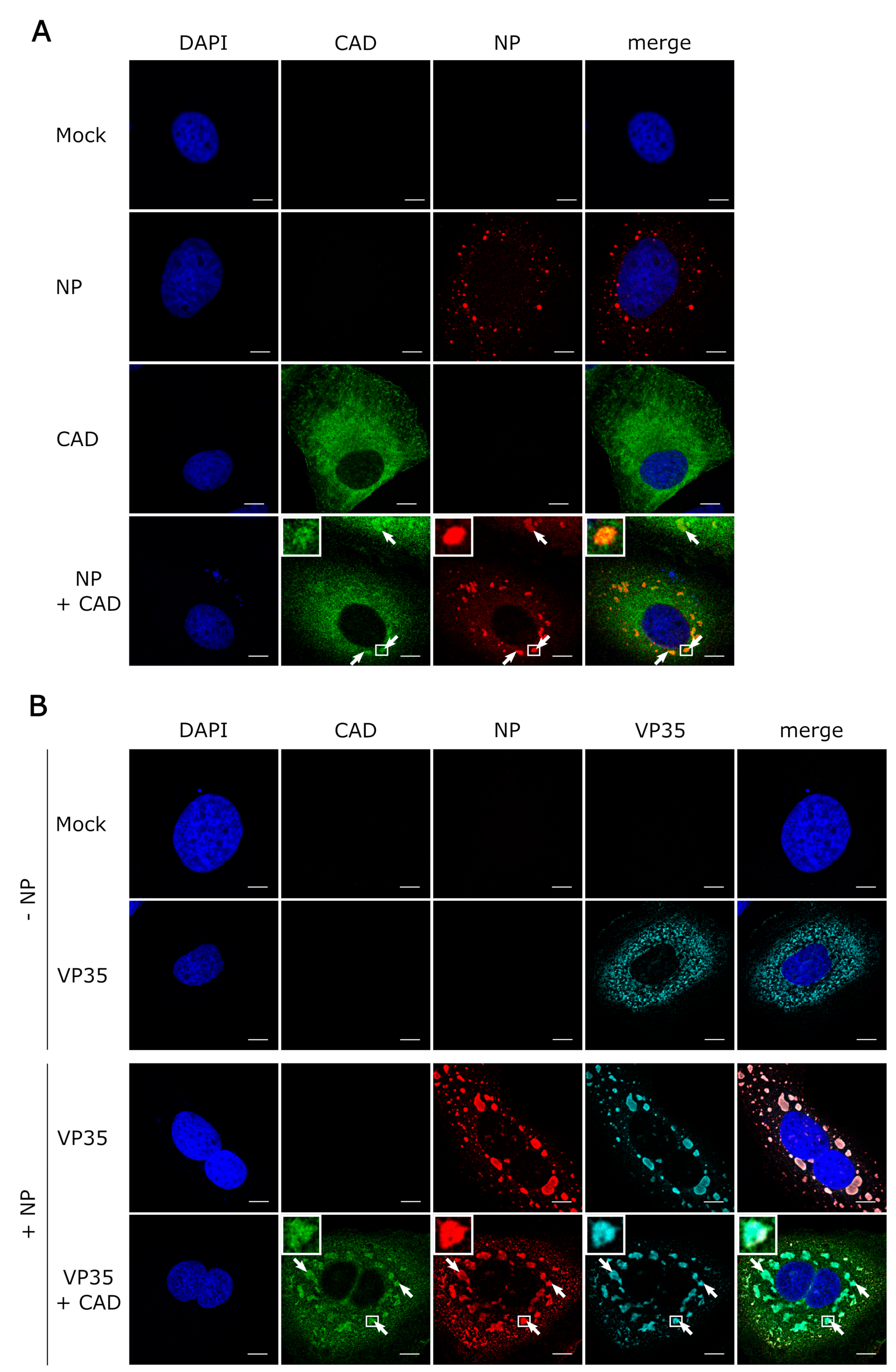
3.3. CAD Colocalizes with NP-Induced Inclusion Bodies
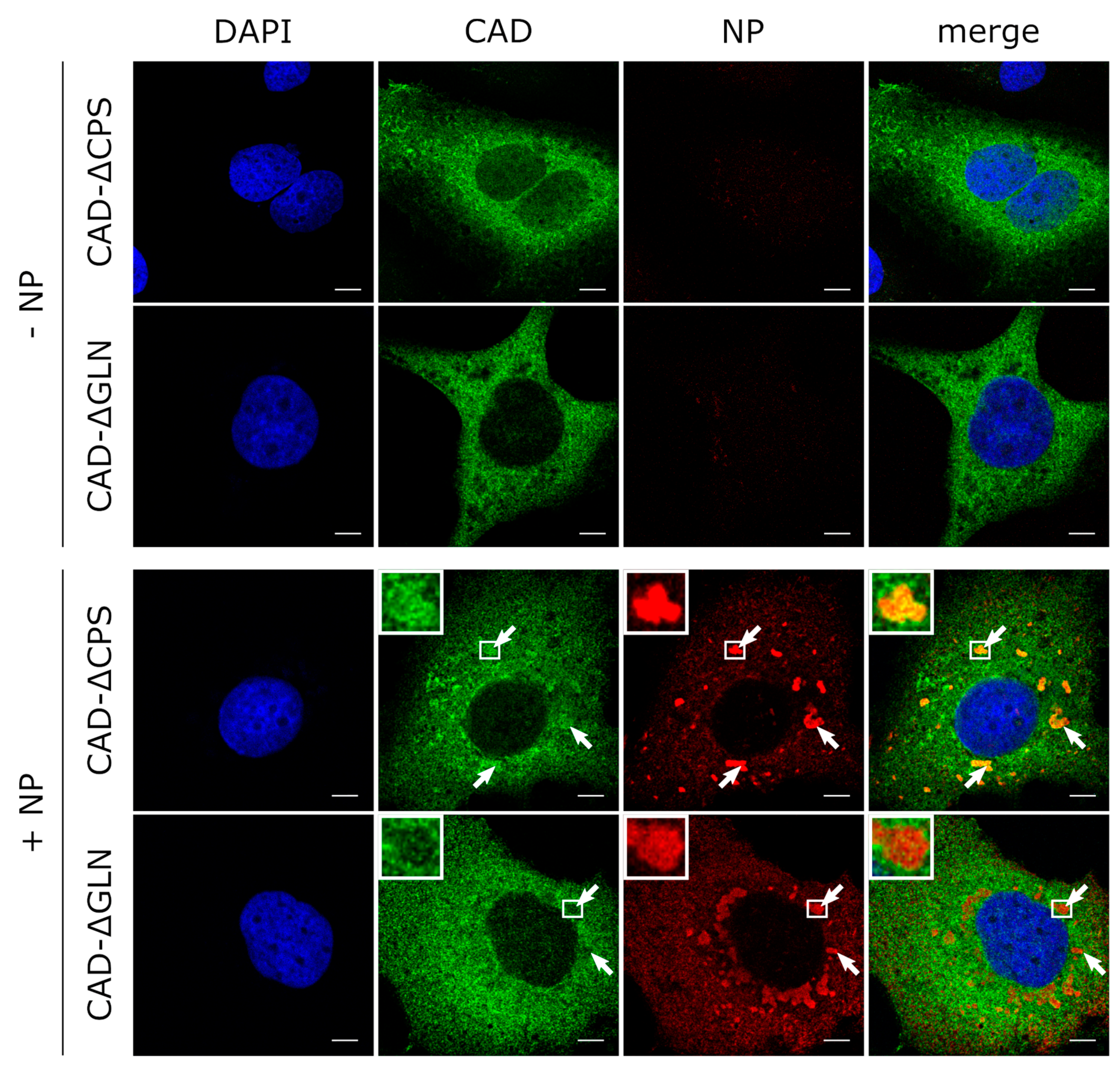
3.4. The GLN Domain of CAD Is Required for its Accumulation in Inclusion Bodies
3.5. CAD Interacts with NP in an RNA-Independent Manner
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Feldmann, H.; Geisbert, T.W. Ebola haemorrhagic fever. Lancet 2011, 377, 849–862. [Google Scholar] [CrossRef]
- Burk, R.; Bollinger, L.; Johnson, J.C.; Wada, J.; Radoshitzky, S.R.; Palacios, G.; Bavari, S.; Jahrling, P.B.; Kuhn, J.H. Neglected filoviruses. FEMS Microbiol. Rev. 2016, 40, 494–519. [Google Scholar] [CrossRef]
- Bharat, T.A.; Noda, T.; Riches, J.D.; Kraehling, V.; Kolesnikova, L.; Becker, S.; Kawaoka, Y.; Briggs, J.A. Structural dissection of Ebola virus and its assembly determinants using cryo-electron tomography. Proc. Natl. Acad. Sci. USA 2012, 109, 4275–4280. [Google Scholar] [CrossRef]
- Ruigrok, R.W.; Crepin, T.; Kolakofsky, D. Nucleoproteins and nucleocapsids of negative-strand RNA viruses. Curr. Opin. Microbiol. 2011, 14, 504–510. [Google Scholar] [CrossRef]
- Becker, S.; Rinne, C.; Hofsass, U.; Klenk, H.D.; Muhlberger, E. Interactions of Marburg virus nucleocapsid proteins. Virology 1998, 249, 406–417. [Google Scholar] [CrossRef]
- Muhlberger, E.; Weik, M.; Volchkov, V.E.; Klenk, H.D.; Becker, S. Comparison of the transcription and replication strategies of marburg virus and Ebola virus by using artificial replication systems. J. Virol. 1999, 73, 2333–2342. [Google Scholar] [CrossRef]
- Weik, M.; Modrof, J.; Klenk, H.D.; Becker, S.; Muhlberger, E. Ebola virus VP30-mediated transcription is regulated by RNA secondary structure formation. J. Virol. 2002, 76, 8532–8539. [Google Scholar] [CrossRef]
- Hoenen, T.; Shabman, R.S.; Groseth, A.; Herwig, A.; Weber, M.; Schudt, G.; Dolnik, O.; Basler, C.F.; Becker, S.; Feldmann, H. Inclusion bodies are a site of ebolavirus replication. J. Virol. 2012, 86, 11779–11788. [Google Scholar] [CrossRef]
- Lier, C.; Becker, S.; Biedenkopf, N. Dynamic phosphorylation of Ebola virus VP30 in NP-induced inclusion bodies. Virology 2017, 512, 39–47. [Google Scholar] [CrossRef]
