The Complete Mitochondrial Genome Sequences of the Philomycus bilineatus (Stylommatophora: Philomycidae) and Phylogenetic Analysis

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Specimen Collection and DNA Extraction
2.2. Sequence Amplification, Assembly, and Gene Annotation
2.3. Sequence Analysis
2.4. Phylogenetic Analysis
3. Results and Discussion
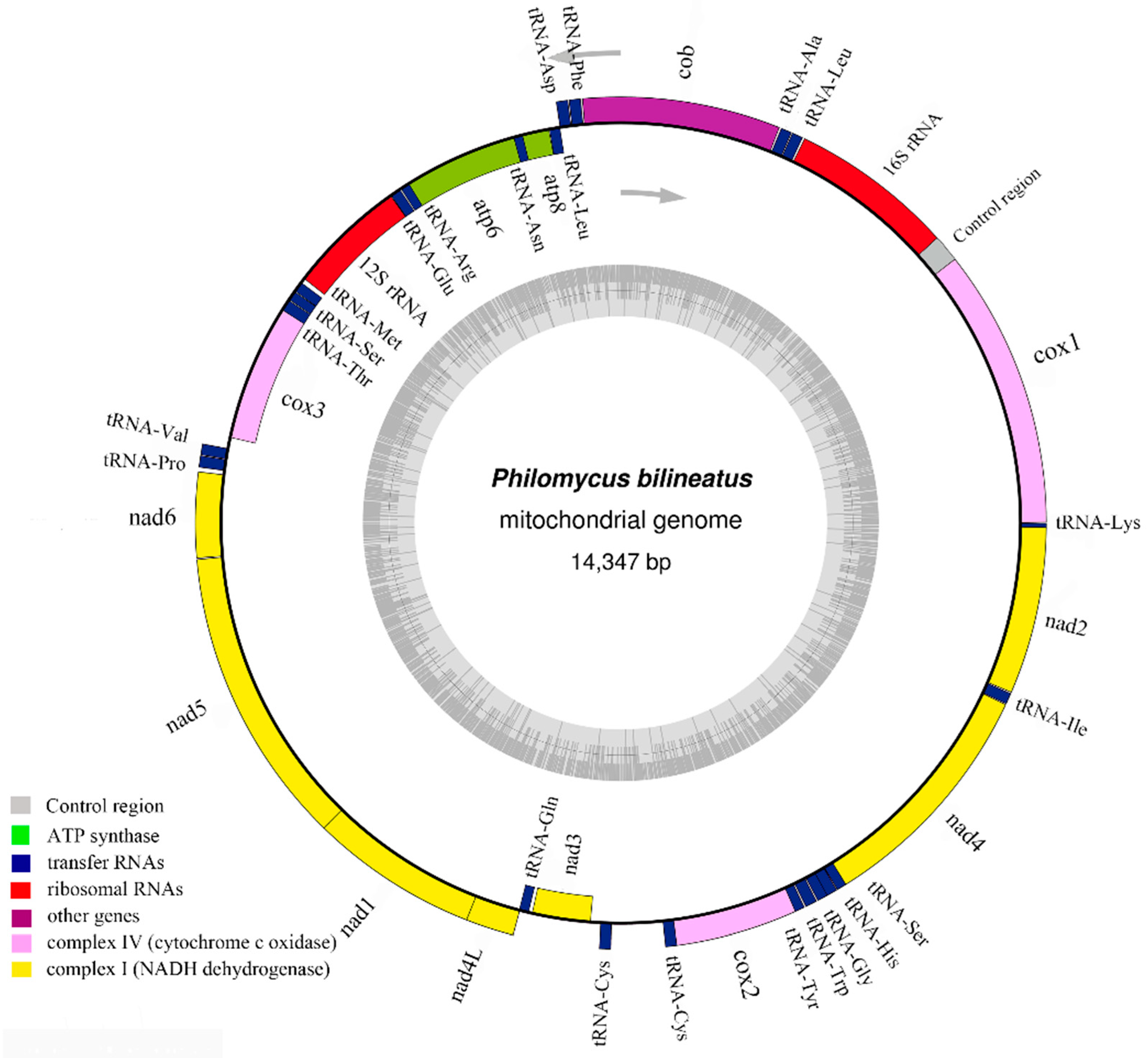
3.1. Mitogenome Sequence Structure Analysis
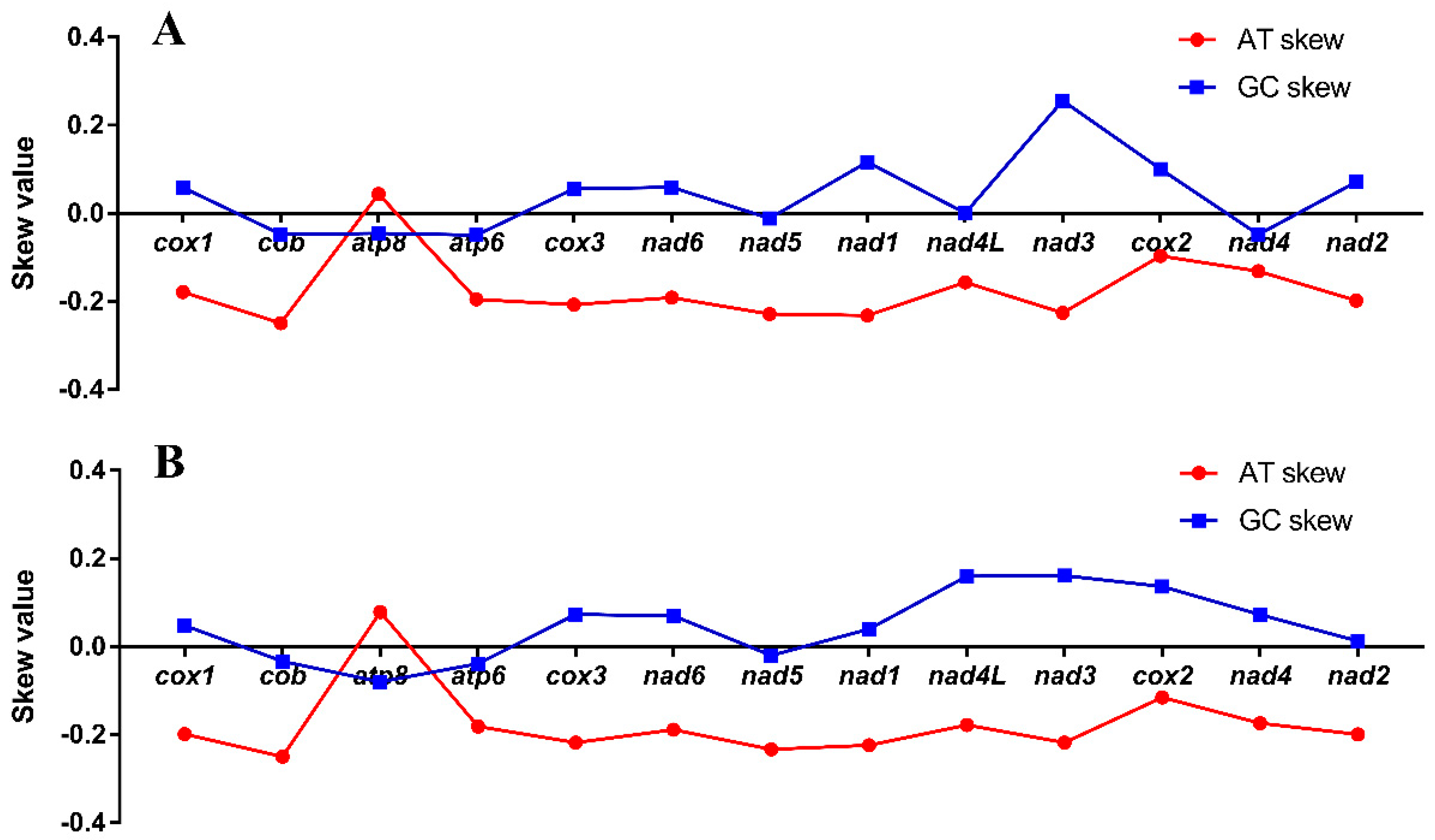
3.2. Skewness, Overlapping, and Intergenic Spacer Regions
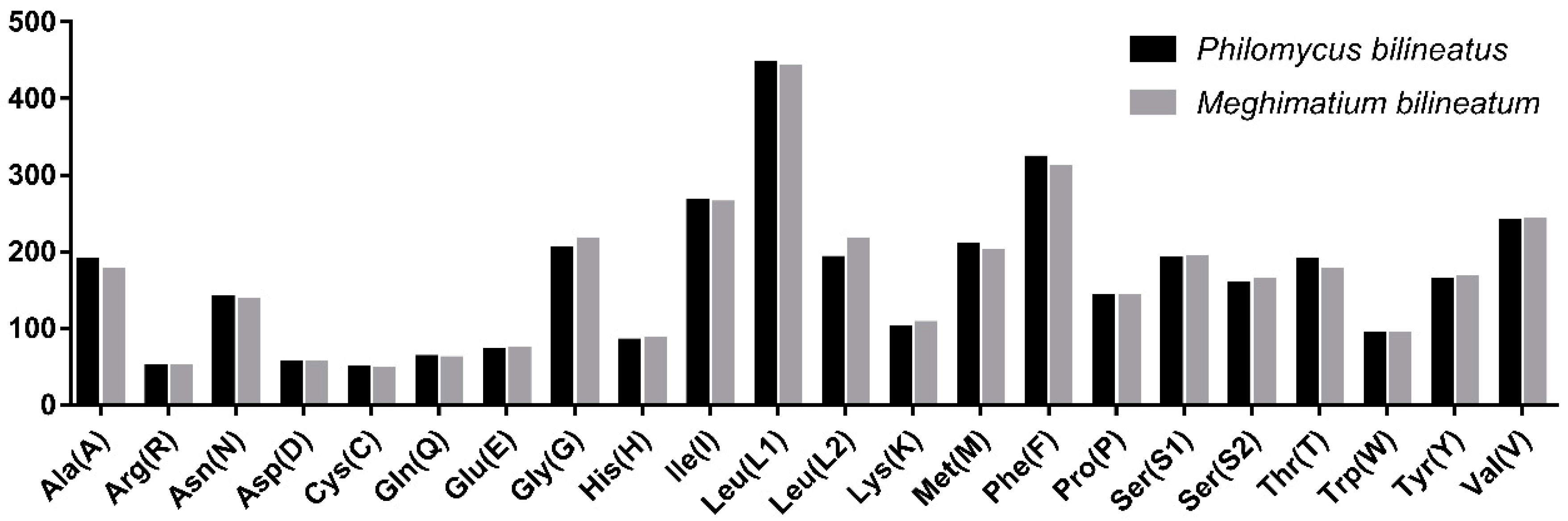
3.3. Protein-Coding Genes
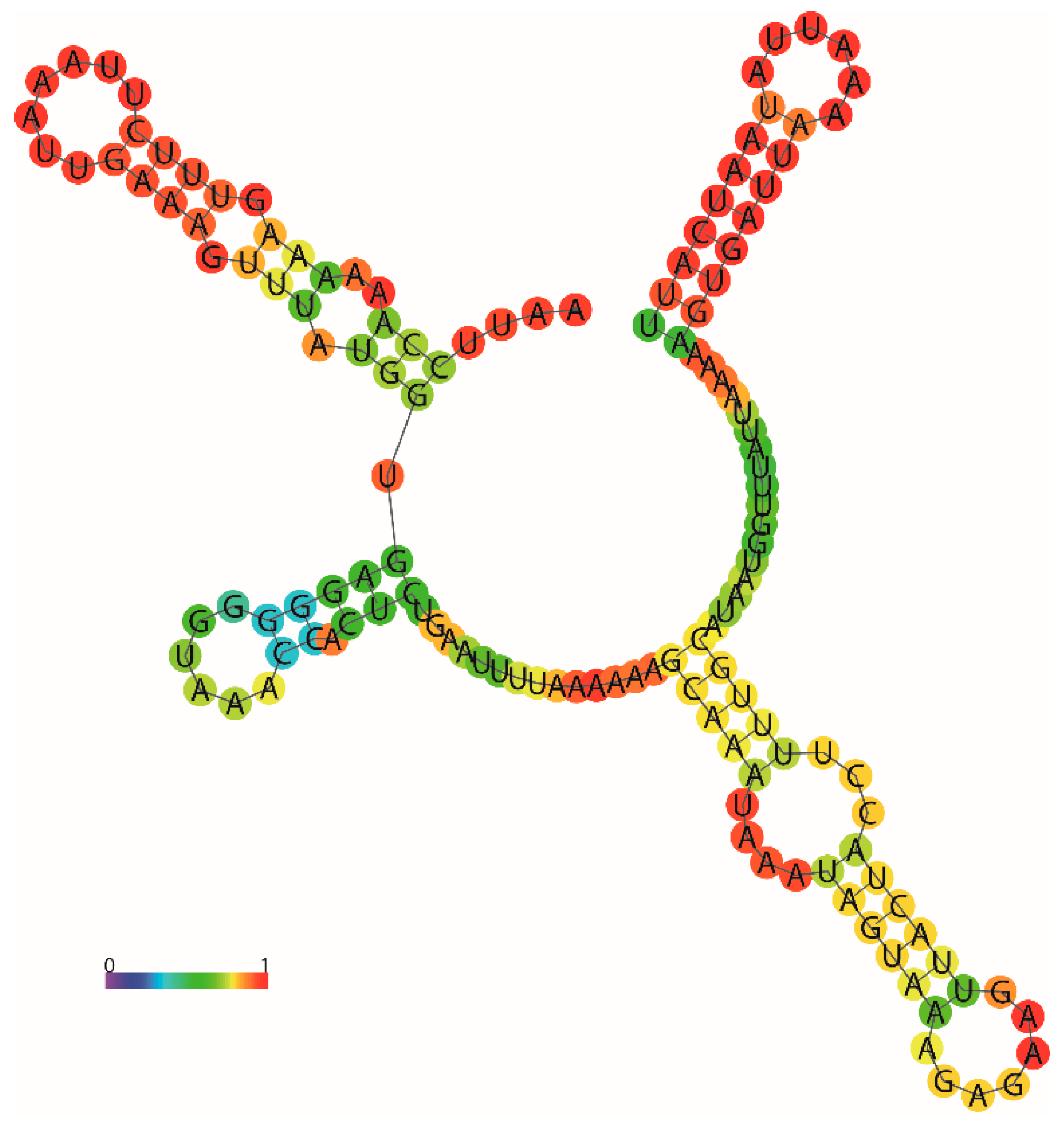
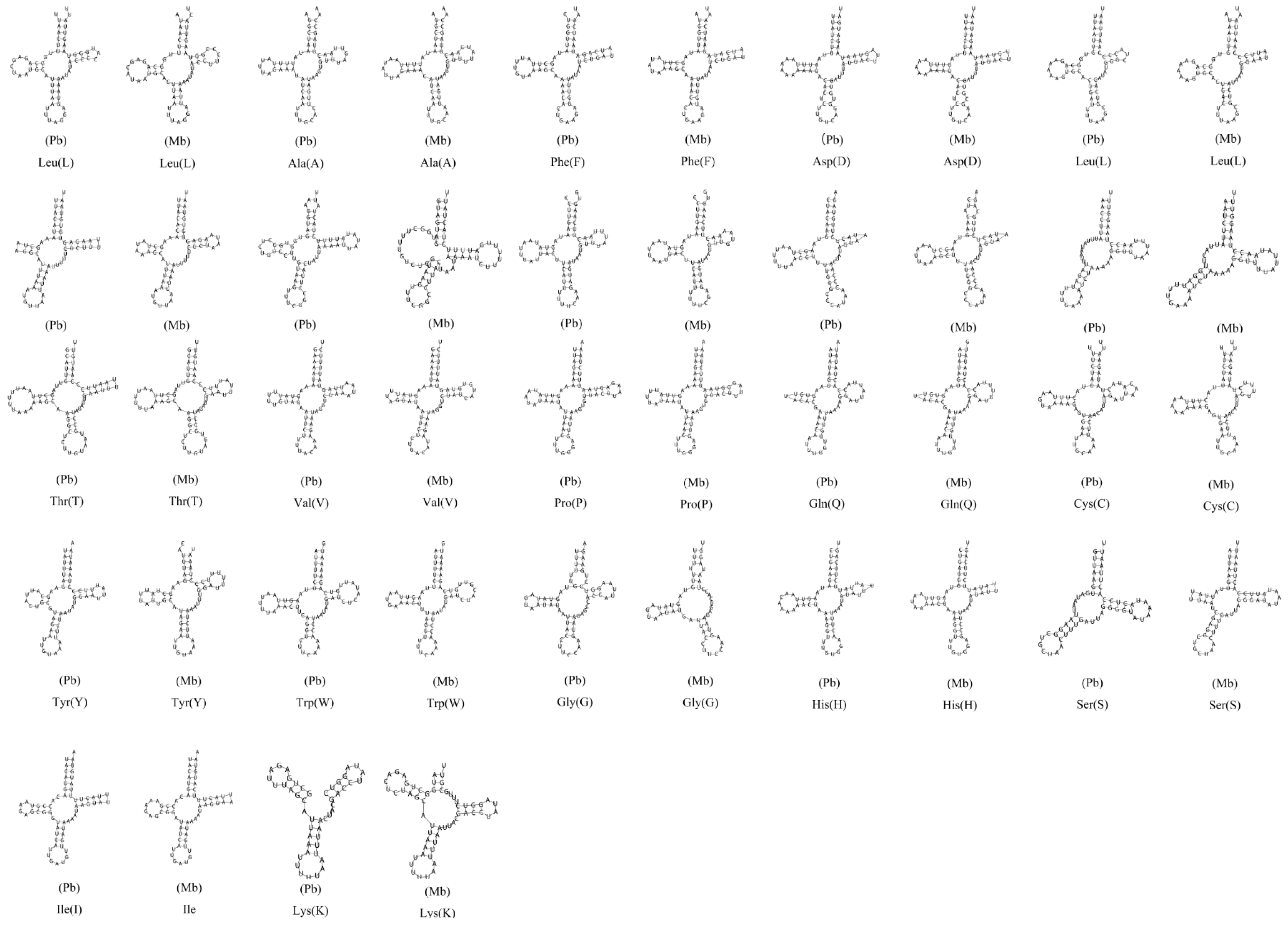
3.4. Transfer RNA Genes
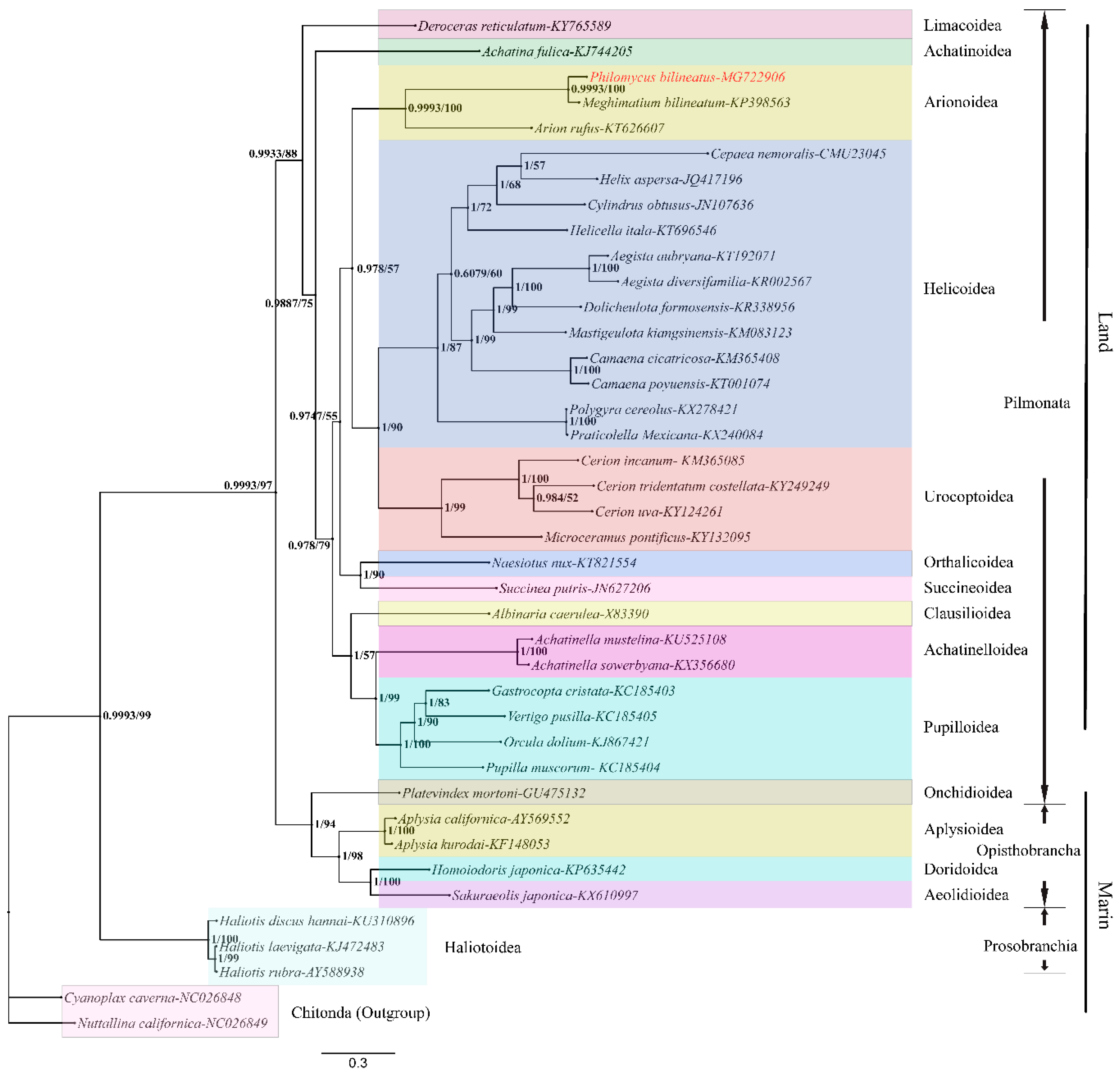
3.5. Phylogenetic Analysis
3.6. Evolution of the Breathing Pattern
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fendt, L.; Röck, A.; Zimmermann, B.; Bodner, M.; Thye, T.; Tschentscher, F.; Owusu-Dabo, E.; Göbel, T.M.; Schneider, P.M.; Parson, W. MtDNA diversity of Ghana: A forensic and phylogeographic view. Forensic Sci. Int. Genet. 2012, 6, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.; Vissing, J. No evidence for paternal inheritance of mtDNA in patients with sporadic mtDNA mutations. J. Neurol. Sci. 2004, 218, 99–101. [Google Scholar] [CrossRef] [PubMed]
- Hurst, G.D.; Jiggins, F.M. Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: The effects of inherited symbionts. Proc. R. Soc. B 2005, 272, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucl. Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.; Soares, P.; Carneiro, J.; Pereira, L.; Richards, M.B.; Samuels, D.C.; Amorim, A. Evidence for variable selective pressures at a large secondary structure of the human mitochondrial DNA control region. Mol. Biol. Evol. 2008, 25, 2759–2770. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nielsen, R.; Hasegawa, M. Models of amino acid substitution and applications to mitochondrial protein evolution. Mol. Biol. Evol. 1998, 15, 1600–1611. [Google Scholar] [CrossRef] [PubMed]
- Ladoukakis, E.D.; Zouros, E. Evolution and inheritance of animal mitochondrial DNA: Rules and exceptions. J. Biol. Res. Thessalon. 2017, 24, 2. [Google Scholar] [CrossRef] [PubMed]
- Weigertab, A.; Golombekc, A.; Gertha, M.; Schwarza, F.; Struckc, T.H.; Bleidornad, C. Evolution of mitochondrial gene order in Annelida. Mol. Phylogenet. Evol. 2016, 94, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wang, Z.; Wei, X.; Hua, S.H.I. Ecological observation on Phiolomycus bilineatus and preliminery study on its damage control. Chin. J. Zool. 1993, 2, 5–8. [Google Scholar]
- Dong, Y.H.; Qian, J.R.; Xu, P.J. occurrence law of Philomycus bilineatus and its prevention. Acta Agric. Jiangxi 2008, 20, 37–38. [Google Scholar]
- Chen, L.; Guo, L.; Wang, Z. Study on inhibitive effect of Limax extracts on Hela and study on the active component. J. Chin. Med. Mater. 2004, 2, 125–127. [Google Scholar]
- Wang, Z.; Wang, H. Pharmacological study of Limax extract on inhibiting H14 cell. J. Chin. Med. Mater. 2004, 1, 33–35. [Google Scholar]
- Kai, J.; Na, L.; Ang, G.; Xu, J.; Ming, W. Research status of harm and control technology of Limas maximus. J. Anhui Agric. Sci. 2011, 39, 7047–7048. [Google Scholar]
