LXRα Regulates ChREBPα Transactivity in a Target Gene-Specific Manner through an Agonist-Modulated LBD-LID Interaction

 , , , ,
, , , ,
Abstract
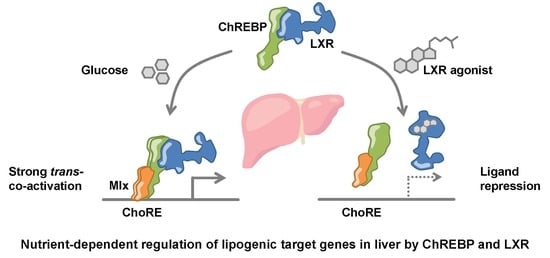
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. ChIP-seq Data Analysis
2.3. Animals and Fasting-Refeeding Experiments
2.4. Mouse Primary Hepatocytes Isolation and Culture
2.5. Cell Culture and Transfection
2.6. Plasmids
2.7. Luciferase Reporter Assay
2.8. Chromatin Immuneprecipitation (ChIP)
2.9. RNA Extraction, cDNA Synthesis and Quantitative RT-PCR
2.10. Co-Immunoprecipitation (CoIP)
2.11. Immunoblotting
2.12. Statistical Analysis
2.13. Gene Set Enrichment Analysis
2.14. Data Availability
3. Results
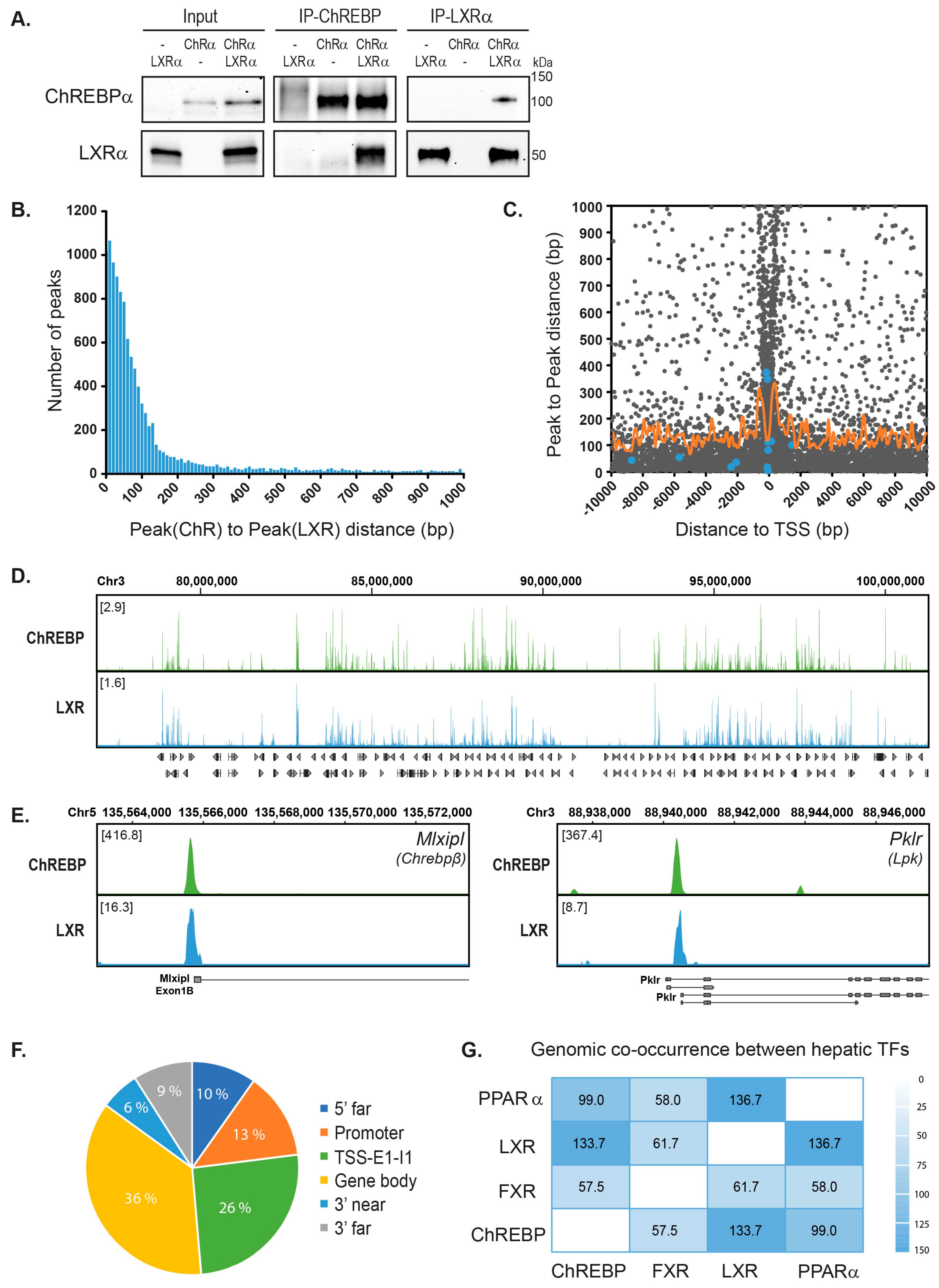
3.1. ChREBPα and LXRα Interact and Show a High Co-Occupancy of the Mouse Liver Genome
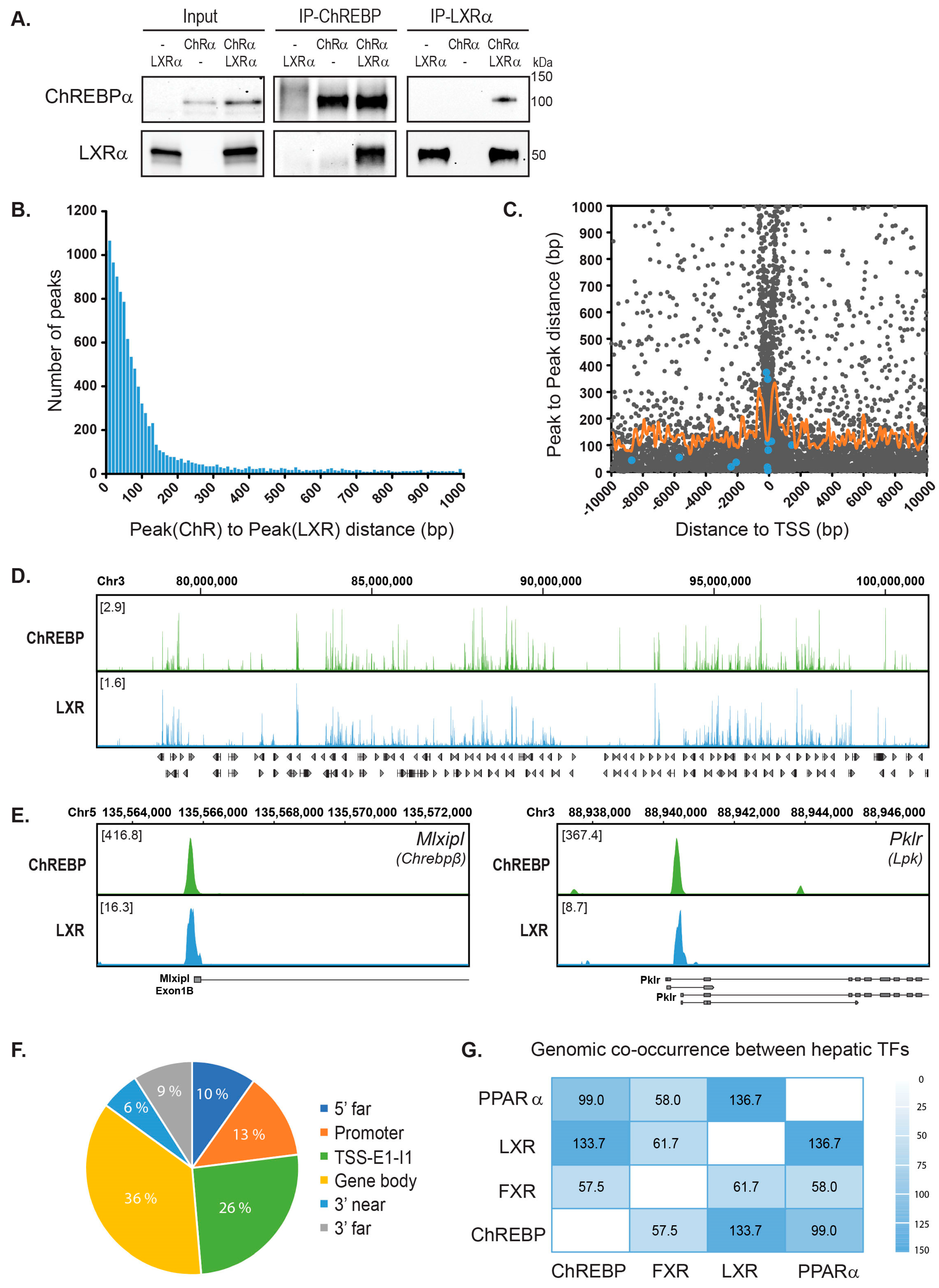
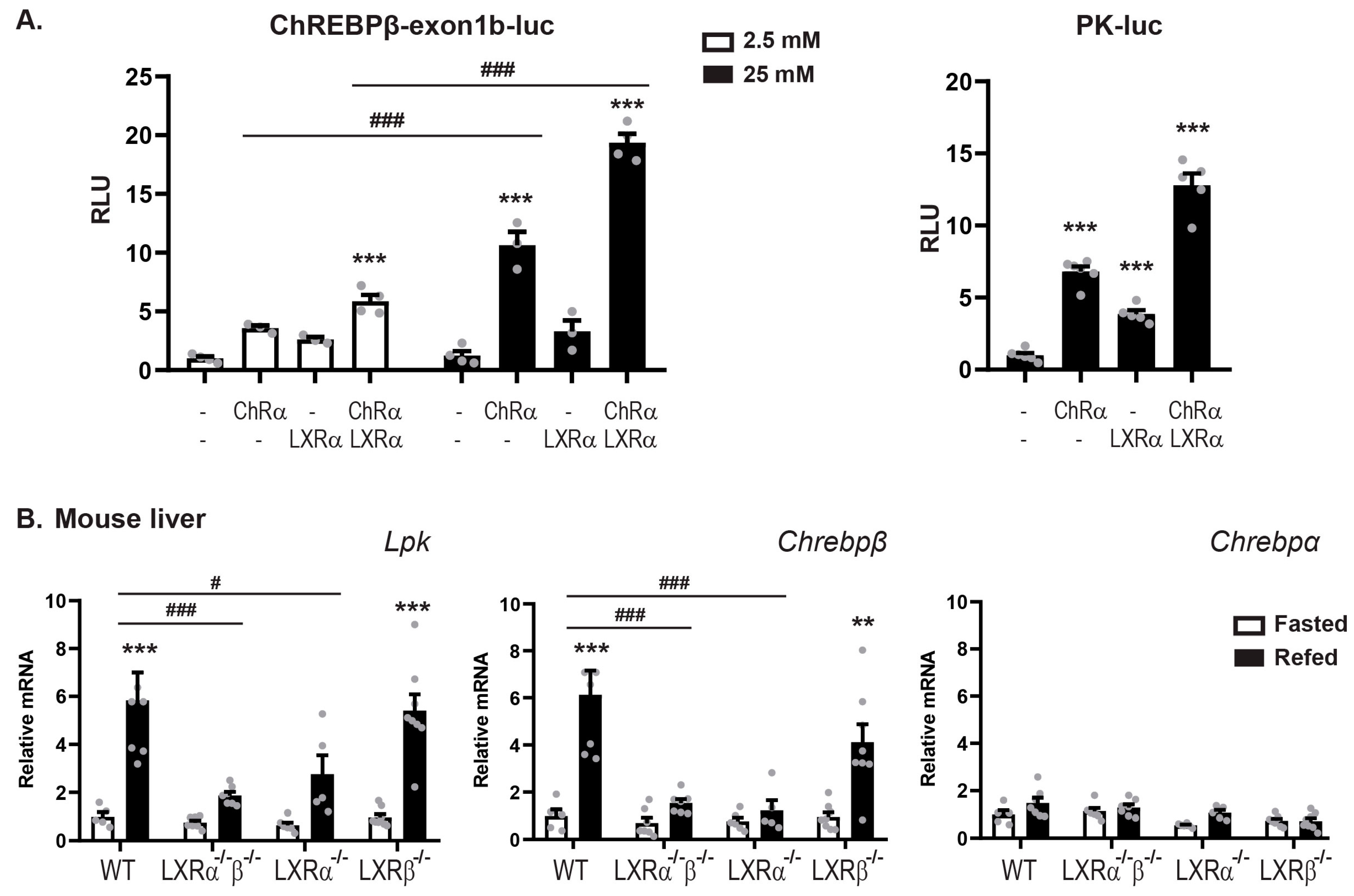
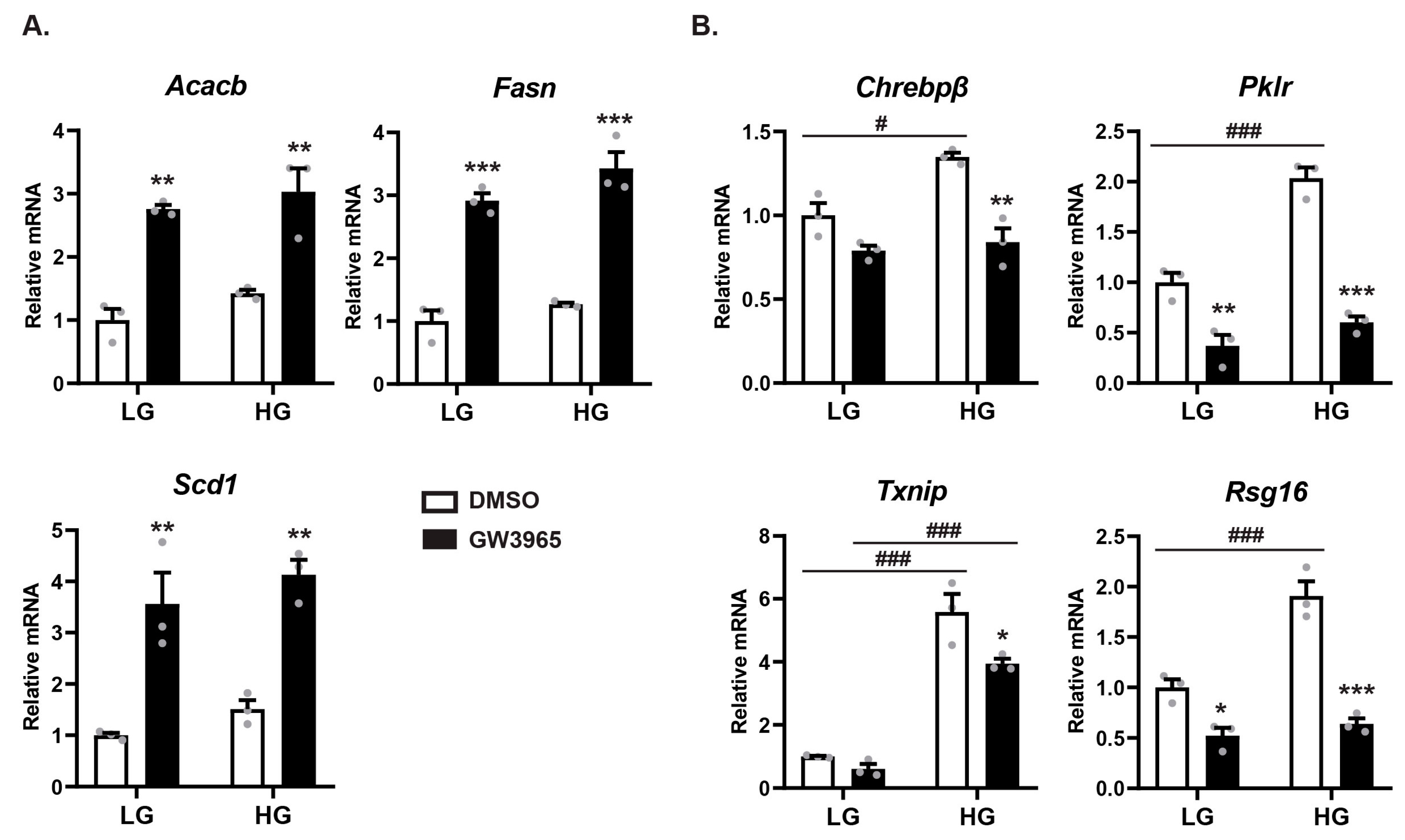
3.2. LXRα and ChREBPα Co-Activates ChREBP Target Genes In Vitro and In Vivo
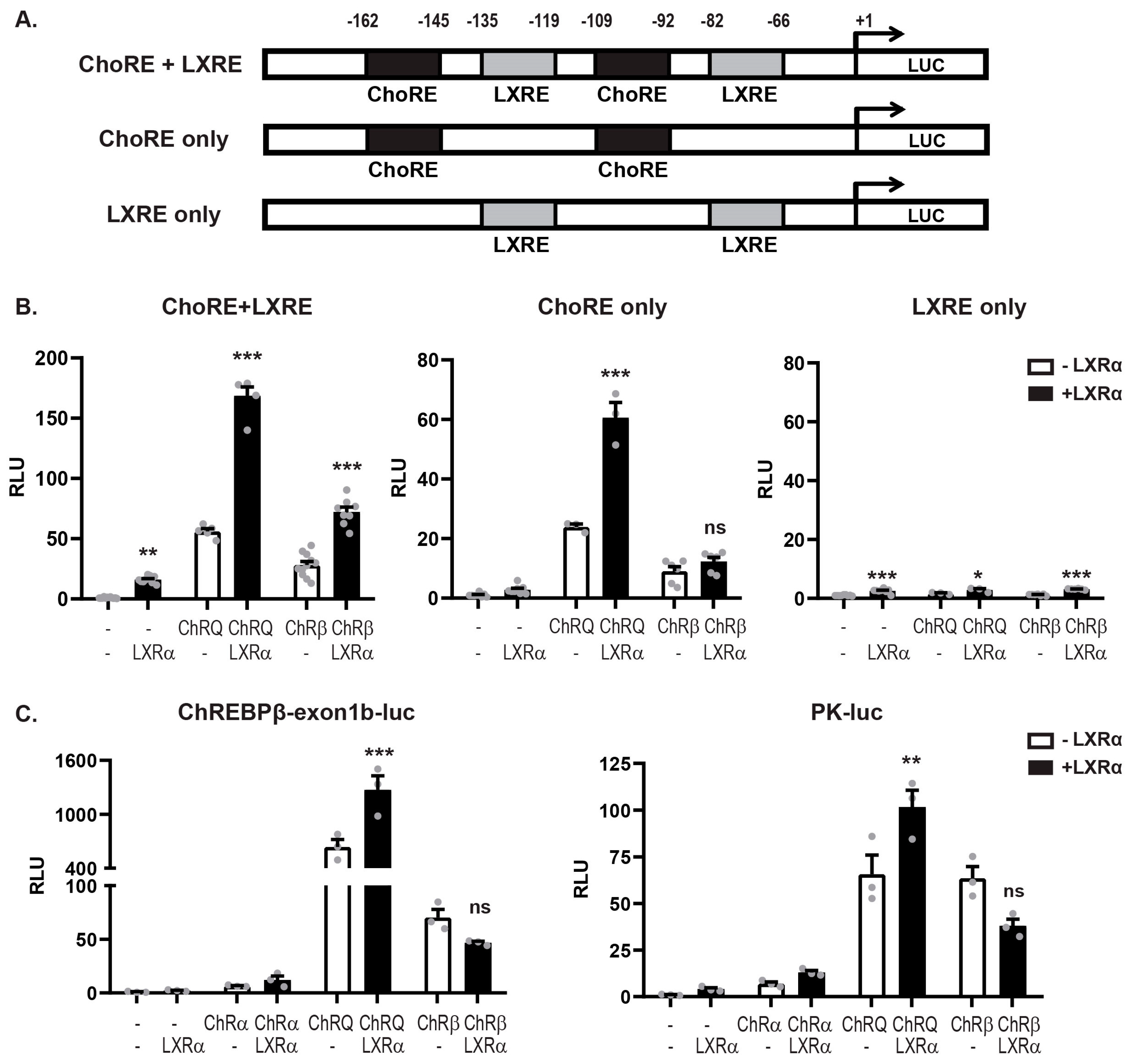
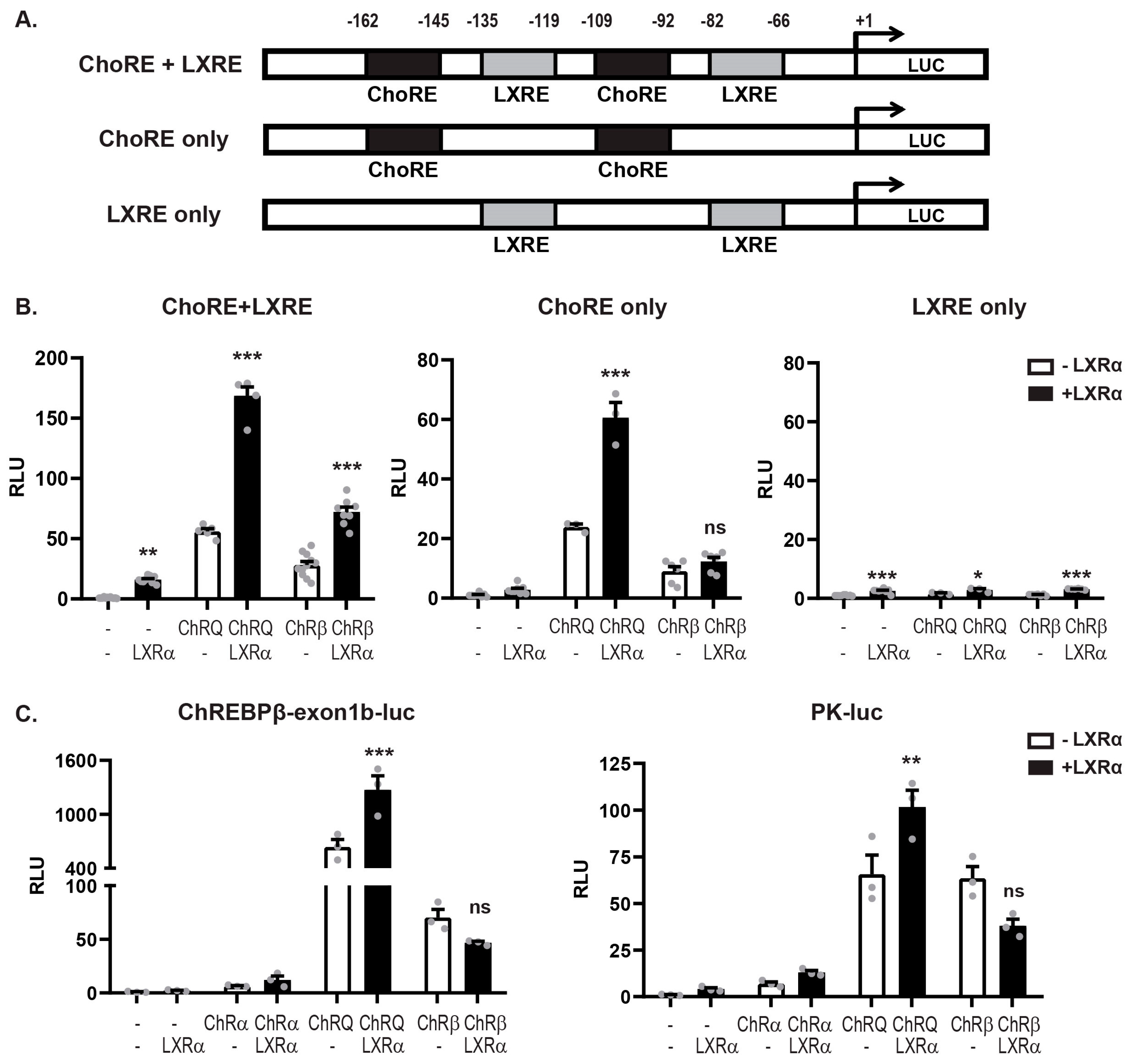
3.3. LXRα:ChREBPα Co-Activation Requires Functional ChoREs But Not LXREs
3.4. LXRα and ChREBPα Interact via Key Activation Domains
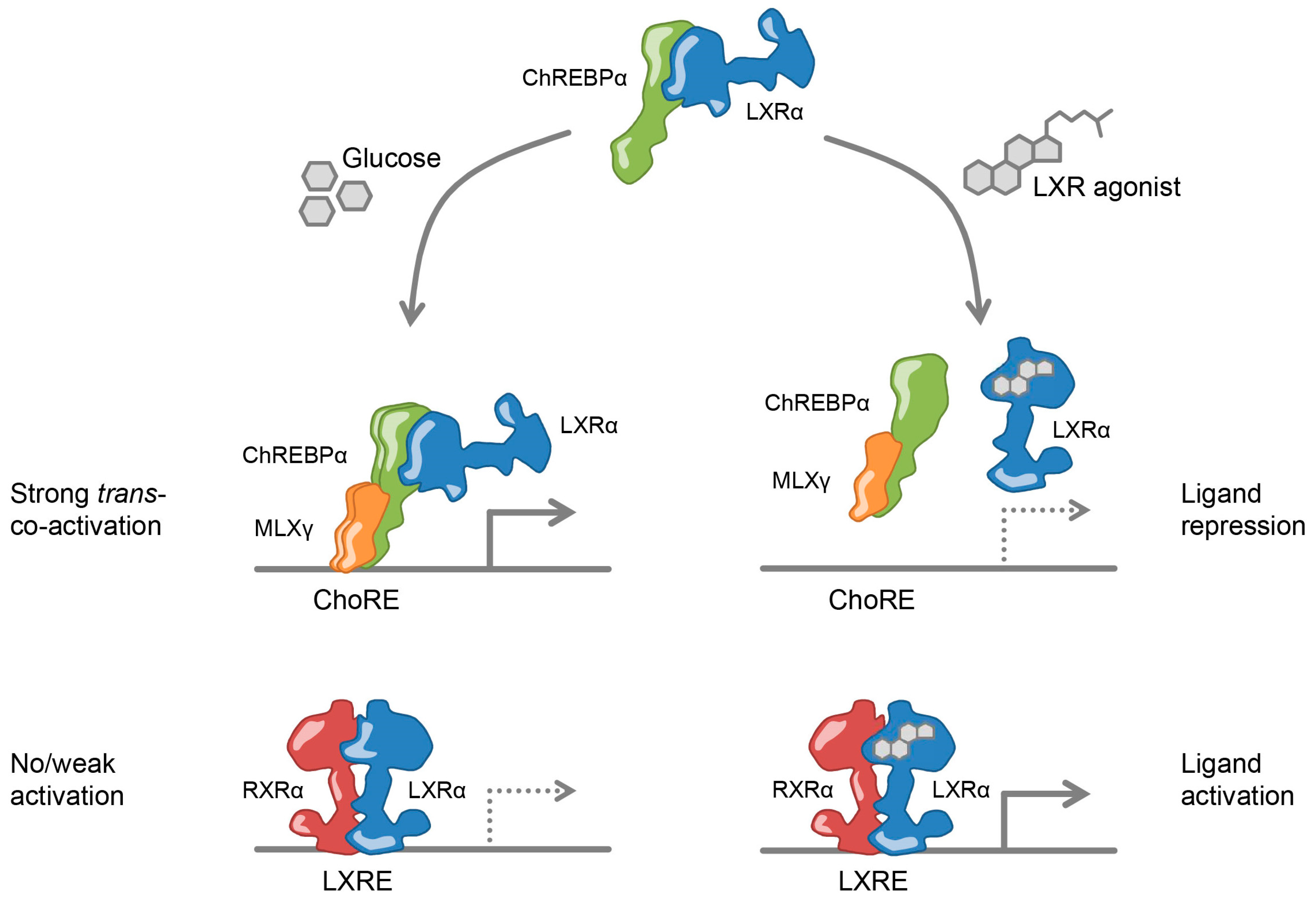
3.5. Ligand-Activated LXRα Represses ChREBPα Activity on ChREBP-Specific Target Genes
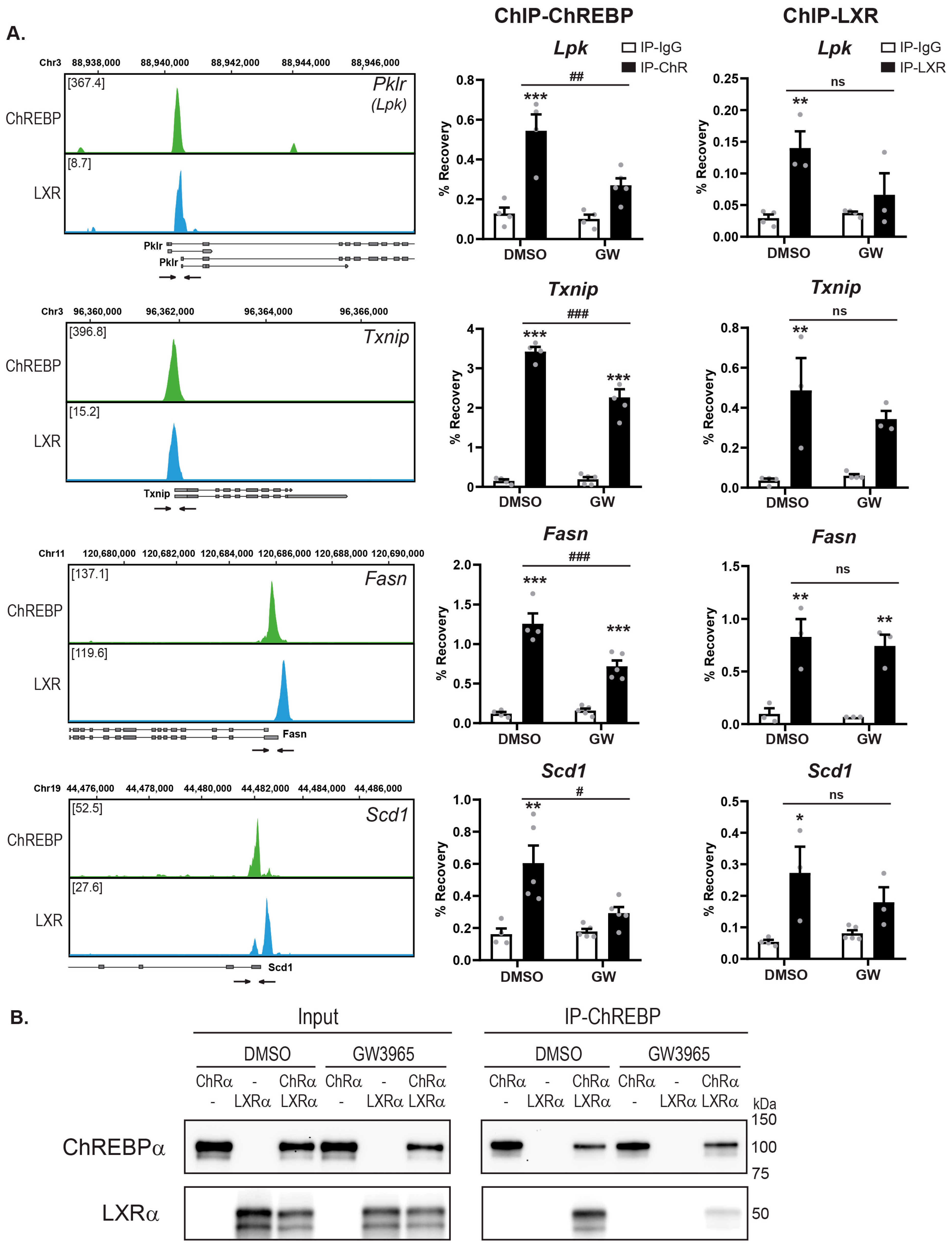
3.6. Ligand-Activated LXRα Reduces ChREBP Binding to Chromatin
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Ballestri, S.; Zona, S.; Targher, G.; Romagnoli, D.; Baldelli, E.; Nascimbeni, F.; Roverato, A.; Guaraldi, G.; Lonardo, A. Nonalcoholic fatty liver disease is associated with an almost twofold increased risk of incident type 2 diabetes and metabolic syndrome. Evidence from a systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2016, 31, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. Nonalcoholic Fatty Liver Disease as a Nexus of Metabolic and Hepatic Diseases. Cell Metab. 2018, 27, 22–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grønning-Wang, L.M.; Bindesbøll, C.; Nebb, H.I. The Role of Liver X Receptor in Hepatic de novo Lipogenesis and Cross-Talk with Insulin and Glucose Signaling. In Lipid Metabolism; Baez, R.V., Ed.; INTECH Open Access: London, UK, 2013; pp. 61–90. [Google Scholar]
- Poupeau, A.; Postic, C. Cross-regulation of hepatic glucose metabolism via ChREBP and nuclear receptors. Biochim. Biophys. Acta 2011, 1812, 995–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakobsson, T.; Treuter, E.; Gustafsson, J.A.; Steffensen, K.R. Liver X receptor biology and pharmacology: New pathways, challenges and opportunities. Trends Pharmacol. Sci. 2012, 33, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Repa, J.J.; Mangelsdorf, D.J. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu. Rev. Cell Dev. Biol. 2000, 16, 459–481. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Breevoort, S.R.; Angdisen, J.; Fu, M.; Schmidt, D.R.; Holmstrom, S.R.; Kliewer, S.A.; Mangelsdorf, D.J.; Schulman, I.G. Liver LXRalpha expression is crucial for whole body cholesterol homeostasis and reverse cholesterol transport in mice. J. Clin. Investig. 2012, 122, 1688–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, J.R.; Tu, H.; Luk, A.; Repa, J.J.; Medina, J.C.; Li, L.; Schwendner, S.; Wang, S.; Thoolen, M.; Mangelsdorf, D.J.; et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000, 14, 2831–2838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laffitte, B.A.; Chao, L.C.; Li, J.; Walczak, R.; Hummasti, S.; Joseph, S.B.; Castrillo, A.; Wilpitz, D.C.; Mangelsdorf, D.J.; Collins, J.L.; et al. Activation of liver X receptor improves glucose tolerance through coordinate regulation of glucose metabolism in liver and adipose tissue. Proc. Natl. Acad. Sci. USA 2003, 100, 5419–5424. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, N.; Bensinger, S.J.; Hong, C.; Beceiro, S.; Bradley, M.N.; Zelcer, N.; Deniz, J.; Ramirez, C.; Diaz, M.; Gallardo, G.; et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 2009, 31, 245–258. [Google Scholar] [CrossRef] [Green Version]
- Willy, P.J.; Umesono, K.; Ong, E.S.; Evans, R.M.; Heyman, R.A.; Mangelsdorf, D.J. LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev. 1995, 9, 1033–1045. [Google Scholar] [CrossRef] [Green Version]
- Janowski, B.A.; Willy, P.J.; Devi, T.R.; Falck, J.R.; Mangelsdorf, D.J. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature 1996, 383, 728–731. [Google Scholar] [CrossRef] [PubMed]
- Svensson, S.; Ostberg, T.; Jacobsson, M.; Norstrom, C.; Stefansson, K.; Hallen, D.; Johansson, I.C.; Zachrisson, K.; Ogg, D.; Jendeberg, L. Crystal structure of the heterodimeric complex of LXRalpha and RXRbeta ligand-binding domains in a fully agonistic conformation. EMBO J. 2003, 22, 4625–4633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.; Li, Y. Structural and functional insights into nuclear receptor signaling. Adv. Drug Deliv. Rev. 2010, 62, 1218–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Li, S.; Wu, J.; Xia, C.; Lala, D.S. Liver X receptors interact with corepressors to regulate gene expression. Mol. Endocrinol. 2003, 17, 1019–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindesboll, C.; Fan, Q.; Norgaard, R.C.; MacPherson, L.; Ruan, H.B.; Wu, J.; Pedersen, T.A.; Steffensen, K.R.; Yang, X.; Matthews, J.; et al. Liver X receptor regulates hepatic nuclear O-GlcNAc signaling and carbohydrate responsive element-binding protein activity. J. Lipid Res. 2015, 56, 771–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Liang, G.; Ou, J.; Goldstein, J.L.; Brown, M.S. Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proc. Natl. Acad. Sci. USA 2004, 101, 11245–11250. [Google Scholar] [CrossRef] [Green Version]
- Anthonisen, E.H.; Berven, L.; Holm, S.; Nygard, M.; Nebb, H.I.; Gronning-Wang, L.M. Nuclear receptor liver X receptor is O-GlcNAc-modified in response to glucose. J. Biol. Chem. 2010, 285, 1607–1615. [Google Scholar] [CrossRef] [Green Version]
- Cha, J.Y.; Repa, J.J. The liver X receptor (LXR) and hepatic lipogenesis. The carbohydrate-response element-binding protein is a target gene of LXR. J. Biol. Chem. 2007, 282, 743–751. [Google Scholar] [CrossRef] [Green Version]
- Repa, J.J.; Liang, G.; Ou, J.; Bashmakov, Y.; Lobaccaro, J.M.; Shimomura, I.; Shan, B.; Brown, M.S.; Goldstein, J.L.; Mangelsdorf, D.J. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. 2000, 14, 2819–2830. [Google Scholar] [CrossRef] [Green Version]
- Filhoulaud, G.; Guilmeau, S.; Dentin, R.; Girard, J.; Postic, C. Novel insights into ChREBP regulation and function. Trends Endocrinol. Metab. 2013, 24, 257–268. [Google Scholar] [CrossRef]
