The Regulation of Homologous Recombination by Helicases


Department of Molecular Biology and Biochemistry, Rutgers, The State University of New Jersey, Center for Advanced Biochemistry and Medicine, 679 Hoes Lane West, Piscataway, NJ 08854, USA
*
Author to whom correspondence should be addressed.
Genes 2020, 11(5), 498; https://doi.org/10.3390/genes11050498
Submission received: 2 April 2020
/
Revised: 29 April 2020
/
Accepted: 29 April 2020
/
Published: 1 May 2020
(This article belongs to the Special Issue DNA Helicases: Mechanisms, Biological Pathways, and Disease Relevance)
Abstract
:Homologous recombination is essential for DNA repair, replication and the exchange of genetic material between parental chromosomes during meiosis. The stages of recombination involve complex reorganization of DNA structures, and the successful completion of these steps is dependent on the activities of multiple helicase enzymes. Helicases of many different families coordinate the processing of broken DNA ends, and the subsequent formation and disassembly of the recombination intermediates that are necessary for template-based DNA repair. Loss of recombination-associated helicase activities can therefore lead to genomic instability, cell death and increased risk of tumor formation. The efficiency of recombination is also influenced by the ‘anti-recombinase’ effect of certain helicases, which can direct DNA breaks toward repair by other pathways. Other helicases regulate the crossover versus non-crossover outcomes of repair. The use of recombination is increased when replication forks and the transcription machinery collide, or encounter lesions in the DNA template. Successful completion of recombination in these situations is also regulated by helicases, allowing normal cell growth, and the maintenance of genomic integrity.
1. Introduction
Homologous recombination (HR) is an essential cellular process, which is required to repair DNA double strand breaks (DSBs), restart collapsed replication forks, and for the rearrangement of genetic information from parental chromosomes during meiosis [1]. Helicases are the enzymes that bind to nucleic acids, and translocate along the molecule, separating base-paired regions using energy from ATP. Helicases have been shown to be necessary to carry out various steps of recombination, and also for regulation of the rate and outcomes of recombination (Table 1). Several of these proteins are classical helicases, which act to unwind single-stranded DNA, whereas others can be classed as translocases, which use their motor activity to move along double-stranded DNA regions [2]. In eukaryotes, recombination proceeds by resection of DNA ends to form a single-stranded region, which is initially bound by Replication Protein A (RPA), and subsequently by RAD51 [1,3]. The nucleoprotein filament loaded with RAD51 invades the homologous DNA regions, creating a displacement loop (D-loop), where sequences from the broken DNA molecule and homologous template pair as ‘heteroduplex’ DNA. A DNA polymerase can subsequently add nucleotides at the free 3′ end, to restore the sequence around the break site. Several competing pathways then complete the HR process by disassembly of recombination intermediates. RAD51-mediated recombination is also essential for DNA replication during S phase, by restoring stalled replication forks, and by restarting replication after fork collapse [4,5,6].
The importance of helicase activities for recombinational repair is demonstrated by the pathology that arises in individuals inheriting mutant copies of helicase genes. Homozygous mutations affecting the RECQ helicases, BLM and WRN, as seen in Bloom Syndrome and Werner Syndrome, respectively, cause defective DNA repair, tumor susceptibility and abnormal development [7,8,9,10]. Understanding the contribution of helicases to recombination is therefore essential to identify how these proteins enable normal growth of the cell.