- Xin, Z.; Velten, J.P.; Oliver, M.J.; Burke, J.J. High-throughput DNA extraction method suitable for PCR. Biotechniques 2003, 34, 820. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.G.; Zhang, D.; Jakovlić, I.; Wang, W.M. Sequencing of the complete mitochondrial genomes of eight freshwater snail species exposes pervasive paraphyly within the Viviparidae family (Caenogastropoda). PLoS ONE 2017, 12, e0181699. [Google Scholar] [CrossRef] [PubMed]
- White, T.R.; Conrad, M.M.; Tseng, R.; Balayan, S.; Golding, R.; Martins, A.M.d.F.; Dayrat, B.A. Ten new complete mitochondrial genomes of pulmonates (Mollusca: Gastropoda) and their impact on phylogenetic relationships. BMC Evol. Biol. 2011, 11, 295. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, J.E.; Guo, J.; Deng, Z.; Luo, H.; Luo, M.; Zhao, B. The complete mitochondrial genome of the giant African snail Achatina fulica (Mollusca: Achatinidae). Mitochondrial DNA Part A 2016, 27, 1622–1624. [Google Scholar] [CrossRef]
- Wang, W. The Molecular Detection of Corynespora Cassiicola on Cucumber by PCR Assay Using DNAman Software and NCBI. In Proceedings of the CCTA 2015: Computer and Computing Technologies in Agriculture IX, Beijing, China, 27–30 September 2015; pp. 248–258. [Google Scholar]
- Rombel, I.; Sykes, K.; Rayner, S.; Johnston, S. ORF-FINDER: A vector for high-throughput gene identification. Gene 2002, 282, 33–41. [Google Scholar] [CrossRef]
- Kumar, V.; Dey, A.; Singh, A. MEGA: A bio computational software for sequence and phylogenetic analysis. Lect. Notes Eng. Comput. Sci. 2009, 2009, 2177. [Google Scholar]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Giribet, G.; Ribera, C. The position of arthropods in the animal kingdom: A search for a reliable outgroup for internal arthropod phylogeny. Mol. Phylogenet. Evol. 1998, 9, 481. [Google Scholar] [CrossRef] [PubMed]
