The Complete Mitochondrial Genome of Platysternon megacephalum peguense and Molecular Phylogenetic Analysis

Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement, Taxon Sampling, and DNA Extraction
2.2. PCR Amplification and Sequencing
2.3. Sequence Assembly, Annotation, and Analysis
2.4. Phylogenetic Analysis and Divergence Time Estimation
3. Results and Discussion
3.1. Sequencing Data and Genome Composition
3.2. PCGs and Codon Usage
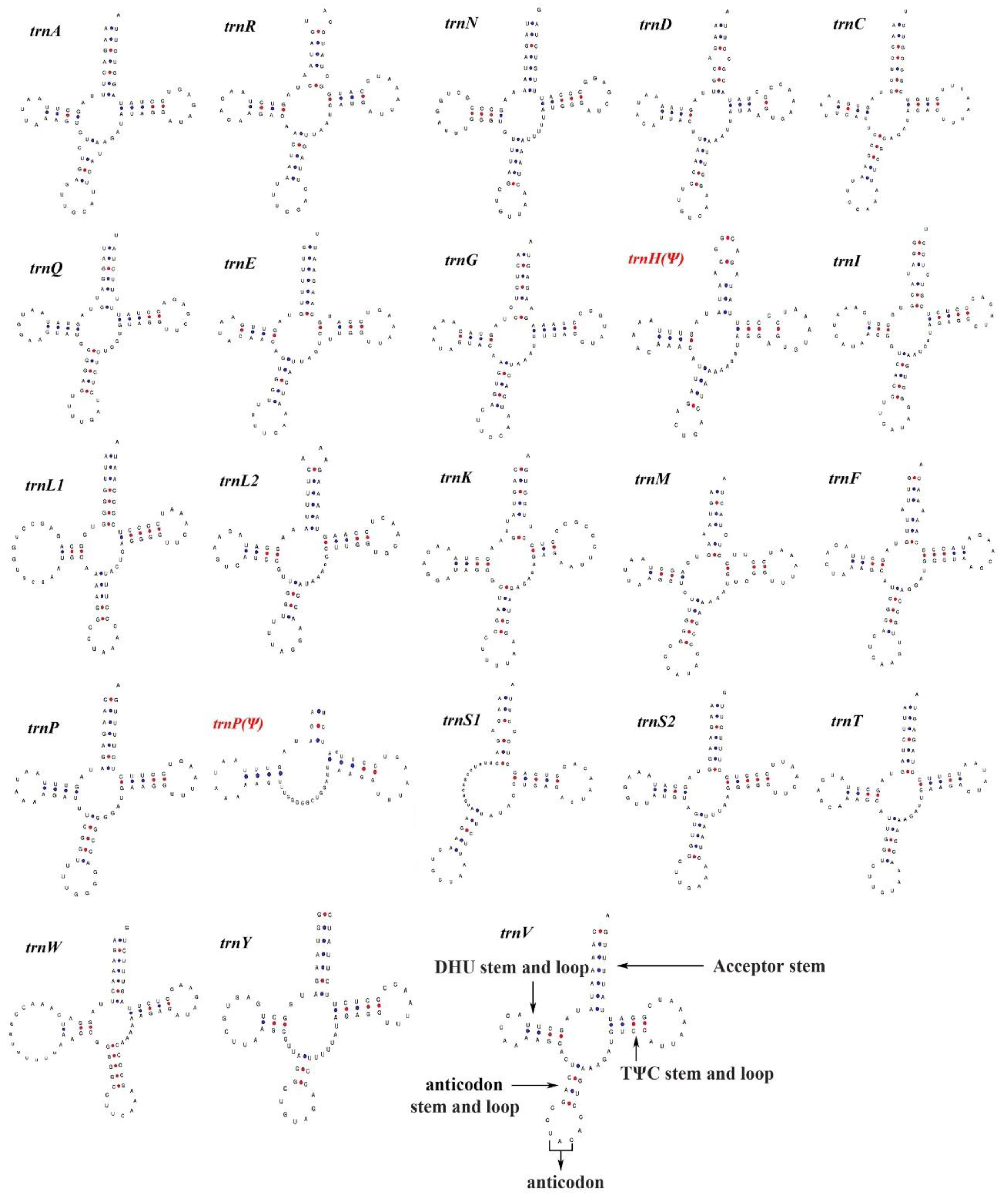
3.3. Transfer RNAs and Control Region
3.4. Gene Rearrangement
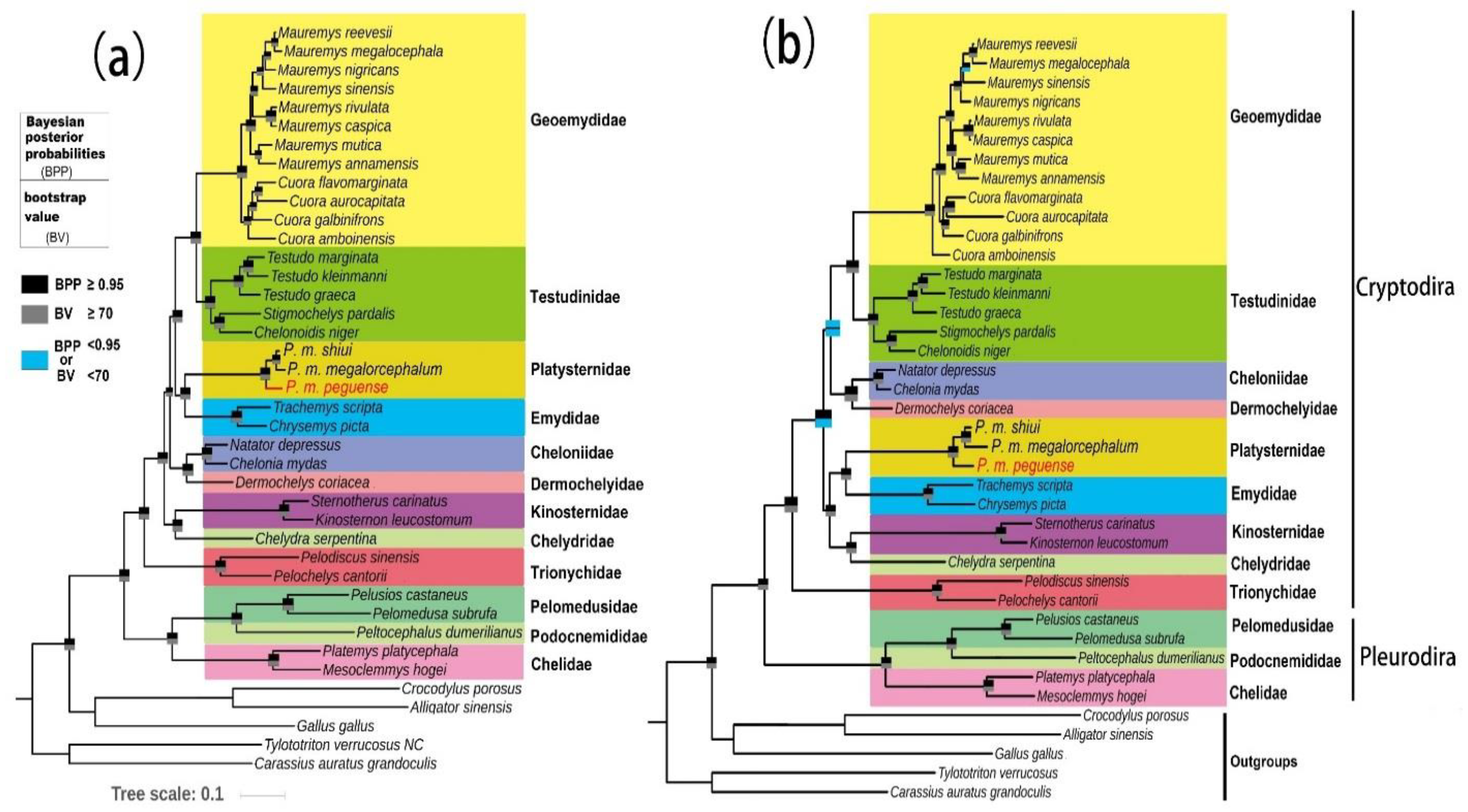
3.5. Phylogenetic Analysis
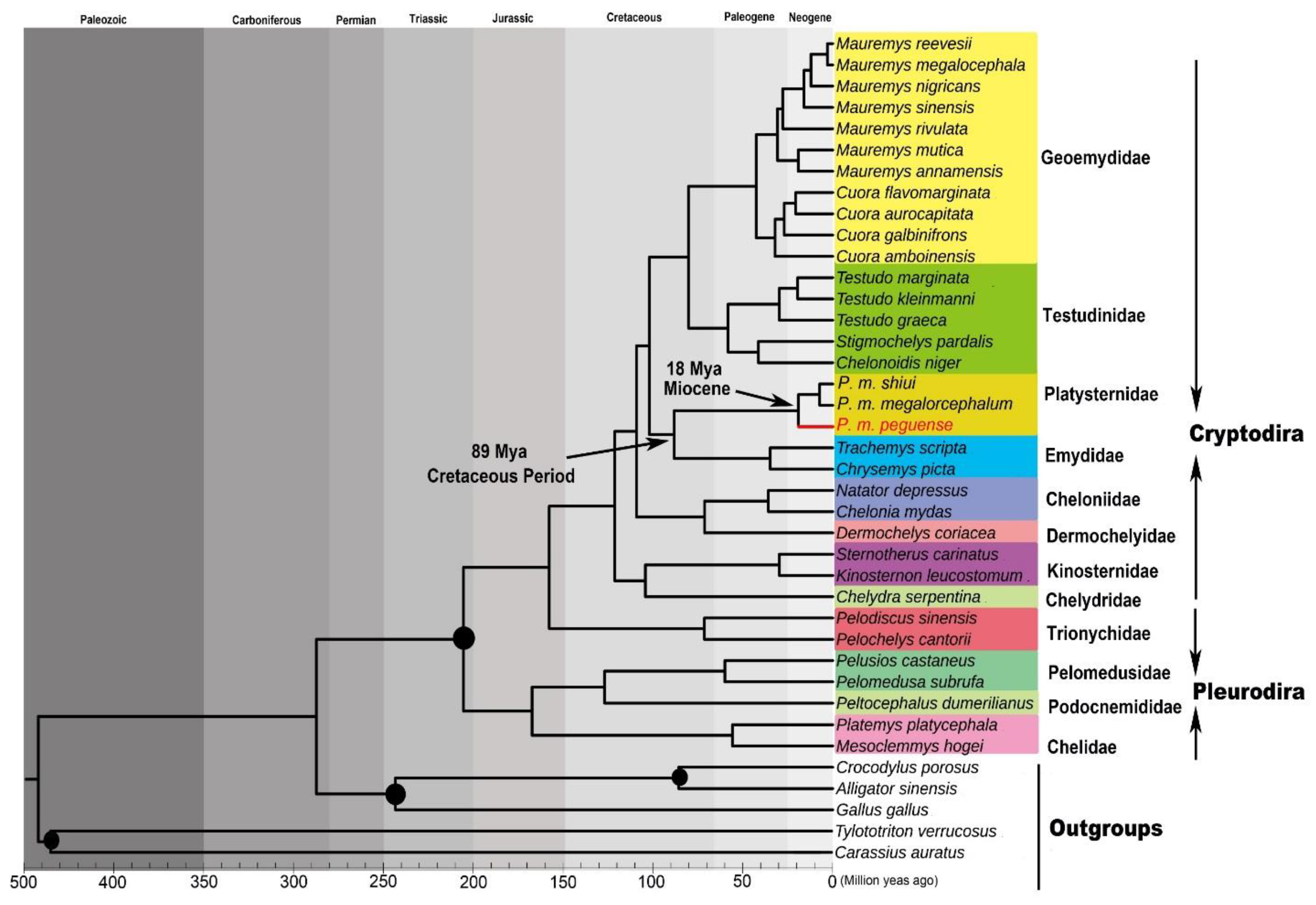
3.6. Divergence Time Estimation
3.7. Genetic Divergence
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Garesse, R.; Vallejo, C.G. Animal mitochondrial biogenesis and function: A regulatory cross-talk between two genomes. Gene 2001, 263, 1–16. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Sorenson, M.D.; Kumazawa, Y.; Mindell, D.P.; Hasegawa, M. Phylogenetic position of turtles among amniotes: Evidence from mitochondrial and nuclear genes. Gene 2000, 259, 139–148. [Google Scholar] [CrossRef]
- Hongyu, M.; Chunyan, M.; Xincang, L.; Zhen, X.; Nana, F.; Lingbo, M. The complete mitochondrial genome sequence and gene organization of the mud crab (Scylla paramamosain) with phylogenetic consideration. Gene 2013, 519, 120–127. [Google Scholar]
- Zardoya, R.; Meyer, A. Cloning and characterization of a microsatellite in the mitochondrial control region of the African side-necked turtle, Pelomedusa subrufa. Gene 1998, 216, 149–153. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Unmack, P.J.; Kuchling, G.; Wang, Y.; Georges, A. Resolution of the enigmatic phylogenetic relationship of the critically endangered Western Swamp Tortoise Pseudemydura umbrina (Pleurodira: Chelidae) using a complete mitochondrial genome. Mol. Phylogenet. Evol. 2017, 115, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.L.; Nie, L.W.; Pu, Y.G. Complete mitochondrial genome of Chinese big-headed turtle, Platysternon megacephalum, with a novel gene organization in vertebrate mtDNA. Gene 2006, 380, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Ernst, C.H.; Laemmerzahl, A.F. Geographic variation in the Asian big-headed turtle, Platysternon megacephalum (Reptilia: Testudines: Platysternidae). Proc. Biol. Soc. Washigton 2002, 18–24. [Google Scholar]