2. Regulation of DNA Double-Strand Break Resection by Helicases
Repair of a DNA double-strand break by HR requires ‘resection’ of the broken DNA end, which involves the production of a single-stranded region at the 3′ end [1]. Resection is considered a key step that commits repair of a DSB to the HR pathway for repair. Both the regulation and mechanism of DSB resection involve the activity of specific helicases (Figure 1). The first steps of end resection involve the MRN complex (MRX in yeast), a heterotrimeric complex of Mre11, Rad50 and Nbs1, which has affinity for broken DNA ends. Association of the CtIP protein with MRN stimulates the nuclease activity of Mre11 to begin short-range resection, and create a small region of single-stranded DNA (ssDNA) at the break site. Certain DNA structures, such as G-quadruplexes, can inhibit the process of resection. Recruitment of the Pif1 helicase aids in unwinding these complex DNA structures, and improves the efficiency of HR [11]. The helicase RECQL4 impacts the process of resection in a cell cycle-dependent manner, by helping the MRN complex recruit CtIP during S and G2 phases of the cell cycle [12]. RECQL4 also suppresses use of HR during the G1 phase of the cell cycle, by favoring the competing NHEJ pathway [13]. In this case, RECQL4 binds to Ku70/Ku80, stabilizing them at the break site, and directing repair toward NHEJ. The RECQ helicase, WRN (Werner syndrome helicase), is also able to promote NHEJ through an interaction with Ku70. In addition to its helicase function, WRN also has exonuclease activity, which can remove modified nucleotides at DSBs to promote NHEJ [14,15,16,17,18]. The helicase SMARCAL1/HARP also promotes NHEJ at this step, by re-annealing DSB ends that have unwound, thereby facilitating the binding of Ku70 [19].
Once short-range end resection is achieved, the single-stranded overhang is lengthened through ‘long-range resection’, to create a more substantial ssDNA substrate for nucleoprotein filament formation and strand invasion [20]. Long-range resection is achieved by two parallel pathways. One of these pathways is dependent on the exonuclease, Exo1, which removes one strand of the DNA duplex in a processive manner in the 5′-3′ direction. The other pathway involves a RECQ helicase working in combination with DNA2. In budding yeast, Sgs1 is the key RECQ helicase for long-range resection, and, in vertebrates, Bloom Syndrome helicase (BLM) provides the equivalent activity [21,22,23,24,25]. This evolutionarily conserved approach to DSB resection involves the unwinding of the DNA duplex by the RECQ helicase, followed by nicking of the resected strand by an endonuclease activity in DNA2. Studies in vitro and in budding yeast have demonstrated that RPA (Replication Protein A) plays an important function in ensuring productive long-range resection [24,26]. The RECQ helicases, BLM and RECQL1, also appear to contribute to resection by stimulating the exonuclease activity of Exo1 at DSBs [24,27,28]. The ability of WRN to substitute for BLM for DNA2-dependent resection has been a matter of some debate. Initial studies indicated that WRN is not able to provide the necessary unwinding activity for resection [23]. Other reports have supported the idea that WRN is also able to mediate DNA2-dependent resection, potentially after activation by phosphorylation by the cell cycle-dependent kinase, CDK1 [29,30,31,32]. In addition to acting as a nuclease, DNA2 also has helicase activity. This helicase activity contributes to resection by removing single-stranded DNA products that are produced by the unwinding and cutting of DSBs by DNA2 with its RECQ helicase partner [33,34]
Several other helicases contribute to regulation of DSB resection. DNA Helicase B (HELB) counteracts unwinding of DSBs by RECQ helicases, using its 5′-3′ ssDNA translocase activity [35]. This repressive effect on resection is exerted during G1 phase of the cell cycle. During the transition to the S/G2 phase of the cell cycle, HELB is exported from the nucleus, allowing increased resection and use of the HR pathway. The yeast SNF2-type ATP-ase, Fun30, and its mammalian ortholog, SMARCAD1, contribute to extensive resection of DSBs [36]. These proteins use ATP-dependent translocase activity to move along chromatin in the vicinity of DSBs, remodeling chromatin to allow extensive resection and normal HR [37,38]. Loss of either Fun30 or SMARCAD1 therefore makes cells hypersensitive to DSBs induced by treatment with the topoisomerase inhibitor, camptothecin. FANCJ is found alongside BLM during long-range resection, and appears to contribute to the efficiency of resection by stabilizing BLM, or by removing obstacles such as complex DNA structures [39,40]. FANCJ also helps recruit CtIP, thereby enabling the initial steps of resection [41]. The MCM8-9 helicase complex is also reported to promote resection [42], although other reports suggest it acts at a later stage of HR [43,44,45].