- Irisarri, I.; Eernisse, D.J.; Zardoya, R. Molecular phylogeny of Acanthochitonina (Mollusca: Polyplacophora: Chitonida): Three new mitochondrial genomes, rearranged gene orders and systematics. J. Nat. Hist. 2014, 48, 2825–2853. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Mark, P.V.D.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Von Haeseler, A.; Schmidt, H.A.; Nguyen, L.-T.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xin, Z.Z.; Zhu, X.Y.; Zhao, X.M.; Wang, Y.; Tang, B.P.; Zhang, H.B.; Zhang, D.Z.; Zhou, C.L.; Liu, Q.N. The complete mitochondrial genome of Euproctis similis (Lepidoptera: Noctuoidea: Erebidae) and phylogenetic analysis. Int. J. Biol. Macromol. 2017, 105, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.P.; Shu, H.Z.; Deng, Z.R.; Jiang, M.S.; Nie, P. Conservation and variation in mitochondrial genomes of gastropods Oncomelania hupensis and Tricula hortensis, intermediate host snails of Schistosoma in China. Mol. Phylogenet. Evol. 2010, 57, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Feldmeyer, B.; Hoffmeier, K.; Pfenninger, M. The complete mitochondrial genome of Radix balthica (Pulmonata, Basommatophora), obtained by low coverage shot gun next generation sequencing. Mol. Phylogenet. Evol. 2010, 57, 1329–1333. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.J.; Martin, R.; Rao, S.; Choi, M.Y. The complete mitochondrial genome of the gray garden slug Deroceras reticulatum (Gastropoda: Pulmonata: Stylommatophora). Mitochondrial DNA Part B 2017, 2, 255–256. [Google Scholar] [CrossRef]