- Crawford, N.G.; Parham, J.F.; Sellas, A.B.; Faircloth, B.C.; Glenn, T.C.; Papenfuss, T.J.; Henderson, J.B.; Hansen, M.H.; Simison, W.B. A phylogenomic analysis of turtles. Mol. Phylogenet. Evol. 2015, 83, 250–257. [Google Scholar] [CrossRef]
- Zheng, C.; Nie, L.; Wang, J.; Zhou, H.; Hou, H.; Wang, H.; Liu, J. Recombination and evolution of duplicate control regions in the mitochondrial genome of the Asian big-headed turtle, Platysternon megacephalum. PLoS ONE 2013, 8, e82854. [Google Scholar] [CrossRef]
- Dayrat, B. Towards integrative taxonomy. Biol. J. Linn. Soc. 2005, 85, 407–417. [Google Scholar] [CrossRef]
- Norman, J.A.; Moritz, C.; Limpus, C.J. Mitochondrial DNA control region polymorphisms: Genetic markers for ecological studies of marine turtles. Mol. Ecol. 2010, 3, 363–373. [Google Scholar] [CrossRef]
- David, C.; Guillaume, J.; Darling, A.E. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 2014, 31, 587–589. [Google Scholar]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.R.; Paul, S. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Peter, S.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, 686–689. [Google Scholar]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [Green Version]
- Hillis, D.M.; Bull, J.J. An Empirical Test of Bootstrapping as a Method for Assessing Confidence in Phylogenetic Analysis. Syst. Biol. 1993, 42, 182–192. [Google Scholar] [CrossRef]
- Kazutaka, K.; Kazuharu, M.; Kei-Ichi, K.; Takashi, M. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar]
- Alexandros, S. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688. [Google Scholar]
- Fredrik, R.; Maxim, T.; Paul, V.D.M.; Ayres, D.L.; Aaron, D.; Sebastian, H.; Bret, L.; Liang, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [Green Version]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [PubMed]