- Boehmann, Y.; Enterlein, S.; Randolf, A.; Muhlberger, E. A reconstituted replication and transcription system for Ebola virus Reston and comparison with Ebola virus Zaire. Virology 2005, 332, 406–417. [Google Scholar] [CrossRef]
- Groseth, A.; Charton, J.E.; Sauerborn, M.; Feldmann, F.; Jones, S.M.; Hoenen, T.; Feldmann, H. The Ebola virus ribonucleoprotein complex: A novel VP30-L interaction identified. Virus Res. 2009, 140, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Pietzsch, C.; Ramanathan, P.; Santos, R.I.; Ilinykh, P.A.; Garcia-Blanco, M.A.; Bukreyev, A.; Bradrick, S.S. Staufen1 Interacts with Multiple Components of the Ebola Virus Ribonucleoprotein and Enhances Viral RNA Synthesis. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; He, Z.; Yuan, Y.; Huang, F.; Luo, B.; Zhang, J.; Pan, T.; Zhang, H.; Zhang, J. Host factor SMYD3 is recruited by Ebola virus nucleoprotein to facilitate viral mRNA transcription. Emerg. Microbes Infect. 2019, 8, 1347–1360. [Google Scholar] [CrossRef] [PubMed]
- Batra, J.; Hultquist, J.F.; Liu, D.; Shtanko, O.; Von Dollen, J.; Satkamp, L.; Jang, G.M.; Luthra, P.; Schwarz, T.M.; Small, G.I.; et al. Protein Interaction Mapping Identifies RBBP6 as a Negative Regulator of Ebola Virus Replication. Cell 2018, 175, 1917–1930. [Google Scholar] [CrossRef]
- Gabriel, G.; Feldmann, F.; Reimer, R.; Thiele, S.; Fischer, M.; Hartmann, E.; Bader, M.; Ebihara, H.; Hoenen, T.; Feldmann, H. Importin-alpha7 Is Involved in the Formation of Ebola Virus Inclusion Bodies but Is Not Essential for Pathogenicity in Mice. J. Infect. Dis. 2015, 212 Suppl 2, S316–S321. [Google Scholar] [CrossRef][Green Version]
- Morwitzer, M.J.; Tritsch, S.R.; Cazares, L.H.; Ward, M.D.; Nuss, J.E.; Bavari, S.; Reid, S.P. Identification of RUVBL1 and RUVBL2 as Novel Cellular Interactors of the Ebola Virus Nucleoprotein. Viruses 2019, 11, 372. [Google Scholar] [CrossRef]
- Kruse, T.; Biedenkopf, N.; Hertz, E.P.T.; Dietzel, E.; Stalmann, G.; Lopez-Mendez, B.; Davey, N.E.; Nilsson, J.; Becker, S. The Ebola Virus Nucleoprotein Recruits the Host PP2A-B56 Phosphatase to Activate Transcriptional Support Activity of VP30. Mol. Cell 2018, 69, 136–145. [Google Scholar] [CrossRef]