- Romero, P.E.; Weigand, A.M.; Pfenninger, M. Positive selection on panpulmonate mitogenomes provide new clues on adaptations to terrestrial life. BMC Evol. Biol. 2016, 16, 164. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Wang, L.; Dai, X.; Shi, Z. Karyotypes in Onchidium struma (Gastropoda: Pulmonata: Systellommatophora). Molluscan Res. 2010, 30, 113–116. [Google Scholar]
- Chen, L.; Xin, W.; He, D.S. Complete mitochondrial genome of Vaginulus alte and Homoiodoris japonica. Mitochondrial DNA Part A 2015, 27, 3454. [Google Scholar]
- Reuter, J.S.; Mathews, D.H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinform. 2010, 11, 129. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Yang, H.; Zhang, C.; Xue, H.; Xia, Y.; Zhang, J.E. Complete mitochondrial genome of the apple snail Pomacea diffusa (Gastropoda, Ampullariidae) with phylogenetic consideration. Mitochondrial DNA Part B 2017, 2, 865–867. [Google Scholar] [CrossRef]
- Lin, G.M.; Xiang, P.; Sampurna, B.P.; Hsiao, C.D. Genome skimming yields the complete mitogenome of Chromodoris annae (Mollusca: Chromodorididae). Mitochondrial DNA Part B 2017, 2, 609–610. [Google Scholar] [CrossRef]
- Rauch, C.; Christa, G.; Vries, J.D.; Woehle, C.; Gould, S.B. Mitochondrial genome assemblies of Elysia timida and Elysia cornigera and the response of mitochondrion-associated metabolism during starvation. Genome Biol. Evol. 2017, 9, 1873. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Zhou, W.C.; Ding, H.L.; Wang, P.; Ai, H.M. The mitochondrial genome of the land snail Cernuella virgata (Da Costa, 1778): The first complete sequence in the family Hygromiidae (Pulmonata, Stylommatophora). Zookeys 2016, 589, 55–69. [Google Scholar]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondrial. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Volatiana, J.A.; Fang, S.; Kinaro, Z.O.; Liu, X. Complete mitochondrial DNA sequences of Saccostrea mordax and Saccostrea cucullata: Genome organization and phylogeny analysis. Mitochondrial DNA Part A 2015, 27, 3024. [Google Scholar] [CrossRef] [PubMed]
- Osca, D.; Irisarri, I.; Todt, C.; Grande, C.; Zardoya, R. The complete mitoc genome of Scutopus ventrolineatus (Mollusca: Chaetodermomorpha) supports the Aculifera hypothesis. BMC Evol. Biol. 2014, 14, 197. [Google Scholar] [CrossRef]
- Giegé, R.; Puglisi, J.D.; Florentz, C. tRNA structure and aminoacylation efficiency. Prog. Nucleic Acid Res. Mol. Biol. 1993, 45, 129–206. [Google Scholar] [PubMed]
- Carroll, D. Gene duplication. Brenners Encycl. Genet. 2013, 25, 185–186. [Google Scholar]
- Sun, B.N.; Wei, L.L.; Shen, H.D.; Wu, H.X.; Wang, D.F. Phylogenetic analysis of euthyneuran gastropods from sea to land mainly based on comparative mitogenomic of four species of Onchidiidae (Mollusca: Gastropoda: Pulmonata). Mitochondrial DNA Part A 2016, 27, 3075. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.W.; Innes, A.J. A functional analysis of the shift from gill- to lung-breathing during the evolution of land crabs (Crustacea, Decapoda). Biol. J. Linn. Soc. 1988, 34, 229–247. [Google Scholar] [CrossRef]
- Brainerd, E.L. The evolution of lung-gill bimodal breathing and the homology of vertebrate respiratory pumps1. Integr. Comp. Biol. 1994, 34, 289–299. [Google Scholar]
