Identification of Potential Biomarkers for Anti-PD-1 Therapy in Melanoma by Weighted Correlation Network Analysis

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Acquisition of Gene Expression Datasets
2.2. Construction of Gene Co-Expression Network
2.3. Identification of Clinically Significant Modules
2.4. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Enrichment Analysis
2.5. Identification of Hub Genes Based on Protein–Protein Interaction (PPI) and Topological Network
2.6. Receiver Operating Characteristic (ROC) Curve Analysis
2.7. Gene Set Variation Analysis of Hub Genes
2.8. Correlation Analysis of Hub Genes and Immune Infiltration Level
2.9. Validation and Survival Analysis of Hub Genes
2.10. Correlation Analysis of Hub Genes and Biomarkers of Anti-PD-1 Therapy
2.11. Random Forest
3. Result
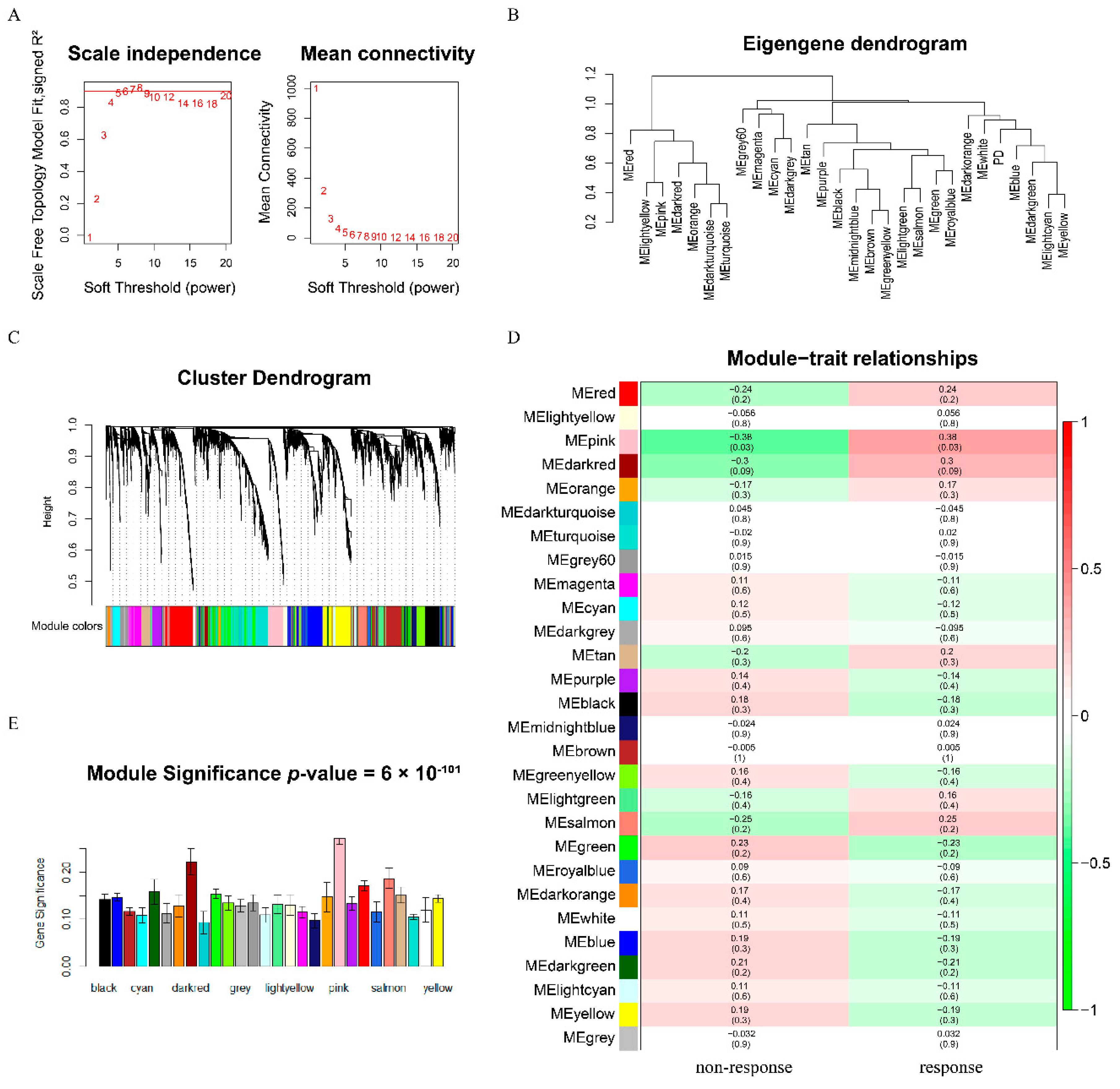
3.1. Construction of Weighted Co-Expression Network and Identification of Key Modules
3.2. Potential Functional Roles of Genes in the Pink Module
3.3. Identification of Hub Genes
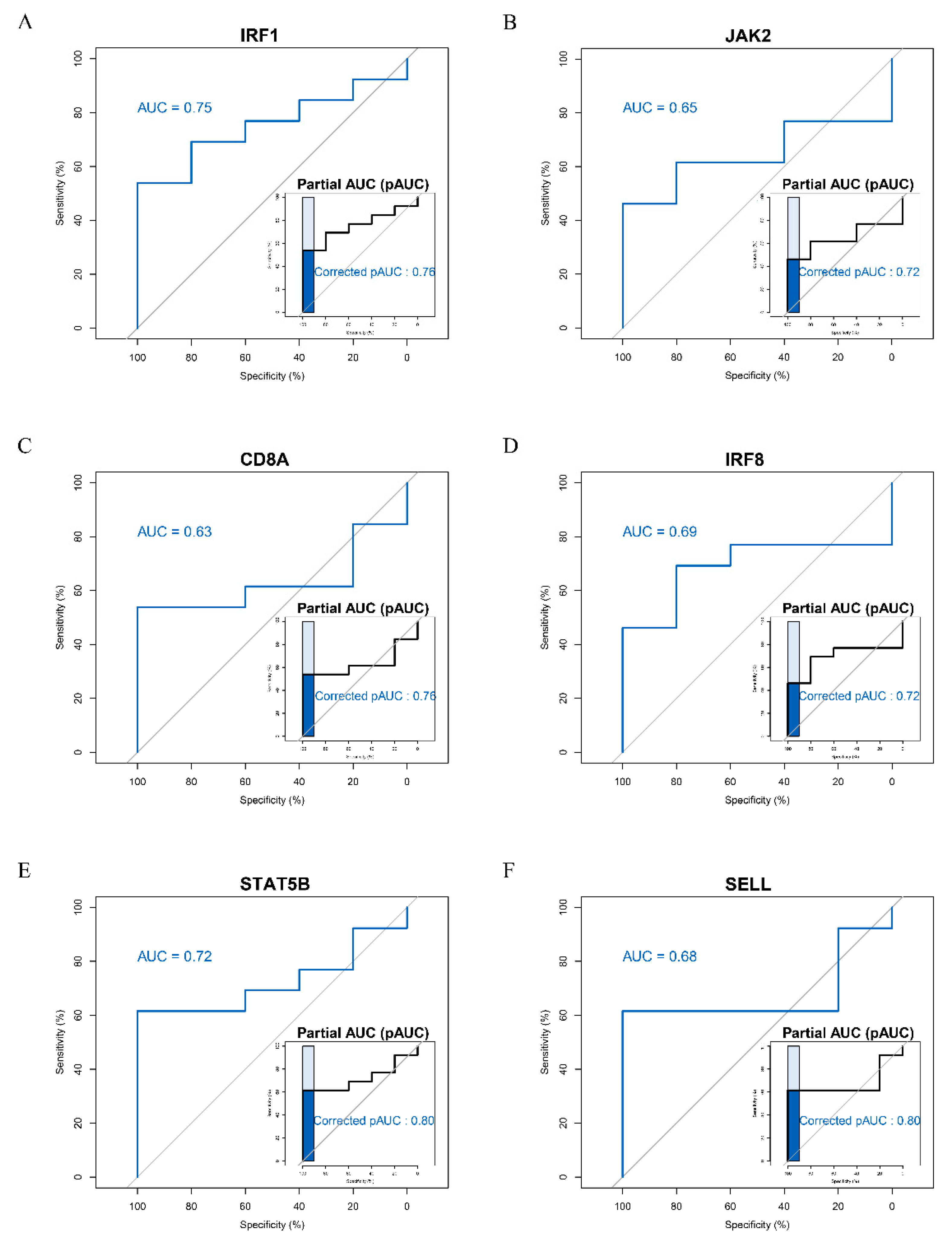
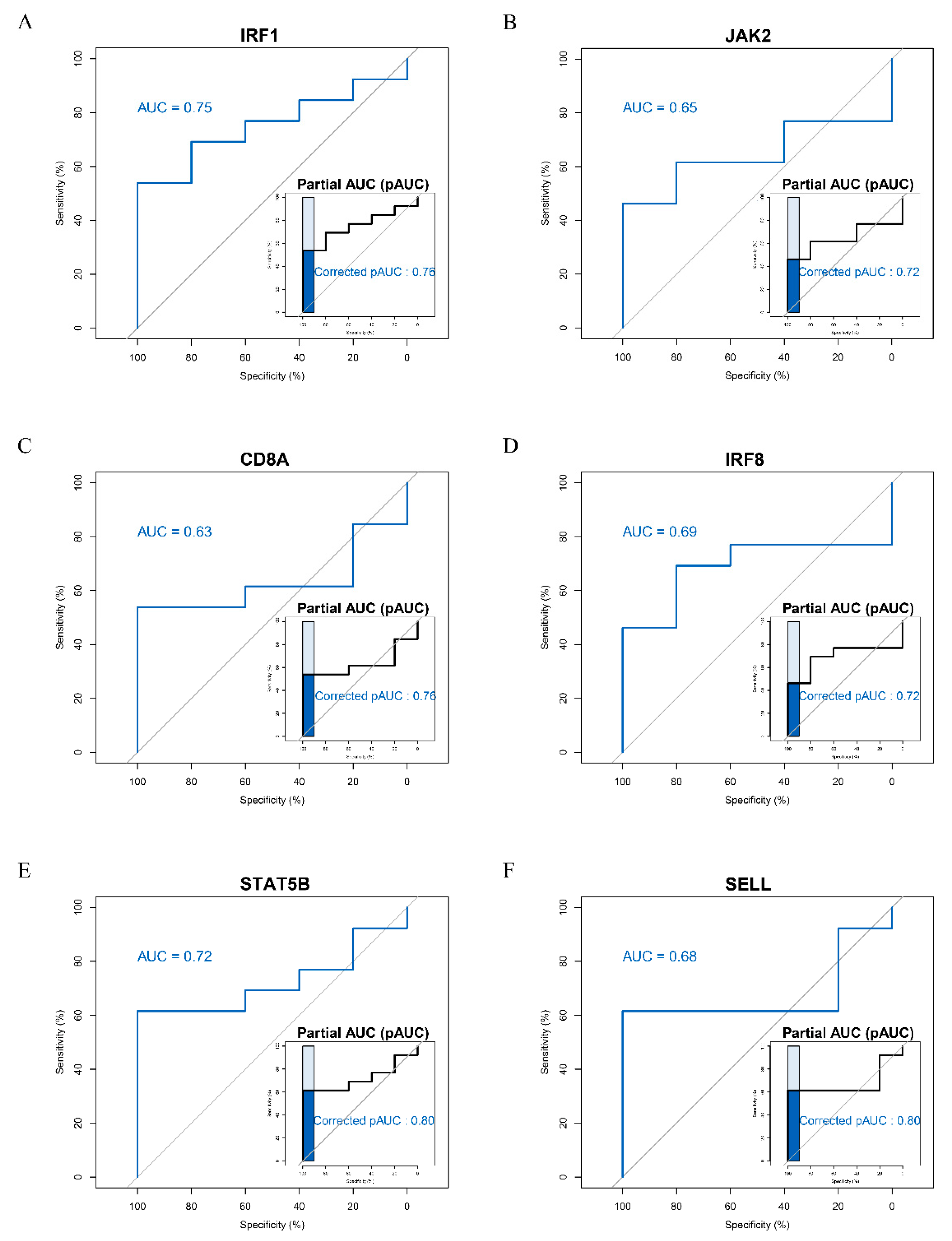
3.4. ROC Curve Analysis of Hub Genes
3.5. Gene Expression of Hub Genes
3.6. Functional Enrichment Analysis by Gene Set Variation Analysis (GSVA)
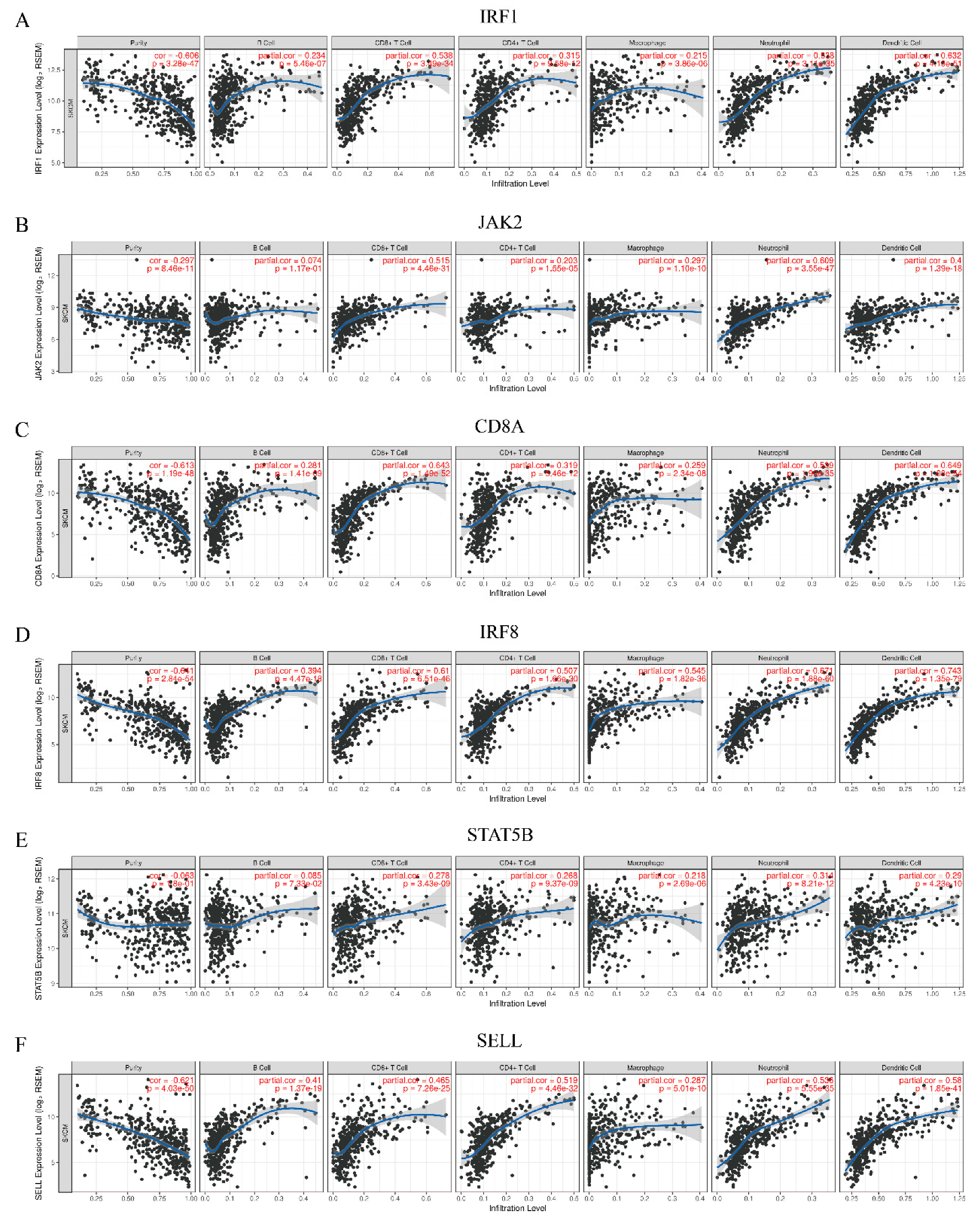
3.7. Analysis of Association Between Hub Genes and Immune Infiltration Level
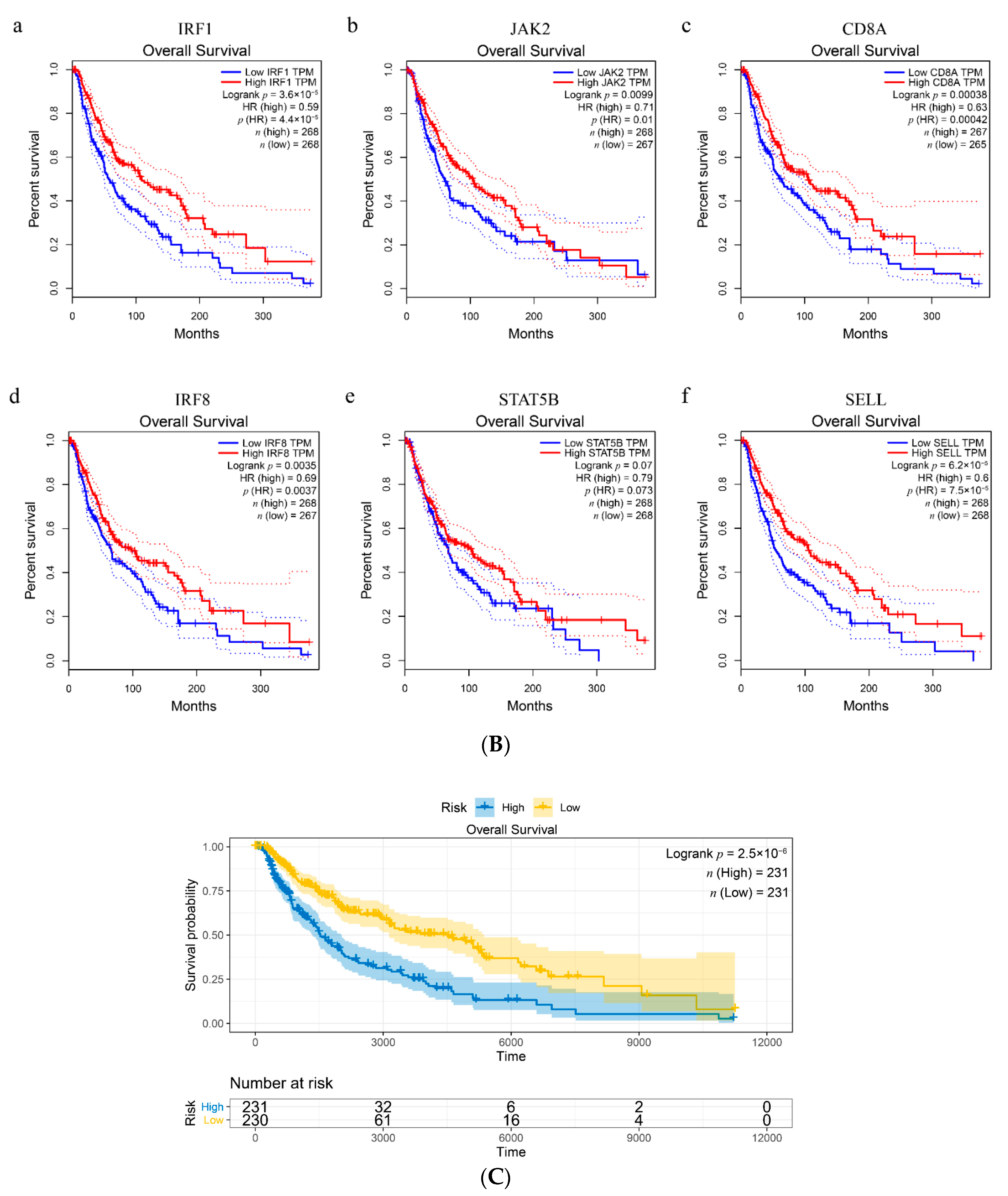
3.8. Validation of Hub Genes in The Cancer Genome Atlas (TCGA) Datasets
3.9. Relationships between Hub Genes and Biomarkers of Anti-PD-1 Therapy
3.10. The Random Forest Model of Hub Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schadendorf, D.; van Akkooi, A.C.J.; Berking, C.; Griewank, K.G.; Gutzmer, R.; Hauschild, A.; Stang, A.; Roesch, A.; Ugurel, S. Melanoma. Lancet 2018, 392, 971–984. [Google Scholar] [CrossRef]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Maleki Vareki, S.; Garrigos, C.; Duran, I. Biomarkers of response to PD-1/PD-L1 inhibition. Crit. Rev. Oncol. Hematol. 2017, 116, 116–124. [Google Scholar] [CrossRef]
- Marrone, K.A.; Ying, W.; Naidoo, J. Immune-related adverse events from immune checkpoint inhibitors. Clin. Pharmacol. Ther. 2016, 100, 242–251. [Google Scholar] [CrossRef]