3. Single-Stranded DNA Binding Protein Displacement
3′ DNA overhangs produced through DSB resection become rapidly coated with single-stranded DNA binding proteins. In prokaryotes, SSB (single-strand binding protein) fulfills this role [1]. In eukaryotes, RPA, which is a heterotrimer of RPA70, RPA32 and RPA14, binds to single-stranded DNA overhangs. For recombination to take place, RPA must be replaced by RAD51, which is equivalent to RecA in prokaryotes. The nucleoprotein filament of single-stranded DNA coated with RAD51 forms a ‘presynaptic filament’, which pairs with homologous DNA regions through strand invasion, allowing formation of a displacement loop (D-loop) and template-based repair. In vertebrate cells, RAD51 paralogs (RAD51B, RAD51C, RAD51D, XRCC2, XRCC3 and DMC1) are also present in recombinogenic nucleoprotein filaments, and ensure efficient and productive recombination [46]. Assembly of the RAD51-containing nucleoprotein filament is therefore an essential step in recombination, and a step at which the rate of recombination can be regulated. Several helicase enzymes regulate the loading and removal of single-strand binding proteins (Figure 2). Prokaryotic UvrD displaces RecA, thereby limiting the rate of HR, and attenuating DNA damage signaling [47]. In budding yeast, the Srs2 helicase likewise acts as an ‘anti-recombinase’, by removing RAD51 from the nucleoprotein filament at resected DSBs [48,49]. Srs2 is also essential for regulating recombination during replication, and is recruited to SUMO-modified PCNA under conditions of replication stress [50,51]. By modulating the rate of HR, Srs2 may help prevent the appearance of defective HR intermediates [52].
Several helicases in vertebrates have been reported to act similarly to UvrD and Srs2, by reducing the rate of HR through displacement of single-stranded DNA binding proteins. A bioinformatic approach identified PCNA-Associated Recombination Inhibitor (PARI), a protein containing a UvrD-type helicase domain, which has a similar domain organization to Srs2, as a potential mammalian anti-recombinase [53]. Loss of PARI is reported to increase RAD51 loading at break sites, causing an elevated level of HR that is associated with genomic instability. PARI appears to stimulate the ATPase activity of RAD51, promoting RAD51 to dissociate from the DNA. The ability of PARI to displace RAD51 is quite weak, however, and only achieved with stoichiometric amounts of protein. The importance of the PARI helicase domain is likewise unclear, because it does not appear to have a functional ATPase activity. The exact mechanism for PARI-mediated regulation of HR is therefore still not fully characterized, and it may play a more important role in regulating other aspects of HR (See Section 4, below). In contrast, the Superfamily 2 helicase, FANCJ (Fanconi Anemia Complementation group J), is able to dissociate DNA complexes in a manner that is clearly dependent on ATP hydrolysis [54]. FANCJ displaces RAD51 from DNA in vitro, thereby reducing the efficiency of RAD51-dependent DNA strand-exchange. F-box DNA helicase 1 (FBH1) was originally shown to repress recombination in S. pombe [55,56]. Anti-recombinase activity of FBH1 was subsequently reported in vertebrates as well, including in human cells [57,58,59]. As with FANCJ, the ability of FBH1 to displace RAD51 is dependent on its helicase activity, which allows the FBH1 protein to move along the nucleoprotein filament, facilitating removal of RAD51. FBH1 also participates in an SCF complex, which ubiquitinates RAD51, leading to RAD51 removal [60].
RECQ helicases, such as Sgs1 and BLM, promote HR by mediating the ‘long-range’ resection of DSBs (see previous section). Several RECQ helicases also have anti-recombinogenic activity, however, which is mediated in part by removal of RAD51 from the presynaptic filament. Single-molecule imaging studies show that Sgs1 from budding yeast can displace RAD51 from DNA [61]. This function is conserved in mammalian species, as in vitro studies have demonstrated that the BLM helicase can also remove RAD51 from DNA [62]. Work in our lab showed that this anti-recombinase activity of BLM plays an important role in regulating the efficiency of HR in cells [63]. Cells that lack the HR factors, BRCA1 or BRCA2, normally show a substantial defect in RAD51 loading at DNA break sites, which results in defective HR and genomic instability. Co-deletion of BLM rescues this HR defect, by allowing increased accumulation of RAD51 at resected DSBs. In addition to BLM, RECQL5, another RECQ helicase, disrupts RAD51 filaments [64,65]. The loss of RECQL5 leads to hyper-recombination, which correlates with genomic instability and cancer susceptibility in Recql5–/– mice.