- Burleson, M.L. Air-Breathing Fishes: Evolution, Diversity and Adaptation. Trans. Am. Fish. Soc. 2011, 184. [Google Scholar] [CrossRef]
- Dickinson, P.S.; Prior, D.J.; Avery, C. The pneumostome rhythm in slugs: A response to dehydration controlled by hemolymph osmolality and peptide hormones. Comp. Biochem. Physiol. Part A Physiol. 1988, 89, 579–585. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Strand | Location | Size (bp) | Intergenic Length | Anti-Codon | Start Codon | Stop Codon |
|---|---|---|---|---|---|---|---|
| cox1 | H | 1–1527 | 1527 | - | - | TTG | TAA |
| CR | H | 1528–1670 | 143 | 0 | - | - | - |
| 16S | H | 1671–2569 | 899 | 0 | - | - | - |
| trnL | H | 2578–2640 | 63 | 8 | TAG | - | - |
| trnA | H | 2643–2705 | 63 | 2 | TGC | - | - |
| cob | H | 2713–3795 | 1083 | 7 | - | ATG | TAA |
| trnF | H | 3802–3867 | 66 | 6 | GAA | - | - |
| trnD | H | 3871–3936 | 66 | 3 | GTC | - | - |
| trnL | L | 3935–3998 | 64 | −2 | TAA | - | - |
| atp8 | L | 3999–4157 | 159 | 0 | - | GTG | TAA |
| trnN | L | 4149–4208 | 60 | −9 | GTT | - | - |
| atp6 | L | 4201–4866 | 666 | −8 | - | ATA | TAA |
| trnR | L | 4852–4918 | 67 | −13 | TCG | - | - |
| trnE | L | 4923–4986 | 64 | 4 | TTC | - | - |
| 12S | L | 4986–5676 | 691 | −1 | - | - | - |
| trnM | L | 5701–5764 | 64 | −6 | CAT | - | - |
| trnS2 | L | 5765–5823 | 59 | 0 | TGA | - | - |
| trnT | L | 5825–5894 | 70 | 1 | TGT | - | - |
| cox3 | L | 5848–6684 | 837 | −47 | - | ATA | TAG |
| trnV | H | 6746–6808 | 63 | 61 | TAC | - | - |
| trnP | H | 6810–6875 | 66 | 1 | TGG | - | - |
| nad6 | H | 6897–7364 | 468 | 21 | - | ATT | TAA |
| nad5 | H | 7368–8999 | 1632 | 3 | - | ATG | TAG |