- Andrews, A. Treating with checkpoint inhibitors-figure $1 million per patient. Am. Health Drug Benefits 2015, 8, 9. [Google Scholar]
- Meng, X.; Huang, Z.; Teng, F.; Xing, L.; Yu, J. Predictive biomarkers in PD-1/PD-L1 checkpoint blockade immunotherapy. Cancer Treat. Rev. 2015, 41, 868–876. [Google Scholar] [CrossRef]
- Yi, M.; Jiao, D.; Xu, H.; Liu, Q.; Zhao, W.; Han, X.; Wu, K. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol. Cancer 2018, 17, 129. [Google Scholar] [CrossRef]
- Teng, F.; Meng, X.; Kong, L.; Yu, J. Progress and challenges of predictive biomarkers of anti PD-1/PD-L1 immunotherapy: A systematic review. Cancer Lett. 2018, 414, 166–173. [Google Scholar] [CrossRef]
- Sunshine, J.C.; Nguyen, P.L.; Kaunitz, G.J.; Cottrell, T.R.; Berry, S.; Esandrio, J.; Xu, H.; Ogurtsova, A.; Bleich, K.B.; Cornish, T.C.; et al. PD-L1 expression in melanoma: A quantitative immunohistochemical antibody comparison. Clin. Cancer Res. 2017, 23, 4938–4944. [Google Scholar] [CrossRef] [Green Version]
- Cottrell, T.R.; Taube, J.M. PD-L1 and emerging biomarkers in immune checkpoint blockade therapy. Cancer J. 2018, 24, 41–46. [Google Scholar] [CrossRef]
- Patel, S.; Osada, T.; Osada, K.; Hurwitz, H.; Lyerly, H.; Morse, M. Modulation of immune system inhibitory checkpoints in colorectal cancer. Curr. Colorectal Cancer Rep. 2013, 9, 391–397. [Google Scholar] [CrossRef]
- Johnson, D.B.; Bordeaux, J.; Kim, J.Y.; Vaupel, C.; Rimm, D.L.; Ho, T.H.; Joseph, R.W.; Daud, A.I.; Conry, R.M.; Gaughan, E.M.; et al. Quantitative spatial profiling of PD-1/PD-L1 interaction and HLA-DR/IDO-1 predicts improved outcomes of anti-PD-1 therapies in metastatic melanoma. Clin. Cancer Res. 2018, 24, 5250–5260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilain, R.E.; Menzies, A.M.; Wilmott, J.S.; Kakavand, H.; Madore, J.; Guminski, A.; Liniker, E.; Kong, B.Y.; Cooper, A.J.; Howle, J.R.; et al. Dynamic changes in PD-L1 expression and immune infiltrates early during treatment predict response to PD-1 blockade in melanoma. Clin. Cancer Res. 2017, 23, 5024–5033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, C.; Jiang, Y.; Li, P.; Wang, Y.; Xue, J.; Li, N.; Li, D.; Wang, R.; Dang, Y.; Hu, Z.; et al. Dynamic change of PD-L1 expression on circulating tumor cells in advanced solid tumor patients undergoing PD-1 blockade therapy. Oncoimmunology 2018, 7, e1438111. [Google Scholar] [CrossRef]