- Takamatsu, Y.; Krahling, V.; Kolesnikova, L.; Halwe, S.; Lier, C.; Baumeister, S.; Noda, T.; Biedenkopf, N.; Becker, S. Serine-Arginine Protein Kinase 1 Regulates Ebola Virus Transcription. mBio 2020, 11. [Google Scholar] [CrossRef]
- Wendt, L.; Brandt, J.; Bodmer, B.S.; Reiche, S.; Schmidt, M.L.; Traeger, S.; Hoenen, T. The Ebola Virus Nucleoprotein Recruits the Nuclear RNA Export Factor NXF1 into Inclusion Bodies to Facilitate Viral Protein Expression. Cells 2020, 9, 187. [Google Scholar] [CrossRef]
- Martin, S.; Chiramel, A.I.; Schmidt, M.L.; Chen, Y.C.; Whitt, N.; Watt, A.; Dunham, E.C.; Shifflett, K.; Traeger, S.; Leske, A.; et al. A genome-wide siRNA screen identifies a druggable host pathway essential for the Ebola virus life cycle. Genome Med. 2018, 10, 58. [Google Scholar] [CrossRef]
- Uebelhoer, L.S.; Albarino, C.G.; McMullan, L.K.; Chakrabarti, A.K.; Vincent, J.P.; Nichol, S.T.; Towner, J.S. High-throughput, luciferase-based reverse genetics systems for identifying inhibitors of Marburg and Ebola viruses. Antiviral Res. 2014, 106, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Nelson, E.V.; Pacheco, J.R.; Hume, A.J.; Cressey, T.N.; Deflube, L.R.; Ruedas, J.B.; Connor, J.H.; Ebihara, H.; Muhlberger, E. An RNA polymerase II-driven Ebola virus minigenome system as an advanced tool for antiviral drug screening. Antiviral Res. 2017, 146, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Coleman, P.F.; Suttle, D.P.; Stark, G.R. Purification from hamster cells of the multifunctional protein that initiates de novo synthesis of pyrimidine nucleotides. J. Biol. Chem. 1977, 252, 6379–6385. [Google Scholar] [PubMed]
- Jones, M.E. Pyrimidine nucleotide biosynthesis in animals: Genes, enzymes, and regulation of UMP biosynthesis. Annu. Rev. Biochem. 1980, 49, 253–279. [Google Scholar] [CrossRef]
- Lee, L.; Kelly, R.E.; Pastra-Landis, S.C.; Evans, D.R. Oligomeric structure of the multifunctional protein CAD that initiates pyrimidine biosynthesis in mammalian cells. Proc. Natl. Acad. Sci. USA 1985, 82, 6802–6806. [Google Scholar] [CrossRef]