The FIGNL1 (Fidgetin-like 1) helicase is required for normal HR, and this regulatory function appears to depend on the ability of FIGNL1 to displace RAD51 [66,67]. FIGNL1 is an AAA+ ATPase helicase, but the ATPase activity does not seem to be required for RAD51 displacement. In recent years, Polθ (DNA Polymerase theta) has also emerged as an important regulator of DNA repair [68]. Polθ has multiple enzymatic activities, including helicase activity. The helicase activity of Polθ is important for directing repair of DSBs toward ‘alternative end-joining’ instead of toward HR. This effect is achieved by removal of RPA from single-stranded regions of DNA by Polθ [69,70]. When Polθ is absent, rates of HR increase, and RAD51 accumulates at increased levels at break sites.
4. Dissolution of D-Loops
After the formation of a RAD51-loaded presynaptic filament, the broken DNA end pairs with a homologous DNA region, and begins a process called strand invasion [1]. The structure formed when an ssDNA filament invades a homologous DNA duplex is referred to as a displacement loop, or D-Loop (Figure 3). The formation of heteroduplex DNA, with the broken DNA molecule paired to the homologous template DNA, allows sequence at the break to be restored by a DNA polymerase enzyme. D-loops can be dissolved and displaced at various points by a number of different helicases which help to prevent erroneous strand invasion or limit the extent of polymerase activity. Yeast Srs2, in addition to displacing RAD51 from resected DNA ends [48,49], can dismantle D-loops, which helps to eliminate nonproductive recombination intermediates [71,72]. The disassembly of D-loops also prevents long-range DNA extension, reducing the frequency of formation of double-Holliday junctions intermediates, which can be resolved as crossovers. The Superfamily 2 (SF2) helicase Mph1 (Mutator Phenotype 1) acts in parallel with Srs2 to promote non-crossover outcomes to recombination [73]. The ability of Mph1 to dissociate D loops is strictly dependent on its helicase activity.
The homologs of Srs2 and Mph1 in mammals have similar D-loop dissociation activity. RTEL1 (Regulator of Telomere Length) is a putative Srs2 homolog that limits recombination by dissociating the D-loop recombination intermediates [74]. This effect of RTEL1 may be particularly important for suppressing recombination at telomeres, because RTEL1-knockout mouse cells show chromosome instability and telomere loss that is associated with embryonic lethality [75]. PARI, like Srs2, can promote non-crossover recombination by unwinding D-loops, which inhibits extension of the synaptic filament by Polymerase δ [76]. This activity of PARI appears to be independent of its UvrD helicase domain, therefore it is not known whether PARI works alone, or potentially in complex with some other factor. Fanconi anemia, a genetic disease that is associated with the loss of leukocytes and cancer predisposition, is caused by mutations in the FANC gene family. One of these genes, FANCM, is homologous to yeast Mph1, although FANCM has nuclease activity in addition to acting as a DNA helicase [77,78]. The nuclease activity of FANCM contributes to its role in repairing DNA damage caused by inter-strand crosslinking agents, but point mutants affecting the helicase domain do not seem to affect this repair pathway [79]. The helicase activity of FANCM instead appears to suppress crossover recombination, thereby preventing the exchange of genetic material between homologous DNA sequences during HR.