- Danilova, L.; Wang, H.; Sunshine, J.; Kaunitz, G.J.; Cottrell, T.R.; Xu, H.; Esandrio, J.; Anders, R.A.; Cope, L.; Pardoll, D.M.; et al. Association of PD-1/PD-L axis expression with cytolytic activity, mutational load, and prognosis in melanoma and other solid tumors. Proc. Natl. Acad. Sci. USA 2016, 113, E7769–E7777. [Google Scholar] [CrossRef] [Green Version]
- Messina, J.L.; Fenstermacher, D.A.; Eschrich, S.; Qu, X.; Berglund, A.E.; Lloyd, M.C.; Schell, M.J.; Sondak, V.K.; Weber, J.S.; Mule, J.J. 12-chemokine gene signature identifies lymph node-like structures in melanoma: Potential for patient selection for immunotherapy? Sci. Rep. 2012, 2, 765. [Google Scholar] [CrossRef] [Green Version]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ-related mrna profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef]
- Ribas, A.; Robert, C.; Hodi, F.S.; Wolchok, J.D.; Joshua, A.; Hwu, W.-J. Association of response to programmed death receptor 1 (PD-1) blockade with pembrolizumab (MK-3475) with an interferon-inflammatory immune gene signature. J. Clin. Oncol. 2015, 33, 3001. [Google Scholar] [CrossRef]
- Varn, F.S.; Wang, Y.; Cheng, C. A B cell-derived gene expression signature associates with an immunologically active tumor microenvironment and response to immune checkpoint blockade therapy. Oncoimmunology 2019, 8, e1513440. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Z.; Shen, J.X.; Pan, Y.; Pu, J.; Li, Y.G.; Shao, X.H.; Wang, W.P. Weighted gene correlation network analysis (WGCNA) detected loss of MAGI2 promotes chronic kidney disease (CKD) by podocyte damage. Cell Physiol. Biochem. 2018, 51, 244–261. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Fu, S.; Lin, X.; Zheng, J.; Pu, J.; Gu, Y.; Deng, W.; Liu, Y.; He, Z.; Liang, W.; et al. miR-92b-3p functions as a key gene in esophageal squamous cell cancer as determined by co-expression analysis. Onco Targets Ther. 2019, 12, 8339–8353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clough, E.; Barrett, T. The gene expression omnibus database. Methods Mol. Biol 2016, 1418, 93–110. [Google Scholar] [PubMed] [Green Version]