- Christopherson, R.I.; Jones, M.E. The overall synthesis of L-5,6-dihydroorotate by multienzymatic protein pyr1-3 from hamster cells. Kinetic studies, substrate channeling, and the effects of inhibitors. J. Biol. Chem. 1980, 255, 11381–11395. [Google Scholar]
- Irvine, H.S.; Shaw, S.M.; Paton, A.; Carrey, E.A. A reciprocal allosteric mechanism for efficient transfer of labile intermediates between active sites in CAD, the mammalian pyrimidine-biosynthetic multienzyme polypeptide. Eur. J. Biochem. 1997, 247, 1063–1073. [Google Scholar] [CrossRef]
- Evans, D.R.; Guy, H.I. Mammalian pyrimidine biosynthesis: Fresh insights into an ancient pathway. J. Biol. Chem. 2004, 279, 33035–33038. [Google Scholar] [CrossRef]
- Sigoillot, F.D.; Berkowski, J.A.; Sigoillot, S.M.; Kotsis, D.H.; Guy, H.I. Cell cycle-dependent regulation of pyrimidine biosynthesis. J. Biol. Chem. 2003, 278, 3403–3409. [Google Scholar] [CrossRef]
- Sigoillot, F.D.; Kotsis, D.H.; Serre, V.; Sigoillot, S.M.; Evans, D.R.; Guy, H.I. Nuclear localization and mitogen-activated protein kinase phosphorylation of the multifunctional protein CAD. J. Biol. Chem. 2005, 280, 25611–25620. [Google Scholar] [CrossRef]
- Chaparian, M.G.; Evans, D.R. Intracellular location of the multidomain protein CAD in mammalian cells. FASEB J. 1988, 2, 2982–2989. [Google Scholar] [CrossRef] [PubMed]
- Hoenen, T.; Jung, S.; Herwig, A.; Groseth, A.; Becker, S. Both matrix proteins of Ebola virus contribute to the regulation of viral genome replication and transcription. Virology 2010, 403, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Shabman, R.S.; Hoenen, T.; Groseth, A.; Jabado, O.; Binning, J.M.; Amarasinghe, G.K.; Feldmann, H.; Basler, C.F. An upstream open reading frame modulates ebola virus polymerase translation and virus replication. PLoS Pathog. 2013, 9, e1003147. [Google Scholar] [CrossRef] [PubMed]