| nad1 | H | 8993–9916 | 924 | −7 | - | ATG | TAA |
| nad4L | H | 9888–10178 | 291 | −19 | ATT | TAG | |
| trnQ | L | 10172–10230 | 59 | −7 | TTG | - | - |
| nad3 | L | 10248–10589 | 342 | 17 | - | ATG | TAA |
| trnC | H | 10644–10708 | 65 | 54 | GCA | - | - |
| trnC | H | 11000–11064 | 65 | 291 | GCA | - | - |
| cox2 | H | 11057–11728 | 672 | −8 | - | ATA | TAG |
| trnY | H | 11722–11780 | 59 | −7 | GTA | - | - |
| trnW | H | 11786–11853 | 68 | 5 | TCA | - | - |
| trnG | H | 11858–11919 | 62 | 4 | TCC | - | - |
| trnH | H | 11913–11977 | 65 | −7 | GTG | - | - |
| trnS1 | H | 11978–12035 | 58 | 0 | GCT | - | - |
| nad4 | H | 12021–13343 | 1323 | −15 | - | ATA | TAA |
| trnI | H | 13345–13407 | 63 | 1 | GAT | - | - |
| nad2 | H | 13411–14325 | 915 | 3 | - | ATG | TAA |
| trnK | H | 14300–14346 | 47 | −26 | TTT | - | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, T.; Xu, G.; Gu, B.; Shi, Y.; Mzuka, H.L.; Shen, H. The Complete Mitochondrial Genome Sequences of the Philomycus bilineatus (Stylommatophora: Philomycidae) and Phylogenetic Analysis. Genes 2019, 10, 198. https://doi.org/10.3390/genes10030198
Yang T, Xu G, Gu B, Shi Y, Mzuka HL, Shen H. The Complete Mitochondrial Genome Sequences of the Philomycus bilineatus (Stylommatophora: Philomycidae) and Phylogenetic Analysis. Genes. 2019; 10(3):198. https://doi.org/10.3390/genes10030198
Chicago/Turabian StyleYang, Tiezhu, Guolyu Xu, Bingning Gu, Yanmei Shi, Hellen Lucas Mzuka, and Heding Shen. 2019. "The Complete Mitochondrial Genome Sequences of the Philomycus bilineatus (Stylommatophora: Philomycidae) and Phylogenetic Analysis" Genes 10, no. 3: 198. https://doi.org/10.3390/genes10030198