- Parkinson, H.; Kapushesky, M.; Shojatalab, M.; Abeygunawardena, N.; Coulson, R.; Farne, A.; Holloway, E.; Kolesnykov, N.; Lilja, P.; Lukk, M.; et al. Arrayexpress—A public database of microarray experiments and gene expression profiles. Nucleic Acids Res. 2007, 35, D747–D750. [Google Scholar] [CrossRef] [PubMed]
- Papatheodorou, I.; Fonseca, N.A.; Keays, M.; Tang, Y.A.; Barrera, E.; Bazant, W.; Burke, M.; Fullgrabe, A.; Fuentes, A.M.; George, N.; et al. Expression atlas: Gene and protein expression across multiple studies and organisms. Nucleic Acids Res. 2018, 46, D246–D251. [Google Scholar] [CrossRef] [PubMed]
- Seymour, L.; Bogaerts, J.; Perrone, A.; Ford, R.; Schwartz, L.H.; Mandrekar, S.; Lin, N.U.; Litiere, S.; Dancey, J.; Chen, A.; et al. Irecist: Guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 2017, 18, e143–e152. [Google Scholar] [CrossRef] [Green Version]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martin-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 2017, 171, 934–949.e916. [Google Scholar] [CrossRef] [Green Version]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. Networkanalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. Cytohubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.C.; Muller, M. Proc: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 2011, 12, 77. [Google Scholar] [CrossRef]
- Hsu, M.J.; Chang, Y.C.; Hsueh, H.M. Biomarker selection for medical diagnosis using the partial area under the ROC curve. BMC Res. Notes 2014, 7, 25. [Google Scholar] [CrossRef] [Green Version]
- Hajian-Tilaki, K. Receiver operating characteristic (ROC) curve analysis for medical diagnostic test evaluation. Caspian J. Intern. Med. 2013, 4, 627–635. [Google Scholar]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 2013, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. Timer: A web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Severson, E.; Pignon, J.C.; Zhao, H.; Li, T.; Novak, J.; Jiang, P.; Shen, H.; Aster, J.C.; Rodig, S.; et al. Comprehensive analyses of tumor immunity: Implications for cancer immunotherapy. Genome Biol. 2016, 17, 174. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitbon, O.; McLaughlin, V.V.; Badesch, D.B.; Barst, R.J.; Black, C.; Galie, N.; Humbert, M.; Rainisio, M.; Rubin, L.J.; Simonneau, G. Survival in patients with class III idiopathic pulmonary arterial hypertension treated with first line oral bosentan compared with an historical cohort of patients started on intravenous epoprostenol. Thorax 2005, 60, 1025–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breiman, L. Random forests. Machine Learning 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Genuer, R.; Poggi, J.; Tuleau-Malot, C. Variable selection using random forests. Pattern Recogn. Lett. 2010, 31, 2225–2236. [Google Scholar] [CrossRef] [Green Version]
- Prat, A.; Navarro, A.; Pare, L.; Reguart, N.; Galvan, P.; Pascual, T.; Martinez, A.; Nuciforo, P.; Comerma, L.; Alos, L.; et al. Immune-related gene expression profiling after PD-1 blockade in non-small cell lung carcinoma, head and neck squamous cell carcinoma, and melanoma. Cancer Res. 2017, 77, 3540–3550. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Hsu, J.; Hodgins, J.J.; Marathe, M.; Nicolai, C.J.; Bourgeois-Daigneault, M.C.; Trevino, T.N.; Azimi, C.S.; Scheer, A.K.; Randolph, H.E.; Thompson, T.W.; et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Investig. 2018, 128, 4654–4668. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [Green Version]
- Dulos, J.; Carven, G.J.; van Boxtel, S.J.; Evers, S.; Driessen-Engels, L.J.; Hobo, W.; Gorecka, M.A.; de Haan, A.F.; Mulders, P.; Punt, C.J.; et al. PD-1 blockade augments Th1 and Th17 and suppresses Th2 responses in peripheral blood from patients with prostate and advanced melanoma cancer. J. Immunother. 2012, 35, 169–178. [Google Scholar] [CrossRef]
- Lu, C.; Talukder, A.; Savage, N.M.; Singh, N.; Liu, K. JAK-STAT-mediated chronic inflammation impairs cytotoxic T lymphocyte activation to decrease anti-PD-1 immunotherapy efficacy in pancreatic cancer. Oncoimmunology 2017, 6, e1291106. [Google Scholar] [CrossRef] [Green Version]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Garris, C.S.; Arlauckas, S.P.; Kohler, R.H.; Trefny, M.P.; Garren, S.; Piot, C.; Engblom, C.; Pfirschke, C.; Siwicki, M.; Gungabeesoon, J.; et al. Successful anti-PD-1 cancer immunotherapy requires T cell-dendritic cell crosstalk involving the cytokines IFN-γ and IL-12. Immunity 2018, 49, 