- Dosreis, M.; Thawornwattana, Y.; Angelis, K.; Telford, M.; Donoghue, P.J.; Yang, Z. Uncertainty in the Timing of Origin of Animals and the Limits of Precision in Molecular Timescales. Curr. Biol. 2015, 25, 2939–2950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, A.; Slice, D.E. The utility of cranial ontogeny for phylogenetic inference: A case study in crocodylians using geometric morphometrics. J. Evol. Biol. 2014, 27, 1078–1092. [Google Scholar] [CrossRef]
- Zheng, Y.; Wiens, J.J. Combining phylogenomic and supermatrix approaches, and a time-calibrated phylogeny for squamate reptiles (lizards and snakes) based on 52 genes and 4162 species. Mol. Phylogenet. Evol. 2016, 94, 537–547. [Google Scholar] [CrossRef]
- Naro-Maciel, E.; Le, M.; Fitzsimmons, N.N.; Amato, G. Evolutionary relationships of marine turtles: A molecular phylogeny based on nuclear and mitochondrial genes. Mol. Phylogenetics Evol. 2008, 49, 659–662. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67. [Google Scholar] [CrossRef]
- Kilpert, F.; Podsiadlowski, L. The complete mitochondrial genome of the common sea slater, Ligia oceanica (Crustacea, Isopoda) bears a novel gene order and unusual control region features. BMC Genom. 2006, 7, 241. [Google Scholar] [CrossRef] [PubMed]
- Lavrov, D.V.; Brown, W.M.; Boore, J.L. A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipede Lithobius forficatus. Proc. Natl. Acad. Sci. USA 2000, 97, 13738–13742. [Google Scholar] [CrossRef] [PubMed]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Cannone, J.J.; Subramanian, S.; Schnare, M.N.; Collett, J.R.; D’Souza, L.M.; Du, Y.; Feng, B.; Nan, L.; Madabusi, L.V.; Müller, K.M. The Comparative RNA Web (CRW) Site: An online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinform. 2002, 3, 2. [Google Scholar]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [PubMed]
- Dörner, M.; Altmann, M.; Pääbo, S.; Mörl, M. Evidence for import of a lysyl-tRNA into marsupial mitochondria. Mol. Biol. Cell 2001, 12, 2688–2698. [Google Scholar] [CrossRef]