- Talukdar, S.; Hillgartner, F.B. The mechanism mediating the activation of acetyl-coenzyme A carboxylase-alpha gene transcription by the liver X receptor agonist T0-901317. J. Lipid Res. 2006, 47, 2451–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitro, N.; Mak, P.A.; Vargas, L.; Godio, C.; Hampton, E.; Molteni, V.; Kreusch, A.; Saez, E. The nuclear receptor LXR is a glucose sensor. Nature 2007, 445, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.L.; Fivush, A.M.; Watson, M.A.; Galardi, C.M.; Lewis, M.C.; Moore, L.B.; Parks, D.J.; Wilson, J.G.; Tippin, T.K.; Binz, J.G.; et al. Identification of a nonsteroidal liver X receptor agonist through parallel array synthesis of tertiary amines. J. Med. Chem. 2002, 45, 1963–1966. [Google Scholar] [CrossRef] [PubMed]
- Fievet, C.; Staels, B. Liver X receptor modulators: Effects on lipid metabolism and potential use in the treatment of atherosclerosis. Biochem. Pharmacol. 2009, 77, 1316–1327. [Google Scholar] [CrossRef] [Green Version]
- Fessler, M.B. The challenges and promise of targeting the Liver X Receptors for treatment of inflammatory disease. Pharmacol. Ther. 2018, 181, 1–12. [Google Scholar] [CrossRef]
- Yamashita, H.; Takenoshita, M.; Sakurai, M.; Bruick, R.K.; Henzel, W.J.; Shillinglaw, W.; Arnot, D.; Uyeda, K. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc. Natl. Acad. Sci. USA 2001, 98, 9116–9121. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Sham, Y.Y.; Walters, K.J.; Towle, H.C. A critical role for the loop region of the basic helix-loop-helix/leucine zipper protein Mlx in DNA binding and glucose-regulated transcription. Nucleic Acids Res. 2007, 35, 35–44. [Google Scholar] [CrossRef]
- Ma, L.; Tsatsos, N.G.; Towle, H.C. Direct role of ChREBP.Mlx in regulating hepatic glucose-responsive genes. J. Biol. Chem. 2005, 280, 12019–12027. [Google Scholar] [CrossRef] [Green Version]
- Shih, H.M.; Liu, Z.; Towle, H.C. Two CACGTG motifs with proper spacing dictate the carbohydrate regulation of hepatic gene transcription. J. Biol. Chem. 1995, 270, 21991–21997. [Google Scholar] [CrossRef] [Green Version]
- Li, M.V.; Chang, B.; Imamura, M.; Poungvarin, N.; Chan, L. Glucose-dependent transcriptional regulation by an evolutionarily conserved glucose-sensing module. Diabetes 2006, 55, 1179–1189. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.N.; O’Callaghan, B.L.; Towle, H.C. Activation and repression of glucose-stimulated ChREBP requires the concerted action of multiple domains within the MondoA conserved region. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E665–E674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFerrin, L.G.; Atchley, W.R. A novel N-terminal domain may dictate the glucose response of Mondo proteins. PLoS ONE 2012, 7, e34803. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Takenoshita, M.; Kabashima, T.; Uyeda, K. Glucose and cAMP regulate the L-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc. Natl. Acad. Sci. USA 2001, 98, 13710–13715. [Google Scholar] [CrossRef] [Green Version]
- Kabashima, T.; Kawaguchi, T.; Wadzinski, B.E.; Uyeda, K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc. Natl. Acad. Sci. USA 2003, 100, 5107–5112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dentin, R.; Tomas-Cobos, L.; Foufelle, F.; Leopold, J.; Girard, J.; Postic, C.; Ferre, P. Glucose 6-phosphate, rather than xylulose 5-phosphate, is required for the activation of ChREBP in response to glucose in the liver. J. Hepatol. 2012, 56, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.A.; Peroni, O.D.; Villoria, J.; Schon, M.R.; Abumrad, N.A.; Bluher, M.; Klein, S.; Kahn, B.B. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 2012, 484, 333–338. [Google Scholar] [CrossRef] [Green Version]
- Sakiyama, H.; Fujiwara, N.; Noguchi, T.; Eguchi, H.; Yoshihara, D.; Uyeda, K.; Suzuki, K. The role of O-linked GlcNAc modification on the glucose response of ChREBP. Biochem. Biophys. Res. Commun. 2010, 402, 784–789. [Google Scholar] [CrossRef]