1148–1161.e1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saravia, D.; Okabe, N.; Park, W.; Kwon, D.; Mezquita, L.; Chae, Y.; Mudad, R.; Jahanzeb, M.; Besse, B.; Suzuki, H.; et al. Neutrophil-lymphocyte-ratio to complement the prediction ability of PD-L1 expression for outcomes in patients with advanced non-small cell lung cancer treated with PD-1/ PD-L1 inhibitors. J. Clin. Oncol. 2018, 36, e15102. [Google Scholar] [CrossRef]
- Gauci, M.L.; Lanoy, E.; Champiat, S.; Caramella, C.; Ammari, S.; Aspeslagh, S.; Varga, A.; Baldini, C.; Bahleda, R.; Gazzah, A.; et al. Long-term survival in patients responding to anti- PD-1/ PD-L1 therapy and disease outcome upon treatment discontinuation. Clin. Cancer Res. 2019, 25, 946–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitt, J.M.; Vetizou, M.; Daillere, R.; Roberti, M.P.; Yamazaki, T.; Routy, B.; Lepage, P.; Boneca, I.G.; Chamaillard, M.; Kroemer, G.; et al. Resistance mechanisms to immune-checkpoint blockade in cancer: Tumor-intrinsic and -extrinsic factors. Immunity 2016, 44, 1255–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishino, M.; Ramaiya, N.H.; Hatabu, H.; Hodi, F.S. Monitoring immune-checkpoint blockade: Response evaluation and biomarker development. Nat. Rev. Clin. Oncol. 2017, 14, 655–668. [Google Scholar] [CrossRef]
- Liu, C.; He, H.; Li, X.; Su, M.A.; Cao, Y. Dynamic metrics-based biomarkers to predict responders to anti-PD-1 immunotherapy. Br. J. Cancer 2019, 120, 346–355. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Wang, Y.; Sun, J.; Tan, T.; Cai, X.; Lin, P.; Tan, Y.; Zheng, B.; Wang, B.; Wang, J.; et al. Role of CXCR3 signaling in response to anti-PD-1 therapy. EBioMedicine 2019, 48, 169–177. [Google Scholar] [CrossRef]
- Chow, M.T.; Ozga, A.J.; Servis, R.L.; Frederick, D.T.; Lo, J.A.; Fisher, D.E.; Freeman, G.J.; Boland, G.M.; Luster, A.D. Intratumoral activity of the CXCR3 chemokine system is required for the efficacy of anti-PD-1 therapy. Immunity 2019, 50, 1498–1512.e1495. [Google Scholar] [CrossRef]