- Kamper, L.; Zierke, L.; Schmidt, M.L.; Muller, A.; Wendt, L.; Brandt, J.; Hartmann, E.; Braun, S.; Holzerland, J.; Groseth, A.; et al. Assessment of the function and intergenus-compatibility of Ebola and Lloviu virus proteins. J. Gen. Virol. 2019, 100, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.L.; Hoenen, T. Characterization of the catalytic center of the Ebola virus L polymerase. PLoS Negl. Trop. Dis. 2017, 11, e0005996. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Xu, L.; Sun, Y.; Nabel, G.J. The assembly of Ebola virus nucleocapsid requires virion-associated proteins 35 and 24 and posttranslational modification of nucleoprotein. Mol. Cell. 2002, 10, 307–316. [Google Scholar] [CrossRef]
- Noda, T.; Hagiwara, K.; Sagara, H.; Kawaoka, Y. Characterization of the Ebola virus nucleoprotein-RNA complex. J. Gen. Virol. 2010, 91, 1478–1483. [Google Scholar] [CrossRef]
- Borawski, J.; Troke, P.; Puyang, X.; Gibaja, V.; Zhao, S.; Mickanin, C.; Leighton-Davies, J.; Wilson, C.J.; Myer, V.; Cornellataracido, I.; et al. Class III phosphatidylinositol 4-kinase alpha and beta are novel host factor regulators of hepatitis C virus replication. J. Virol. 2009, 83, 10058–10074. [Google Scholar] [CrossRef]
- Ortiz-Riano, E.; Ngo, N.; Devito, S.; Eggink, D.; Munger, J.; Shaw, M.L.; de la Torre, J.C.; Martinez-Sobrido, L. Inhibition of arenavirus by A3, a pyrimidine biosynthesis inhibitor. J. Virol. 2014, 88, 878–889. [Google Scholar] [CrossRef]
- Katsafanas, G.C.; Grem, J.L.; Blough, H.A.; Moss, B. Inhibition of vaccinia virus replication by N-(phosphonoacetyl)-L-aspartate: Differential effects on viral gene expression result from a reduced pyrimidine nucleotide pool. Virology 1997, 236, 177–187. [Google Scholar] [CrossRef]
- Warren, T.K.; Jordan, R.; Lo, M.K.; Ray, A.S.; Mackman, R.L.; Soloveva, V.; Siegel, D.; Perron, M.; Bannister, R.; Hui, H.C.; et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 2016, 531, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Luthra, P.; Naidoo, J.; Pietzsch, C.A.; De, S.; Khadka, S.; Anantpadma, M.; Williams, C.G.; Edwards, M.R.; Davey, R.A.; Bukreyev, A.; et al. Inhibiting pyrimidine biosynthesis impairs Ebola virus replication through depletion of nucleoside pools and activation of innate immune responses. Antiviral Res. 2018, 158, 288–302. [Google Scholar] [CrossRef] [PubMed]