- Guinez, C.; Filhoulaud, G.; Rayah-Benhamed, F.; Marmier, S.; Dubuquoy, C.; Dentin, R.; Moldes, M.; Burnol, A.F.; Yang, X.; Lefebvre, T.; et al. O-GlcNAcylation increases ChREBP protein content and transcriptional activity in the liver. Diabetes 2011, 60, 1399–1413. [Google Scholar] [CrossRef] [Green Version]
- Fan, Q.; Norgaard, R.C.; Bindesboll, C.; Lucas, C.; Dalen, K.T.; Babaie, E.; Itkonen, H.M.; Matthews, J.; Nebb, H.I.; Gronning-Wang, L.M. LXRalpha Regulates Hepatic ChREBPalpha Activity and Lipogenesis upon Glucose, but Not Fructose Feeding in Mice. Nutrients 2017, 9, 678. [Google Scholar] [CrossRef] [Green Version]
- Ducheix, S.; Montagner, A.; Polizzi, A.; Lasserre, F.; Regnier, M.; Marmugi, A.; Benhamed, F.; Bertrand-Michel, J.; Mselli-Lakhal, L.; Loiseau, N.; et al. Dietary oleic acid regulates hepatic lipogenesis through a liver X receptor-dependent signaling. PLoS ONE 2017, 12, e0181393. [Google Scholar] [CrossRef] [Green Version]
- Poungvarin, N.; Chang, B.; Imamura, M.; Chen, J.; Moolsuwan, K.; Sae-Lee, C.; Li, W.; Chan, L. Genome-Wide Analysis of ChREBP Binding Sites on Male Mouse Liver and White Adipose Chromatin. Endocrinology 2015, 156, 1982–1994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boergesen, M.; Pedersen, T.A.; Gross, B.; van Heeringen, S.J.; Hagenbeek, D.; Bindesboll, C.; Caron, S.; Lalloyer, F.; Steffensen, K.R.; Nebb, H.I.; et al. Genome-wide profiling of liver X receptor, retinoid X receptor, and peroxisome proliferator-activated receptor alpha in mouse liver reveals extensive sharing of binding sites. Mol. Cell. Biol. 2012, 32, 852–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amemiya, H.M.; Kundaje, A.; Boyle, A.P. The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci. Rep. 2019, 9, 9354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furio-Tari, P.; Conesa, A.; Tarazona, S. RGmatch: Matching genomic regions to proximal genes in omics data integration. BMC Bioinformatics 2016, 17, 427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandve, G.K.; Gundersen, S.; Rydbeck, H.; Glad, I.K.; Holden, L.; Holden, M.; Liestol, K.; Clancy, T.; Ferkingstad, E.; Johansen, M.; et al. The Genomic HyperBrowser: Inferential genomics at the sequence level. Genome Biol. 2010, 11, R121. [Google Scholar] [CrossRef] [Green Version]
- Alberti, S.; Schuster, G.; Parini, P.; Feltkamp, D.; Diczfalusy, U.; Rudling, M.; Angelin, B.; Bjorkhem, I.; Pettersson, S.; Gustafsson, J.A. Hepatic cholesterol metabolism and resistance to dietary cholesterol in LXRbeta-deficient mice. J. Clin. Investig. 2001, 107, 565–573. [Google Scholar] [CrossRef] [Green Version]
- Schuster, G.U.; Parini, P.; Wang, L.; Alberti, S.; Steffensen, K.R.; Hansson, G.K.; Angelin, B.; Gustafsson, J.A. Accumulation of foam cells in liver X receptor-deficient mice. Circulation 2002, 106, 1147–1153. [Google Scholar] [CrossRef] [Green Version]
- Faul, F.; Erdfelder, E.; Lang, A.G.; Buchner, A. G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods 2007, 39, 175–191. [Google Scholar] [CrossRef]
- Arnesen, H.; Haj-Yasein, N.N.; Tungen, J.E.; Soedling, H.; Matthews, J.; Paulsen, S.M.; Nebb, H.I.; Sylte, I.; Hansen, T.V.; Saether, T. Molecular modelling, synthesis, and biological evaluations of a 3,5-disubstituted isoxazole fatty acid analogue as a PPARalpha-selective agonist. Bioorg. Med. Chem. 2019, 27, 4059–4068. [Google Scholar] [CrossRef]
