The Detrimental Action of Adenosine on Glutamate-Induced Cytotoxicity in PC12 Cells Can Be Shifted towards a Neuroprotective Role through A1AR Positive Allosteric Modulation

 , ,
, ,  ,
,
Abstract
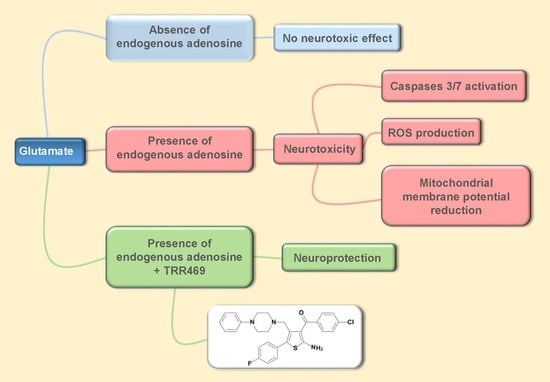
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. Apoptosis Evaluation
2.4. Activation of Caspases 3/7
2.5. ROS Production Evaluation
2.6. Mitochondrial Membrane Potential Evaluation
2.7. Statistical Analysis
3. Results
3.1. Adenosine Is Necessary for Glutamate Cytotoxic Effect in PC12 Cells
3.2. The Detrimental Action of Endogenous Adenosine Is Mitigated by Blocking A2A and A2BARs and Further Exacerbated by Blocking A1ARs
3.3. Enhancing the Effect of Endogenous Adenosine on A1ARs through PAM TRR469 Abrogates the Cytotoxic Action of Glutamate
3.4. TRR469 Prevented Glutamate-Induced Caspase 3/7 Activation
3.5. TRR469 Prevented ROS Production and Mitochondrial Membrane Potential Reduction Induced by Glutamate
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflug. Arch. Eur. J. Physiol. 2010, 460, 525–542. [Google Scholar] [CrossRef] [PubMed]
- Gladstone, D.J.; Black, S.E.; Hakim, A.M. Toward Wisdom From Failure: Lessons From Neuroprotective Stroke Trials and New Therapeutic Directions. Stroke 2002, 33, 2123–2136. [Google Scholar] [CrossRef] [PubMed]
- Muir, K. Glutamate-based therapeutic approaches: clinical trials with NMDA antagonists. Curr. Opin. Pharmacol. 2006, 6, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidou, C.; Turski, L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 2002, 1, 383–386. [Google Scholar] [CrossRef]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef] [Green Version]
- Albrecht, P.; Lewerenz, J.; Dittmer, S.; Noack, R.; Maher, P.; Methner, A. Mechanisms of oxidative glutamate toxicity: the glutamate/cystine antiporter system xc- as a neuroprotective drug target. CNS Neurol. Disord. Drug Targets 2010, 9, 373–382. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Merighi, S.; Varani, K. Adenosine as a Multi-Signalling Guardian Angel in Human Diseases: When, Where and How Does it Exert its Protective Effects? Trends Pharmacol. Sci. 2016, 37, 419–434. [Google Scholar] [CrossRef]
- Cunha, R.A. How does adenosine control neuronal dysfunction and neurodegeneration? J. Neurochem. 2016, 139, 1019–1055. [Google Scholar] [CrossRef]
- Serpa, A.; Pinto, I.; Bernardino, L.; Cascalheira, J.F. Combined neuroprotective action of adenosine A1 and cannabinoid CB1 receptors against NMDA-induced excitotoxicity in the hippocampus. Neurochem. Int. 2015, 87, 106–109. [Google Scholar] [CrossRef]
- Hartwick, A.T.E.; Lalonde, M.R.; Barnes, S.; Baldridge, W.H. Adenosine A1-receptor modulation of glutamate-induced calcium influx in rat retinal ganglion cells. Invest. Ophthalmol. Vis. Sci. 2004, 45, 3740–3748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martire, A.; Lambertucci, C.; Pepponi, R.; Ferrante, A.; Benati, N.; Buccioni, M.; Dal Ben, D.; Marucci, G.; Klotz, K.-N.; Volpini, R.; et al. Neuroprotective potential of adenosine A1 receptor partial agonists in experimental models of cerebral ischemia. J. Neurochem. 2019, 149, 211–230. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.-S.-W.; Cao, Z.-P.; Shang, Y.-J.; Liu, Q.-Y.; Wu, B.-Y.; Liu, W.-X.; Li, C.-H. Neuroprotection of cordycepin in NMDA-induced excitotoxicity by modulating adenosine A1 receptors. Eur. J. Pharmacol. 2019, 853, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pathological overproduction: the bad side of adenosine. Br. J. Pharmacol. 2017, 174, 1945–1960. [Google Scholar] [CrossRef] [Green Version]