- Deans, R.M.; Morgens, D.W.; Okesli, A.; Pillay, S.; Horlbeck, M.A.; Kampmann, M.; Gilbert, L.A.; Li, A.; Mateo, R.; Smith, M.; et al. Parallel shRNA and CRISPR-Cas9 screens enable antiviral drug target identification. Nat. Chem. Biol. 2016, 12, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Kota, K.P.; Benko, J.G.; Mudhasani, R.; Retterer, C.; Tran, J.P.; Bavari, S.; Panchal, R.G. High content image based analysis identifies cell cycle inhibitors as regulators of Ebola virus infection. Viruses 2012, 4, 1865–1877. [Google Scholar] [CrossRef] [PubMed]
- Angeletti, P.C.; Engler, J.A. Adenovirus preterminal protein binds to the CAD enzyme at active sites of viral DNA replication on the nuclear matrix. J. Virol. 1998, 72, 2896–2904. [Google Scholar] [CrossRef] [PubMed]
- Fredman, J.N.; Engler, J.A. Adenovirus precursor to terminal protein interacts with the nuclear matrix in vivo and in vitro. J. Virol. 1993, 67, 3384–3395. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brandt, J.; Wendt, L.; Bodmer, B.S.; Mettenleiter, T.C.; Hoenen, T. The Cellular Protein CAD is Recruited into Ebola Virus Inclusion Bodies by the Nucleoprotein NP to Facilitate Genome Replication and Transcription. Cells 2020, 9, 1126. https://doi.org/10.3390/cells9051126
Brandt J, Wendt L, Bodmer BS, Mettenleiter TC, Hoenen T. The Cellular Protein CAD is Recruited into Ebola Virus Inclusion Bodies by the Nucleoprotein NP to Facilitate Genome Replication and Transcription. Cells. 2020; 9(5):1126. https://doi.org/10.3390/cells9051126
Chicago/Turabian StyleBrandt, Janine, Lisa Wendt, Bianca S. Bodmer, Thomas C. Mettenleiter, and Thomas Hoenen. 2020. "The Cellular Protein CAD is Recruited into Ebola Virus Inclusion Bodies by the Nucleoprotein NP to Facilitate Genome Replication and Transcription" Cells 9, no. 5: 1126. https://doi.org/10.3390/cells9051126
APA StyleBrandt, J., Wendt, L., Bodmer, B. S., Mettenleiter, T. C., & Hoenen, T. (2020). The Cellular Protein CAD is Recruited into Ebola Virus Inclusion Bodies by the Nucleoprotein NP to Facilitate Genome Replication and Transcription. Cells, 9(5), 1126. https://doi.org/10.3390/cells9051126