- Tan, T.H.P.; Roland, P.; Anne, C.; Al, I.; André, S. tRNAs in Trypanosoma brucei: Genomic organization, expression, and mitochondrial import. Mol. Cell. Biol. 2002, 22, 3707–3717. [Google Scholar] [CrossRef]
- Schirtzinger, E.E.; Tavares, E.S.; Gonzales, L.A.; Eberhard, J.R.; Miyaki, C.Y.; Sanchez, J.J.; Hernandez, A.; Müeller, H.; Graves, G.R.; Fleischer, R.C. Multiple independent origins of mitochondrial control region duplications in the order Psittaciformes. Mol. Phylogenet. Evol. 2012, 64, 342–356. [Google Scholar] [CrossRef] [Green Version]
- Shao, R.; Barker, S.C.; Mitani, H.; Aoki, Y.; Fukunaga, M. Evolution of duplicate control regions in the mitochondrial genomes of metazoa: A case study with Australasian Ixodes ticks. Mol. Biol. Evol. 2005, 22, 620–629. [Google Scholar] [CrossRef]
- Parham, J.F.; Feldman, C.R.; Boore, J.L. The complete mitochondrial genome of the enigmatic bigheaded turtle (Platysternon): Description of unusual genomic features and the reconciliation of phylogenetic hypotheses based on mitochondrial and nuclear DNA. BMC Evol. Biol. 2006, 6, 11. [Google Scholar] [CrossRef]
- Mindell, D.P.; Sorenson, M.D.; Dimcheff, D.E. An extra nucleotide is not translated in mitochondrial ND3 of some birds and turtles. Mol. Biol. Evol. 1998, 15, 1568–1571. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ma, F.; Xiao, B.; Zheng, J.; Yuan, X.; Tang, M.; Li, Q. The complete mitochondrial genomes sequences of Asio flammeus and Asio otus and comparative analysis. Sci. China C Life Sci. 2004, 47, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.D.; Beckenbach, A.T. Recoding of Translation in Turtle Mitochondrial Genomes: Programmed Frameshift Mutations and Evidence of a Modified Genetic Code. J. Mol. Evol. 2008, 67, 682–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barley, A.J.; Spinks, P.Q.; Thomson, R.C.; Shaffer, H.B. Fourteen nuclear genes provide phylogenetic resolution for difficult nodes in the turtle tree of life. Mol. Phylogenet. Evol. 2010, 55, 1189–1194. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.G.; Sterli, J.; Moreira, F.R.R.; Schrago, C.G. Multilocus phylogeny and statistical biogeography clarify the evolutionary history of major lineages of turtles. Mol. Phylogenet. Evol. 2017, 113, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Giribet, G.; Ribera, C. A Review of Arthropod Phylogeny: New Data Based on Ribosomal DNA Sequences and Direct Character Optimization. Cladistics-Int. J. Willi Hennig Soc. 2000, 16, 204–231. [Google Scholar] [CrossRef]
- Giribet, G.; Edgecombe, G.D.; Wheeler, W.C. Arthropod phylogeny based on eight molecular loci and morphology. Nature 2001, 413, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Luan, Y.; Mallatt, J.M.; Xie, R.; Yang, Y.; Yin, W. The Phylogenetic Positions of Three Basal-Hexapod Groups (Protura, Diplura, and Collembola) Based on Ribosomal RNA Gene Sequences. Mol. Biol. Evol. 2005, 22, 1579–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lourenço, J.M.; Claude, J.; Galtier, N.; Chiari, Y. Dating cryptodiran nodes: Origin and diversification of the turtle superfamily Testudinoidea. Mol. Phylogenet. Evol. 2012, 62, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Novacek, M.J. 100 Million Years of Land Vertebrate Evolution: The Cretaceous-Early Tertiary Transition. Ann. Mo. Bot. Gard. 1999, 86, 230–258. [Google Scholar] [CrossRef]
- Erbacher, J.; Huber, B.T.; Norris, R.D.; Markey, M. Increased thermohaline stratification as a possible cause for an ocean anoxic event in the Cretaceous period. Nature 2001, 409, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Zhisheng, A.; Kutzbach, J.E.; Prell, W.L.; Porter, S.C. Evolution of Asian monsoons and phased uplift of the Himalaya-Tibetan plateau since Late Miocene times. Nature 2001, 411, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Favre, A.; Päckert, M.; Pauls, S.U.; Jähnig, S.C.; Uhl, D.; Michalak, I.; Muellner-Riehl, A.N. The role of the uplift of the Qinghai-Tibetan Plateau for the evolution of Tibetan biotas. Biol. Rev. Camb. Philos. Soc. 2015, 90, 236–253. [Google Scholar] [CrossRef] [PubMed]