- Nakabayashi, H.; Taketa, K.; Miyano, K.; Yamane, T.; Sato, J. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res. 1982, 42, 3858–3863. [Google Scholar]
- Weedon-Fekjaer, M.S.; Dalen, K.T.; Solaas, K.; Staff, A.C.; Duttaroy, A.K.; Nebb, H.I. Activation of LXR increases acyl-CoA synthetase activity through direct regulation of ACSL3 in human placental trophoblast cells. J. Lipid Res. 2010, 51, 1886–1896. [Google Scholar] [CrossRef] [Green Version]
- Thompson, K.S.; Towle, H.C. Localization of the carbohydrate response element of the rat L-type pyruvate kinase gene. J. Biol. Chem. 1991, 266, 8679–8682. [Google Scholar]
- Collier, J.J.; Zhang, P.; Pedersen, K.B.; Burke, S.J.; Haycock, J.W.; Scott, D.K. c-Myc and ChREBP regulate glucose-mediated expression of the L-type pyruvate kinase gene in INS-1-derived 832/13 cells. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E48–E56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshikawa, T.; Shimano, H.; Amemiya-Kudo, M.; Yahagi, N.; Hasty, A.H.; Matsuzaka, T.; Okazaki, H.; Tamura, Y.; Iizuka, Y.; Ohashi, K.; et al. Identification of liver X receptor-retinoid X receptor as an activator of the sterol regulatory element-binding protein 1c gene promoter. Molecul. Cell. Biol. 2001, 21, 2991–3000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindesbøll, C.; Grønning-Wang, L.M. Liver X receptors connect nuclear O-GlcNAc signaling to hepatic glucose utilization and lipogenesis. Recept. Clin. Investig. 2015, 2, e897. [Google Scholar]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herwig, R.; Hardt, C.; Lienhard, M.; Kamburov, A. Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat. Protoc. 2016, 11, 1889–1907. [Google Scholar] [CrossRef] [PubMed]
- Koudritsky, M.; Domany, E. Positional distribution of human transcription factor binding sites. Nucleic Acids Res. 2008, 36, 6795–6805. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Lu, J.; Kulbokas, E.J.; Golub, T.R.; Mootha, V.; Lindblad-Toh, K.; Lander, E.S.; Kellis, M. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature 2005, 434, 338–345. [Google Scholar] [CrossRef]
- Chong, H.K.; Infante, A.M.; Seo, Y.K.; Jeon, T.I.; Zhang, Y.; Edwards, P.A.; Xie, X.; Osborne, T.F. Genome-wide interrogation of hepatic FXR reveals an asymmetric IR-1 motif and synergy with LRH-1. Nucleic Acids Res. 2010, 38, 6007–6017. [Google Scholar] [CrossRef] [Green Version]
- Stender, J.D.; Kim, K.; Charn, T.H.; Komm, B.; Chang, K.C.; Kraus, W.L.; Benner, C.; Glass, C.K.; Katzenellenbogen, B.S. Genome-wide analysis of estrogen receptor alpha DNA binding and tethering mechanisms identifies Runx1 as a novel tethering factor in receptor-mediated transcriptional activation. Mol. Cell. Biol. 2010, 30, 3943–3955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ijssennagger, N.; Janssen, A.W.F.; Milona, A.; Ramos Pittol, J.M.; Hollman, D.A.A.; Mokry, M.; Betzel, B.; Berends, F.J.; Janssen, I.M.; van Mil, S.W.C.; et al. Gene expression profiling in human precision cut liver slices in response to the FXR agonist obeticholic acid. J. Hepatol. 2016, 64, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, S.; Dagestad Rand, K.; Grytten, I.; Ferkingstad, E.; Domanska, D.; Holden, L.; Gheorghe, M.; Mathelier, A.; Glad, I.; Kjetil Sandve, G. Beware the Jaccard: The choice of similarity measure is important and non-trivial in genomic colocalisation analysis. Brief. Bioinform. 2019. [Google Scholar] [CrossRef] [PubMed]