Of the RecQ helicases, yeast Sgs1 and mammalian BLM have been shown to have the ability to dissolve D-loops. Sgs1 acts as part of the Sgs1-Top3-Rmi1 complex to negatively regulate D-loops [80], and BLM forms an equivalent complex with TOPOIIIa-RMI1-RMI2 [81]. BLM therefore appears to counteract recombination at two potential steps, by displacing RAD51 from resected DNA ends as discussed in the previous section, and by dissociating D-loops. It is not clear which of these activities is most important for the contribution of BLM to maintenance of genomic integrity, and preventing the tumor susceptibility that is characteristic of Bloom Syndrome. WRN, the helicase that is mutated in Werner’s Syndrome, is required for genomic integrity and normal HR [82]. Genomic instability in WRN-deficient cells can be partially suppressed by the overexpression of a dominant-negative form of RAD51, suggesting that WRN normally resolves toxic recombination intermediates [83]. Biochemical evidence indicates that WRN degrades D-loops through branch migration dependent on its helicase domain, and by targeting 3′ strand-invaded DNA ends with its exonuclease activity [84,85,86]. RECQL1 can likewise dissociate D-loops using its branch migration activity [87]. At least three mammalian RECQ helicases therefore counteract formation of D-loops, although the relative importance of these activities for recombination in cells is not fully established.
5. Activities of Helicases in the Postsynaptic Stages of HR
After the formation of a stable D-loop, the synaptic filament is paired as heteroduplex DNA with a homologous region from another chromosome or chromatid. The next stages determine the outcome of recombination, which can proceed through either a ‘synthesis dependent strand annealing’ (SDSA) pathway or through the ‘double-strand break repair’ pathway (DSBR) [1]. SDSA involves unwinding the D-loop intermediate formed after strand invasion, and always produces non-crossover products. Helicases that can disrupt D-loops therefore tend to promote SDSA as a mechanism for completing HR. The DSBR pathway involves extended synthesis of DNA within the D-loop, second-end capture, and formation of ‘double-Holliday junction’ structures (Figure 4). The resolution of double-Holliday junctions produces a mixture of crossover products, in which regions from the broken DNA molecule and template duplex become spliced together, in addition to non-crossover products. The frequency with which these pathways are used varies in different cell types. A mixture of crossover and non-crossover recombination products are produced during the repair of Spo11-mediated DNA breaks in meiosis, but non-crossover outcomes are favored in somatic cells [88,89].
The MCM8-9 complex is proposed to act at the D-loop to unwind the template DNA, and allow the paired heteroduplex DNA to be extended by a polymerase [90]. MCM8 and MCM9 form a helicase complex that is paralagous to the MCM2-7 complex, which forms the primary eukaryotic replicative helicase [91]. MCM8-9 can mediate normal S phase DNA replication when MCM2-7 is absent [92], but is principally involved in HR-associated DNA replication [43,44,45]. Mouse knockouts of MCM8 or MCM9 are sterile, demonstrating the essential role of these factors in meiotic recombination during gametogenesis. The loss of MCM8 and MCM9 also disrupts HR in somatic cells, leading to hypersensitivity to DNA damage, genomic instability, and tumor predisposition [93,94]. MCM8-9 may act in a parallel pathway with the HELQ helicase to mediate HR after strand invasion, because cells lacking both HELQ and the MCM8-9 loading factor, HROB, show a very severe defect in HR and persistent RAD51 loading [90]. HELQ is a 3′-5′ helicase, which is required for normal DNA repair and for the survival of germ cell progenitors [95,96,97]. Work in C. elegans and mice has supported a model in which HELQ acts after RAD51 loading to enable HR [98,99]. In the absence of HELQ, RAD51 loads at recombination sites, but remains bound there, indicating that HELQ may be required for disassembly of RAD51 from the postsynaptic filament to complete recombination. The importance of HELQ for normal DNA repair is demonstrated by the high tumor incidence seen in HELQ–/– mice.