- Lei, F.; Qu, Y.; Song, G. Species diversification and phylogeographical patterns of birds in response to the uplift of the Qinghai-Tibet Plateau and Quaternary glaciations. Curr. Zool. 2014, 60, 149161. [Google Scholar] [CrossRef]
- Wei-Wei, Z.; Yang, W.; Jinzhong, F.; Yong-Biao, X.; Jie-Qiong, J.; Li, D.; Mi-Sook, M.; Jing, C.; Ya-Ping, Z. Speciation in the Rana chensinensis species complex and its relationship to the uplift of the Qinghai-Tibetan Plateau. Mol. Ecol. 2012, 21, 960–973. [Google Scholar]
- Herler, J.; Bogorodsky, S.V.; Suzuki, T. Four new species of coral gobies (Teleostei: Gobiidae: Gobiodon), with comments on their relationships within the genus. Zootaxa 2013, 3709, 301–329. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Direction | Location | Size (bp) | Anticodon | Start Codon | Stop Codon | Intergenic Nucleotides |
|---|---|---|---|---|---|---|---|
| trnF | + | 1–70 | 70 | GAA | 0 | ||
| rrnS | + | 71–1028 | 958 | −1 | |||
| trnV | + | 1028–1096 | 69 | TAC | 21 | ||
| rrnL | + | 1118–2702 | 1585 | −1 | |||
| trnL2 | + | 2702–2777 | 76 | TAA | 0 | ||
| nad1 | + | 2778–3749 | 972 | ATG | TAG | −1 | |
| trnI | + | 3749–3819 | 71 | GAT | 131 | ||
| trnH (Ψ) | + | 3951–4017 | 67 | CTG | 0 | ||
| trnS1 | + | 4018–4083 | 66 | GCT | −1 | ||
| trnL1 | + | 4083–4154 | 72 | TAG | 1 | ||
| nad5 | + | 4156–5976 | 1821 | ATG | TAA | 135 | |
| trnT | + | 6112–6182 | 71 | TGT | 1 | ||
| trnP | − | 6184–6253 | 70 | TGG | 15 | ||
| CR2 | 6267–7553 | 1286 | −1 | ||||
| trnQ | − | 7553–7623 | 71 | TTG | −1 | ||
| trnM | + | 7623–7691 | 69 | CAT | 0 | ||
| nad2 | + | 7692–8732 | 1041 | ATG | TAG | −2 | |
| trnW | + | 8731–8809 | 79 | TCA | 1 | ||
| trnA | − | 8811–8879 | 69 | TGC | 1 | ||
| trnN | − | 8881–8954 | 74 | GTT | 3 | ||
| OL | + | 8958–8984 | 27 | −2 | |||
| trnC | − | 8983–9048 | 66 | GCA | 0 | ||
| trnY | − | 9049–9119 | 71 | GTA | 1 | ||
| cox1 | + | 9121–10,668 | 1548 | GTG | AGG | −9 | |
| trnS2 | − | 10,660–10,730 | 71 | TGA | 3 | ||
| trnD | + | 10,734–10,803 | 70 | GTC | 0 | ||
| cox2 | + | 10,804–11,490 | 687 | ATG | TAG | 1 | |
| trnK | + | 11,492–11,563 | 72 | TTT | 1 | ||
| atp8 | + | 11,565–11,732 | 168 | ATG | TAA | −16 | |
| atp6 | + | 11717–12,399 | 683 | ATG | TA(A) | 18 | |