- Caron, S.; Huaman Samanez, C.; Dehondt, H.; Ploton, M.; Briand, O.; Lien, F.; Dorchies, E.; Dumont, J.; Postic, C.; Cariou, B.; et al. Farnesoid X receptor inhibits the transcriptional activity of carbohydrate response element binding protein in human hepatocytes. Mol. Cell. Biol. 2013, 33, 2202–2211. [Google Scholar] [CrossRef] [Green Version]
- Trabelsi, M.S.; Daoudi, M.; Prawitt, J.; Ducastel, S.; Touche, V.; Sayin, S.I.; Perino, A.; Brighton, C.A.; Sebti, Y.; Kluza, J.; et al. Farnesoid X receptor inhibits glucagon-like peptide-1 production by enteroendocrine L cells. Nat. Commun. 2015, 6, 7629. [Google Scholar] [CrossRef] [Green Version]
- Ide, T.; Shimano, H.; Yoshikawa, T.; Yahagi, N.; Amemiya-Kudo, M.; Matsuzaka, T.; Nakakuki, M.; Yatoh, S.; Iizuka, Y.; Tomita, S.; et al. Cross-talk between peroxisome proliferator-activated receptor (PPAR) alpha and liver X receptor (LXR) in nutritional regulation of fatty acid metabolism. II. LXRs suppress lipid degradation gene promoters through inhibition of PPAR signaling. Mol. Endocrinol. 2003, 17, 1255–1267. [Google Scholar] [CrossRef] [Green Version]
- Molvaersmyr, A.K.; Saether, T.; Gilfillan, S.; Lorenzo, P.I.; Kvaloy, H.; Matre, V.; Gabrielsen, O.S. A SUMO-regulated activation function controls synergy of c-Myb through a repressor-activator switch leading to differential p300 recruitment. Nucleic Acids Res. 2010, 38, 4970–4984. [Google Scholar] [CrossRef]
- De Bosscher, K.; Beck, I.M.; Dejager, L.; Bougarne, N.; Gaigneaux, A.; Chateauvieux, S.; Ratman, D.; Bracke, M.; Tavernier, J.; Vanden Berghe, W.; et al. Selective modulation of the glucocorticoid receptor can distinguish between transrepression of NF-kappaB and AP-1. Cell. Mol. Life Sci. 2014, 71, 143–163. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Pascual, G.; Glass, C.K. Peroxisome proliferator-activated receptor gamma-dependent repression of the inducible nitric oxide synthase gene. Mol. Cell. Biol. 2000, 20, 4699–4707. [Google Scholar] [CrossRef] [Green Version]
- Ghisletti, S.; Huang, W.; Ogawa, S.; Pascual, G.; Lin, M.E.; Willson, T.M.; Rosenfeld, M.G.; Glass, C.K. Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARgamma. Mol. Cell 2007, 25, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Ghisletti, S.; Saijo, K.; Gandhi, M.; Aouadi, M.; Tesz, G.J.; Zhang, D.X.; Yao, J.; Czech, M.P.; Goode, B.L.; et al. Coronin 2A mediates actin-dependent de-repression of inflammatory response genes. Nature 2011, 470, 414–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Fang, B.; Emmett, M.J.; Damle, M.; Sun, Z.; Feng, D.; Armour, S.M.; Remsberg, J.R.; Jager, J.; Soccio, R.E.; et al. GENE REGULATION. Discrete functions of nuclear receptor Rev-erbalpha couple metabolism to the clock. Science 2015, 348, 1488–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, S.F.; Larsen, B.D.; Loft, A.; Nielsen, R.; Madsen, J.G.; Mandrup, S. Acute TNF-induced repression of cell identity genes is mediated by NFkappaB-directed redistribution of cofactors from super-enhancers. Genome Res. 2015, 25, 1281–1294. [Google Scholar] [CrossRef] [Green Version]
- Step, S.E.; Lim, H.W.; Marinis, J.M.; Prokesch, A.; Steger, D.J.; You, S.H.; Won, K.J.; Lazar, M.A. Anti-diabetic rosiglitazone remodels the adipocyte transcriptome by redistributing transcription to PPARgamma-driven enhancers. Genes Dev. 2014, 28, 1018–1028. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Shimano, H.; Nakagawa, Y.; Ide, T.; Yahagi, N.; Matsuzaka, T.; Nakakuki, M.; Takahashi, A.; Suzuki, H.; Sone, H.; et al. SREBP-1 interacts with hepatocyte nuclear factor-4 alpha and interferes with PGC-1 recruitment to suppress hepatic gluconeogenic genes. J. Biol. Chem. 2004, 279, 12027–12035. [Google Scholar] [CrossRef] [Green Version]