- Reynier, F.; Petit, F.; Paye, M.; Turrel-Davin, F.; Imbert, P.E.; Hot, A.; Mougin, B.; Miossec, P. Importance of correlation between gene expression levels: Application to the type I interferon signature in rheumatoid arthritis. PLoS ONE 2011, 6, e24828. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.P.; Kurzrock, R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Giulietti, M.; Occhipinti, G.; Principato, G.; Piva, F. Identification of candidate mirna biomarkers for pancreatic ductal adenocarcinoma by weighted gene co-expression network analysis. Cell Oncol. (Dordr.) 2017, 40, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Heinzel, S.; Marchingo, J.M.; Horton, M.B.; Hodgkin, P.D. The regulation of lymphocyte activation and proliferation. Curr. Opin. Immunol. 2018, 51, 32–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smithy, J.W.; Moore, L.M.; Pelekanou, V.; Rehman, J.; Gaule, P.; Wong, P.F.; Neumeister, V.M.; Sznol, M.; Kluger, H.M.; Rimm, D.L. Nuclear IRF-1 expression as a mechanism to assess “capability” to express PD-L1 and response to PD-1 therapy in metastatic melanoma. J. Immunother. Cancer 2017, 5, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Xiao, L.-S.; Dong, Z.-Y.; Li, W.-W.; Wang, K.-Y.; Wu, D.-H.; Liu, L. Potential predictive value of JAK2 expression for pan-cancer response to PD-1 blockade immunotherapy. Transl. Cancer Res. 2018, 7, 462–471. [Google Scholar] [CrossRef]
- Kioussis, D.; Ellmeier, W. Chromatin and CD4, CD8A and CD8B gene expression during thymic differentiation. Nat. Rev. Immunol. 2002, 2, 909–919. [Google Scholar] [CrossRef]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B.; et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017, 545, 60–65. [Google Scholar] [CrossRef] [Green Version]
- Fumet, J.D.; Richard, C.; Ledys, F.; Klopfenstein, Q.; Joubert, P.; Routy, B.; Truntzer, C.; Gagne, A.; Hamel, M.A.; Guimaraes, C.F.; et al. Prognostic and predictive role of CD8 and PD-L1 determination in lung tumor tissue of patients under anti-PD-1 therapy. Br. J. Cancer 2018, 119, 950–960. [Google Scholar] [CrossRef]
- Schiavoni, G.; Gabriele, L.; Mattei, F. The dual role of IRF8 in cancer immunosurveillance. Oncoimmunology 2013, 2, e25476. [Google Scholar] [CrossRef] [Green Version]