| cox3 | + | 12,418–13,202 | 785 | ATG | TA(A) | −1 | |
| trnG | + | 13,202–13,271 | 70 | TCC | 0 | ||
| nad3-a | + | 13,272–13,445 | 174 | ATG | --- | 1 | |
| nad3-b | + | 13,447–13,621 | 175 | --- | T(AA) | 0 | |
| trnR | + | 13,622–13,690 | 69 | TCG | 0 | ||
| nad4l | + | 13,691–13,987 | 297 | ATG | TAA | −7 | |
| nad4 | + | 13,981–15,358 | 1378 | ATG | T(AA) | 640 | |
| nad6 | − | 15,999–16,523 | 525 | ATG | AGG | 0 | |
| trnE | − | 16,524–16,591 | 68 | TTC | 4 | ||
| cob | + | 16,596–17,762 | 1167 | ATG | TAA | 48 | |
| trnP (Ψ) | − | 17,811–17,872 | 62 | --- | 17 | ||
| CR1 | 17,890–19,093 | 1203 | 0 |
| Species | Total Size (bp) | Complete Mitogenome | tRNAs | PCGs | rRNAs | CRs | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A + T% | AT-skew | GC-skew | A + T% | AT-skew | GC-skew | A + T% | AT-skew | GC-skew | A + T% | AT-skew | GC-skew | A + T% | AT-skew | GC-skew | ||
| P. m. peguense | 19,093 | 61.2 | 0.106 | −0.335 | 61.9 | 0.040 | 0.031 | 59.9 | 0.034 | −0.313 | 60.2 | 0.236 | −0.149 | 66.3 | 0.011 | −0.344 |
| P. m. megalorcephalum | 19,196 | 61.5 | 0.106 | −0.323 | 61.9 | 0.048 | 0.020 | 59.9 | 0.034 | −0.313 | 60.0 | 0.236 | −0.149 | 68.1 | −0.008 | −0.223 |
| P. m. shiui | 19,043 | 61.4 | 0.107 | −0.329 | 62.3 | 0.071 | −0.053 | 59.7 | 0.033 | −0.311 | 60.3 | 0.237 | −0.147 | 66.9 | −0.006 | −0.256 |
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | P. m. peguense | |||||||||||||||||
| 2 | P. m. megalorcephalum | 0.068 | ||||||||||||||||
| 3 | P. m. shiui | 0.059 | 0.024 | |||||||||||||||
| 4 | M. annamensis | 0.257 | 0.266 | 0.255 | ||||||||||||||
| 5 | M. megalocephala | 0.268 | 0.271 | 0.259 | 0.094 | |||||||||||||
| 6 | M. mutica | 0.255 | 0.260 | 0.250 | 0.058 | 0.086 | ||||||||||||
| 7 | M. reevesii | 0.261 | 0.264 | 0.253 | 0.087 | 0.051 | 0.080 | |||||||||||
| 8 | M. nigricans | 0.260 | 0.265 | 0.253 | 0.089 | 0.049 | 0.081 | 0.040 | ||||||||||
| 9 | M. rivulata | 0.253 | 0.261 | 0.251 | 0.081 | 0.082 | 0.079 | 0.076 | 0.074 | |||||||||
| 10 | M. sinensis | 0.260 | 0.266 | 0.256 | 0.090 | 0.055 | 0.081 | 0.049 | 0.047 | 0.077 | ||||||||