- Ponugoti, B.; Fang, S.; Kemper, J.K. Functional interaction of hepatic nuclear factor-4 and peroxisome proliferator-activated receptor-gamma coactivator 1alpha in CYP7A1 regulation is inhibited by a key lipogenic activator, sterol regulatory element-binding protein-1c. Mol. Endocrinol. 2007, 21, 2698–2712. [Google Scholar] [CrossRef]
- Flotho, A.; Melchior, F. Sumoylation: A regulatory protein modification in health and disease. Annu. Rev. Biochem. 2013, 82, 357–385. [Google Scholar] [CrossRef]
- Abdul-Wahed, A.; Guilmeau, S.; Postic, C. Sweet Sixteenth for ChREBP: Established Roles and Future Goals. Cell Metab. 2017, 26, 324–341. [Google Scholar] [CrossRef]
- Becares, N.; Gage, M.C.; Pineda-Torra, I. Posttranslational Modifications of Lipid-Activated Nuclear Receptors: Focus on Metabolism. Endocrinology 2017, 158, 213–225. [Google Scholar] [CrossRef] [Green Version]
- Hardiville, S.; Hart, G.W. Nutrient regulation of signaling, transcription, and cell physiology by O-GlcNAcylation. Cell Metab. 2014, 20, 208–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poungvarin, N.; Lee, J.K.; Yechoor, V.K.; Li, M.V.; Assavapokee, T.; Suksaranjit, P.; Thepsongwajja, J.J.; Saha, P.K.; Oka, K.; Chan, L. Carbohydrate response element-binding protein (ChREBP) plays a pivotal role in beta cell glucotoxicity. Diabetologia 2012, 55, 1783–1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, G.; Chen, J.; Xu, G.; Shalev, A. Islet ChREBP-beta is increased in diabetes and controls ChREBP-alpha and glucose-induced gene expression via a negative feedback loop. Mol. Metab. 2016, 5, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Siersbaek, R.; Nielsen, R.; John, S.; Sung, M.H.; Baek, S.; Loft, A.; Hager, G.L.; Mandrup, S. Extensive chromatin remodelling and establishment of transcription factor ‘hotspots’ during early adipogenesis. EMBO J. 2011, 30, 1459–1472. [Google Scholar] [CrossRef] [Green Version]
- Mora, A.; Sandve, G.K.; Gabrielsen, O.S.; Eskeland, R. In the loop: Promoter-enhancer interactions and bioinformatics. Brief. Bioinform. 2016, 17, 980–995. [Google Scholar] [CrossRef] [Green Version]
- West, A.G.; Gaszner, M.; Felsenfeld, G. Insulators: Many functions, many mechanisms. Genes Dev. 2002, 16, 271–288. [Google Scholar] [CrossRef] [Green Version]
- Siersbaek, R.; Madsen, J.G.S.; Javierre, B.M.; Nielsen, R.; Bagge, E.K.; Cairns, J.; Wingett, S.W.; Traynor, S.; Spivakov, M.; Fraser, P.; et al. Dynamic Rewiring of Promoter-Anchored Chromatin Loops during Adipocyte Differentiation. Mol. Cell 2017, 66, 420–435. [Google Scholar] [CrossRef] [Green Version]
- Paulsen, J.; Liyakat Ali, T.M.; Nekrasov, M.; Delbarre, E.; Baudement, M.O.; Kurscheid, S.; Tremethick, D.; Collas, P. Long-range interactions between topologically associating domains shape the four-dimensional genome during differentiation. Nat. Genet. 2019, 51, 835–843. [Google Scholar] [CrossRef]
- Vakoc, C.R.; Letting, D.L.; Gheldof, N.; Sawado, T.; Bender, M.A.; Groudine, M.; Weiss, M.J.; Dekker, J.; Blobel, G.A. Proximity among distant regulatory elements at the beta-globin locus requires GATA-1 and FOG-1. Mol. Cell 2005, 17, 453–462. [Google Scholar] [CrossRef]
- Dekker, J.; Marti-Renom, M.A.; Mirny, L.A. Exploring the three-dimensional organization of genomes: Interpreting chromatin interaction data. Nat. Rev. Genet. 2013, 14, 390–403. [Google Scholar] [CrossRef] [Green Version]