- Muhitch, J.B.; Hoffend, N.C.; Azabdaftari, G.; Miller, A.; Bshara, W.; Morrison, C.D.; Schwaab, T.; Abrams, S.I. Tumor-associated macrophage expression of interferon regulatory factor-8 (IRF8) is a predictor of progression and patient survival in renal cell carcinoma. J. Immunother. Cancer 2019, 7, 155. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, J.M.M.M.; Heim, L.; Springel, R.; Mittler, S.; Gafencu, D.; Hartmann, A.; Sirbu, H.; Finotto, S. The role of signal transducer and activator of transcription protein 5 (STAT5) in non-small cell lung cancer (NSCLC). J. Immunol. 2016, 196, 73.18. [Google Scholar]
- Verdeil, G.; Lawrence, T.; Schmitt-Verhulst, A.M.; Auphan-Anezin, N. Targeting STAT3 and STAT5 in tumor-associated immune cells to improve immunotherapy. Cancers (Basel) 2019, 11, 1832. [Google Scholar] [CrossRef] [Green Version]
- Majri, S.S.; Fritz, J.M.; Villarino, A.V.; Zheng, L.; Kanellopoulou, C.; Chaigne-Delalande, B.; Gronholm, J.; Niemela, J.E.; Afzali, B.; Biancalana, M.; et al. STAT5B: A differential regulator of the life and death of CD4(+) effector memory T cells. J. Immunol. 2018, 200, 110–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivetic, A.; Hoskins Green, H.L.; Hart, S.J. L-selectin: A major regulator of leukocyte adhesion, migration and signaling. Front. Immunol. 2019, 10, 1068. [Google Scholar] [CrossRef] [Green Version]
- Watson, H.A.; Durairaj, R.R.P.; Ohme, J.; Alatsatianos, M.; Almutairi, H.; Mohammed, R.N.; Vigar, M.; Reed, S.G.; Paisey, S.J.; Marshall, C.; et al. L-selectin enhanced T cells improve the efficacy of cancer immunotherapy. Front. Immunol. 2019, 10, 1321. [Google Scholar] [CrossRef]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Chai, Z.; Li, Y.; Long, F.; Hao, Y.; Pan, G.; Liu, M.; Li, B. Identification of Potential Biomarkers for Anti-PD-1 Therapy in Melanoma by Weighted Correlation Network Analysis. Genes 2020, 11, 435. https://doi.org/10.3390/genes11040435
Wang X, Chai Z, Li Y, Long F, Hao Y, Pan G, Liu M, Li B. Identification of Potential Biomarkers for Anti-PD-1 Therapy in Melanoma by Weighted Correlation Network Analysis. Genes. 2020; 11(4):435. https://doi.org/10.3390/genes11040435
Chicago/Turabian StyleWang, Xuanyi, Zixuan Chai, Yinghong Li, Fei Long, Youjin Hao, Guizhi Pan, Mingwei Liu, and Bo Li. 2020. "Identification of Potential Biomarkers for Anti-PD-1 Therapy in Melanoma by Weighted Correlation Network Analysis" Genes 11, no. 4: 435. https://doi.org/10.3390/genes11040435