| 11 | T. graeca | 0.245 | 0.253 | 0.242 | 0.198 | 0.198 | 0.194 | 0.194 | 0.193 | 0.190 | 0.195 | |||||||
| 12 | T. kleinmanni | 0.251 | 0.255 | 0.245 | 0.201 | 0.201 | 0.195 | 0.197 | 0.194 | 0.197 | 0.198 | 0.089 | ||||||
| 13 | T. marginata | 0.246 | 0.253 | 0.244 | 0.201 | 0.202 | 0.195 | 0.196 | 0.193 | 0.193 | 0.196 | 0.082 | 0.071 | |||||
| 14 | C. amboinensis | 0.259 | 0.266 | 0.256 | 0.118 | 0.121 | 0.114 | 0.115 | 0.114 | 0.113 | 0.114 | 0.194 | 0.198 | 0.194 | ||||
| 15 | C. aurocapitata | 0.277 | 0.286 | 0.276 | 0.127 | 0.123 | 0.126 | 0.119 | 0.122 | 0.121 | 0.121 | 0.218 | 0.222 | 0.219 | 0.116 | |||
| 16 | C. flavomarginata | 0.260 | 0.268 | 0.258 | 0.110 | 0.112 | 0.107 | 0.107 | 0.109 | 0.105 | 0.111 | 0.193 | 0.201 | 0.196 | 0.099 | 0.085 | ||
| 17 | C. galbinifrons | 0.257 | 0.262 | 0.251 | 0.106 | 0.110 | 0.106 | 0.105 | 0.105 | 0.102 | 0.105 | 0.192 | 0.194 | 0.195 | 0.092 | 0.096 | 0.083 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, H.; Li, H.; Huang, A.; Ni, Q.; Yao, Y.; Xu, H.; Zeng, B.; Li, Y.; Wei, Z.; Yu, G.; et al. The Complete Mitochondrial Genome of Platysternon megacephalum peguense and Molecular Phylogenetic Analysis. Genes 2019, 10, 487. https://doi.org/10.3390/genes10070487
Luo H, Li H, Huang A, Ni Q, Yao Y, Xu H, Zeng B, Li Y, Wei Z, Yu G, et al. The Complete Mitochondrial Genome of Platysternon megacephalum peguense and Molecular Phylogenetic Analysis. Genes. 2019; 10(7):487. https://doi.org/10.3390/genes10070487
Chicago/Turabian StyleLuo, Hongdi, Haijun Li, An Huang, Qingyong Ni, Yongfang Yao, Huailiang Xu, Bo Zeng, Ying Li, Zhimin Wei, Guohua Yu, and et al. 2019. "The Complete Mitochondrial Genome of Platysternon megacephalum peguense and Molecular Phylogenetic Analysis" Genes 10, no. 7: 487. https://doi.org/10.3390/genes10070487
APA StyleLuo, H., Li, H., Huang, A., Ni, Q., Yao, Y., Xu, H., Zeng, B., Li, Y., Wei, Z., Yu, G., & Zhang, M. (2019). The Complete Mitochondrial Genome of Platysternon megacephalum peguense and Molecular Phylogenetic Analysis. Genes, 10(7), 487. https://doi.org/10.3390/genes10070487