- Naik, S.U.; Wang, X.; Da Silva, J.S.; Jaye, M.; Macphee, C.H.; Reilly, M.P.; Billheimer, J.T.; Rothblat, G.H.; Rader, D.J. Pharmacological activation of liver X receptors promotes reverse cholesterol transport in vivo. Circulation 2006, 113, 90–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, R.; Ou, Z.; Ruan, X.; Gong, J. Role of liver X receptors in cholesterol efflux and inflammatory signaling (review). Mol. Med. Rep. 2012, 5, 895–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furbee, J.W., Jr.; Francone, O.; Parks, J.S. In vivo contribution of LCAT to apolipoprotein B lipoprotein cholesteryl esters in LDL receptor and apolipoprotein E knockout mice. J. Lipid Res. 2002, 43, 428–437. [Google Scholar] [PubMed]
- Xie, C.; Woollett, L.A.; Turley, S.D.; Dietschy, J.M. Fatty acids differentially regulate hepatic cholesteryl ester formation and incorporation into lipoproteins in the liver of the mouse. J. Lipid Res. 2002, 43, 1508–1519. [Google Scholar] [CrossRef] [Green Version]
- Repa, J.J.; Turley, S.D.; Lobaccaro, J.A.; Medina, J.; Li, L.; Lustig, K.; Shan, B.; Heyman, R.A.; Dietschy, J.M.; Mangelsdorf, D.J. Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers. Science 2000, 289, 1524–1529. [Google Scholar] [CrossRef]
- Moore, T.W.; Mayne, C.G.; Katzenellenbogen, J.A. Minireview: Not picking pockets: Nuclear receptor alternate-site modulators (NRAMs). Mol. Endocrinol. 2010, 24, 683–695. [Google Scholar] [CrossRef] [Green Version]
- Leung, C.H.; Chan, D.S.; Ma, V.P.; Ma, D.L. DNA-binding small molecules as inhibitors of transcription factors. Med. Res. Rev. 2013, 33, 823–846. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway Name | Reactome ID | Candidates | p-Value | LXR | ChREBP |
|---|---|---|---|---|---|
| Metabolism | 1430728 | 302 (15.4%) | 3.6 × 10−27 | × | × |
| Metabolism of lipids | 556833 | 113 (17.1%) | 4.6 × 10−13 | × | × |
| Metabolism of amino acids and Derivatives | 71291 | 61 (18.0%) | 2.2 × 10−8 | ||
| Glucose metabolism | 70326 | 24 (26.7%) | 3.9 × 10−7 | × | × |
| γ-carboxylation, transport and N- terminal cleavage of proteins | 159854 | 8 (72.7%) | 3.9 × 10−7 | ||
| Formation of Fibrin Clot | 140877 | 14 (35.9%) | 2.2 × 10−6 | ||
| Fatty acid metabolism | 8978868 | 36 (19.5%) | 2.7 × 10−6 | × | × |
| γ-carboxylation of protein precursors | 159740 | 7 (70.0%) | 3.3 × 10−6 | ||
| Removal of N-terminal propeptides from γ-carboxylated proteins | 159782 | 7 (70.0%) | 3.3 × 10−6 | ||
| Gluconeogenesis | 70263 | 12 (35.3%) | 1.5 × 10−5 | × | × |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Q.; Nørgaard, R.C.; Grytten, I.; Ness, C.M.; Lucas, C.; Vekterud, K.; Soedling, H.; Matthews, J.; Lemma, R.B.; Gabrielsen, O.S.; et al. LXRα Regulates ChREBPα Transactivity in a Target Gene-Specific Manner through an Agonist-Modulated LBD-LID Interaction. Cells 2020, 9, 1214. https://doi.org/10.3390/cells9051214
Fan Q, Nørgaard RC, Grytten I, Ness CM, Lucas C, Vekterud K, Soedling H, Matthews J, Lemma RB, Gabrielsen OS, et al. LXRα Regulates ChREBPα Transactivity in a Target Gene-Specific Manner through an Agonist-Modulated LBD-LID Interaction. Cells. 2020; 9(5):1214. https://doi.org/10.3390/cells9051214
Chicago/Turabian StyleFan, Qiong, Rikke Christine Nørgaard, Ivar Grytten, Cecilie Maria Ness, Christin Lucas, Kristin Vekterud, Helen Soedling, Jason Matthews, Roza Berhanu Lemma, Odd Stokke Gabrielsen, and et al. 2020. "LXRα Regulates ChREBPα Transactivity in a Target Gene-Specific Manner through an Agonist-Modulated LBD-LID Interaction" Cells 9, no. 5: 1214. https://doi.org/10.3390/cells9051214