- Pedata, F.; Pugliese, A.M.; Coppi, E.; Dettori, I.; Maraula, G.; Cellai, L.; Melani, A. Adenosine A2A Receptors Modulate Acute Injury and Neuroinflammation in Brain Ischemia. Mediat. Inflamm. 2014, 2014, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Pedata, F.; Dettori, I.; Coppi, E.; Melani, A.; Fusco, I.; Corradetti, R.; Pugliese, A.M. Purinergic signalling in brain ischemia. Neuropharmacology 2016, 104, 105–130. [Google Scholar] [CrossRef]
- Fusco, I.; Ugolini, F.; Lana, D.; Coppi, E.; Dettori, I.; Gaviano, L.; Nosi, D.; Cherchi, F.; Pedata, F.; Giovannini, M.G.; et al. The Selective Antagonism of Adenosine A2B Receptors Reduces the Synaptic Failure and Neuronal Death Induced by Oxygen and Glucose Deprivation in Rat CA1 Hippocampus in Vitro. Front. Pharmacol. 2018, 9, 399. [Google Scholar] [CrossRef] [Green Version]
- Galvao, J.; Elvas, F.; Martins, T.; Cordeiro, M.F.; Ambrósio, A.F.; Santiago, A.R. Adenosine A3 receptor activation is neuroprotective against retinal neurodegeneration. Exp. Eye Res. 2015, 140, 65–74. [Google Scholar] [CrossRef]
- Zhang, M.; Hu, H.; Zhang, X.; Lu, W.; Lim, J.; Eysteinsson, T.; Jacobson, K.A.; Laties, A.M.; Mitchell, C.H. The A3 adenosine receptor attenuates the calcium rise triggered by NMDA receptors in retinal ganglion cells. Neurochem. Int. 2010, 56, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Kiesman, W.F.; Elzein, E.; Zablocki, J. A1 adenosine receptor antagonists, agonists, and allosteric enhancers. Handb. Exp. Pharm. 2009, 25–58. [Google Scholar]
- Chen, J.-F.; Eltzschig, H.K.; Fredholm, B.B. Adenosine receptors as drug targets--what are the challenges? Nat. Rev. Drug Discov. 2013, 12, 265–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romagnoli, R.; Baraldi, P.G.; Carrion, M.D.; Cara, C.L.; Cruz-Lopez, O.; Salvador, M.K.; Preti, D.; Tabrizi, M.A.; Shryock, J.C.; Moorman, A.R.; et al. Structure-activity relationships of 2-amino-3-aroyl-4-[(4-arylpiperazin-1-yl)methyl]thiophenes. Part 2: Probing the influence of diverse substituents at the phenyl of the arylpiperazine moiety on allosteric enhancer activity at the A1 adenosine receptor. Bioorg. Med. Chem. 2012, 20, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, R.; Baraldi, P.G.; Carrion, M.D.; Cara, C.L.; Cruz-Lopez, O.; Salvador, M.K.; Preti, D.; Tabrizi, M.A.; Moorman, A.R.; Vincenzi, F.; et al. Synthesis and biological evaluation of 2-amino-3-(4-chlorobenzoyl)-4-[(4-arylpiperazin-1-yl)methyl]-5-substituted-thiophenes. effect of the 5-modification on allosteric enhancer activity at the A1 adenosine receptor. J. Med. Chem. 2012, 55, 7719–7735. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, R.; Baraldi, P.G.; Carrion, M.D.; Lopez Cara, C.; Kimatrai Salvador, M.; Preti, D.; Aghazadeh Tabrizi, M.; Moorman, A.R.; Vincenzi, F.; Borea, P.A.; et al. Synthesis and biological effects of novel 2-amino-3-(4-chlorobenzoyl)-4-substituted thiophenes as allosteric enhancers of the A1 adenosine receptor. Eur. J. Med. Chem. 2013, 67, 409–427. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; IJzerman, A.P.; Massink, A.; Cruz-Lopez, O.; Lopez-Cara, L.C.; Saponaro, G.; Preti, D.; Aghazadeh Tabrizi, M.; Baraldi, S.; et al. Synthesis and biological evaluation of novel allosteric enhancers of the A1 adenosine receptor based on 2-amino-3-(4′-chlorobenzoyl)-4-substituted-5-arylethynyl thiophene. J. Med. Chem. 2014, 57, 7673–7686. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Carrion, M.D.; Cruz-Lopez, O.; Cara, C.L.; Saponaro, G.; Preti, D.; Tabrizi, M.A.; Baraldi, S.; Moorman, A.R.; et al. Synthesis and biological evaluation of novel 2-amino-3-aroyl-4-neopentyl-5-substituted thiophene derivatives as allosteric enhancers of the A₁ adenosine receptor. Bioorg. Med. Chem. 2014, 22, 148–166. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Lopez-Cara, C.; Cruz-Lopez, O.; Moorman, A.R.; Massink, A.; IJzerman, A.P.; Vincenzi, F.; Borea, P.A.; Varani, K. Synthesis and biological evaluation of a new series of 2-amino-3-aroyl thiophene derivatives as agonist allosteric modulators of the A1 adenosine receptor. A position-dependent effect study. Eur. J. Med. Chem. 2015, 101, 185–204. [Google Scholar] [CrossRef]
- Vincenzi, F.; Targa, M.; Romagnoli, R.; Merighi, S.; Gessi, S.; Baraldi, P.G.; Borea, P.A.; Varani, K. TRR469, a potent A(1) adenosine receptor allosteric modulator, exhibits anti-nociceptive properties in acute and neuropathic pain models in mice. Neuropharmacology 2014, 81, 6–14. [Google Scholar] [CrossRef]
- Vincenzi, F.; Ravani, A.; Pasquini, S.; Merighi, S.; Gessi, S.; Romagnoli, R.; Baraldi, P.G.; Borea, P.A.; Varani, K. Positive allosteric modulation of A1 adenosine receptors as a novel and promising therapeutic strategy for anxiety. Neuropharmacology 2016, 111, 283–292. [Google Scholar] [CrossRef]
- Vincenzi, F.; Borea, P.A.; Varani, K. Anxiolytic properties of A1 adenosine receptor PAMs. Oncotarget 2017, 8, 7216–7217. [Google Scholar] [CrossRef] [Green Version]
- Vincenzi, F.; Ravani, A.; Pasquini, S.; Merighi, S.; Gessi, S.; Setti, S.; Cadossi, R.; Borea, P.A.; Varani, K. Pulsed Electromagnetic Field Exposure Reduces Hypoxia and Inflammation Damage in Neuron-Like and Microglial Cells. J. Cell. Physiol. 2017, 232, 1200–1208. [Google Scholar] [CrossRef] [PubMed]
- Kritis, A.A.; Stamoula, E.G.; Paniskaki, K.A.; Vavilis, T.D. Researching glutamate—Induced cytotoxicity in different cell lines: A comparative/collective analysis/study. Front. Cell. Neurosci. 2015, 9, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olloquequi, J.; Cornejo-Córdova, E.; Verdaguer, E.; Soriano, F.X.; Binvignat, O.; Auladell, C.; Camins, A. Excitotoxicity in the pathogenesis of neurological and psychiatric disorders: Therapeutic implications. J. Psychopharmacol. 2018, 32, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Wang, Y.; Qin, Z. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haskó, G.; Szabó, C.; Németh, Z.H.; Kvetan, V.; Pastores, S.M.; Vizi, E.S. Adenosine receptor agonists differentially regulate IL-10, TNF-alpha, and nitric oxide production in RAW 264.7 macrophages and in endotoxemic mice. J. Immunol. 1996, 157, 4634–4640. [Google Scholar] [PubMed]
- Haskó, G.; Kuhel, D.G.; Chen, J.F.; Schwarzschild, M.A.; Deitch, E.A.; Mabley, J.G.; Marton, A.; Szabó, C. Adenosine inhibits IL-12 and TNF-[alpha] production via adenosine A2a receptor-dependent and independent mechanisms. Faseb J. 2000, 14, 2065–2074. [Google Scholar] [CrossRef] [Green Version]
- Varani, K.; Vincenzi, F.; Tosi, A.; Gessi, S.; Casetta, I.; Granieri, G.; Fazio, P.; Leung, E.; MacLennan, S.; Granieri, E.; et al. A2A adenosine receptor overexpression and functionality, as well as TNF-alpha levels, correlate with motor symptoms in Parkinson’s disease. Faseb J. 2010, 24, 587–598. [Google Scholar] [CrossRef]
- Dias, R.B.; Ribeiro, J.A.; Sebastião, A.M. Enhancement of AMPA currents and GluR1 membrane expression through PKA-coupled adenosine A(2A) receptors. Hippocampus 2012, 22, 276–291. [Google Scholar] [CrossRef]
- Mouro, F.M.; Rombo, D.M.; Dias, R.B.; Ribeiro, J.A.; Sebastião, A.M. Adenosine A2A receptors facilitate synaptic NMDA currents in CA1 pyramidal neurons. Br. J. Pharmacol. 2018, 175, 4386–4397. [Google Scholar] [CrossRef] [Green Version]
- Tschammer, N. Allosteric Modulators of the Class A G Protein Coupled Receptors. Adv. Exp. Med. Biol. 2016, 917, 185–207. [Google Scholar]
- Bartuzi, D.; Kaczor, A.A.; Matosiuk, D. Opportunities and Challenges in the Discovery of Allosteric Modulators of GPCRs. Methods Mol. Biol. 2018, 1705, 297–319. [Google Scholar] [PubMed]
- May, L.T.; Leach, K.; Sexton, P.M.; Christopoulos, A. Allosteric modulation of G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 1–51. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-J.; Kang, Y.-J.; Shin, S.-A.; Bak, D.-H.; Lee, J.W.; Lee, K.B.; Yoo, Y.C.; Kim, D.-K.; Lee, B.H.; Kim, D.W.; et al. Phlorofucofuroeckol Improves Glutamate-Induced Neurotoxicity through Modulation of Oxidative Stress-Mediated Mitochondrial Dysfunction in PC12 Cells. PLos ONE 2016, 11, e0163433. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Yuan, X.; Zeng, H.-W.; Liu, R.-H.; Zhang, W.-D. Protective effect of cinnamaldehyde against glutamate-induced oxidative stress and apoptosis in PC12 cells. Eur. J. Pharmacol. 2017, 815, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Bak, D.-H.; Kim, H.D.; Kim, Y.O.; Park, C.G.; Han, S.-Y.; Kim, J.-J. Neuroprotective effects of 20(S)-protopanaxadiol against glutamate-induced mitochondrial dysfunction in PC12 cells. Int. J. Mol. Med. 2016, 37, 378–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vincenzi, F.; Pasquini, S.; Gessi, S.; Merighi, S.; Romagnoli, R.; Borea, P.A.; Varani, K. The Detrimental Action of Adenosine on Glutamate-Induced Cytotoxicity in PC12 Cells Can Be Shifted towards a Neuroprotective Role through A1AR Positive Allosteric Modulation. Cells 2020, 9, 1242. https://doi.org/10.3390/cells9051242
Vincenzi F, Pasquini S, Gessi S, Merighi S, Romagnoli R, Borea PA, Varani K. The Detrimental Action of Adenosine on Glutamate-Induced Cytotoxicity in PC12 Cells Can Be Shifted towards a Neuroprotective Role through A1AR Positive Allosteric Modulation. Cells. 2020; 9(5):1242. https://doi.org/10.3390/cells9051242
Chicago/Turabian StyleVincenzi, Fabrizio, Silvia Pasquini, Stefania Gessi, Stefania Merighi, Romeo Romagnoli, Pier Andrea Borea, and Katia Varani. 2020. "The Detrimental Action of Adenosine on Glutamate-Induced Cytotoxicity in PC12 Cells Can Be Shifted towards a Neuroprotective Role through A1AR Positive Allosteric Modulation" Cells 9, no. 5: 1242. https://doi.org/10.3390/cells9051242