RAD54 and ATRX are two members of the SWI/SNF2 class of SF2 helicases, which have important functions in regulation of events at several stages of recombination including those taking place after strand invasion [100,101]. RAD54 stimulates strand invasion of RAD51-loaded DNA into a homologous dsDNA duplex [102], and stabilizes RAD51 nucleoprotein filaments, but it can also displace RAD51 from DNA, dependent on its ATPase activity [103,104]. RAD54 mediates branch migration of Holliday junctions, and promotes resolution of Holliday junctions through stimulation of the Mus81 nuclease [105,106]. ATRX (alpha thalassemia/mental retardation syndrome X-linked) has an N-terminal chromatin-binding domain, and a C-terminal helicase domain [100]. Mutations in either of these domains cause a developmental disorder characterized by growth defects and intellectual disabilities, although mutations in the helicase domain tend to have a milder effect [107]. The chromatin-binding domain of ATRX helps load the variant histone H3.3 at repair sites, and the loss of this activity appears to contribute to DNA repair defects in ATRX-deficient cells. Loss of ATRX causes persistence of DNA breaks, and abnormal HR, as measured by cellular reporter assays [108]. RAD51 loads normally in ATRX-deficient cells, but a greater amount of DNA synthesis takes place after strand invasion, and crossover products from HR are suppressed. ATRX therefore appears to increase the use of the DSBR pathway for HR, instead of the SDSA pathway [3]. RAD54 and ATRX do not act as typical dsDNA-unwinding helicases. Instead, their helicase domains provide ATP-dependent translocase activity, allowing the proteins to move along DNA to regulate HR. The S. cerevisiae Mer3 helicase also promotes crossover formation in meiosis through the DSBR pathway, by stabilizing D-loops to promote extension of heteroduplex DNA in the 3′–5′ direction [109]. The mouse homolog of Mer3, HFM1, is required for crossover recombination during meiosis [110]. HFM1-knockout mice therefore show a failure to complete spermatogenesis and are infertile.
In yeast and mammals, RECQ helicases play vital roles at the late stages of HR by mediating branch migration and subsequent ‘dissolution’ of Holliday junctions. Holliday junctions can be ‘resolved’ by structure-specific nucleases, which make nicks around the Holliday junction, leading to formation of a mixture of crossover and non-crossover products [111]. In contrast, dissolution of Holliday junctions produces only non-crossover products, therefore RECQ helicases and their associated factors are very important regulators of the outcome of recombination. Branch migration refers to directed unwinding and re-annealing of DNA regions around the Holliday junction, to move the Holliday junction or alter the extent of heteroduplex DNA. This activity is carried out by Sgs1 in yeast, and BLM in mammalian cells [112,113]. Branch migration of double-Holliday junction intermediates can bring the two Holliday junctions together, to form a ‘hemicatenane’ structure that can be dismantled by Sgs1 or BLM in complex with several other proteins [80,114,115,116,117]. The protein complex for the dissolution of Holliday junctions has been called the ‘dissolvasome’, and is made up of Sgs1-Top3-Rmi1 in yeast, and BLM-TOPIIIa-RMI1-RMI2 in mammals. In particular, the topoisomerase activity provided by Top3/TOPIII allows nicking and re-ligation of the heteroduplex DNA in the hemicatenane to restore intact, separate DNA molecules. Through this process, Sgs1 and BLM greatly reduce the frequency of crossover recombination. Cells lacking BLM, such as those from Bloom Syndrome patients, show a much higher rate of crossovers, which can be demonstrated experimentally by the sister chromatid exchange assay, which allows exchanges of chromosome regions to be quantified by differential staining [118]. In yeast, proteins of the SIC/ZMM family promote crossovers during meiosis, by counteracting the ability of Sgs1 to dissolve Holliday junctions [119]. This indicates that regulation of helicases and helicase-containing complexes is essential to ensure appropriate outcomes of recombination.
In addition to BLM and Sgs1, other helicases are active in the final stages of HR. Deletion of either RECQL1 or RECQL5 is reported to increase the frequency of sister chromatid exchanges in BLM-knockout cells, suggesting that these alternative RECQ helicases act as a backup for BLM in suppressing crossover recombination [120]. As discussed previously, RAD54, RECQL1 and WRN have branch migration activity [84,87,105]. Branch migration activity has also been reported for SMARCAL1, ZRANB3 and FANCM [121,122,123,124], although these helicases may have specialized roles at replication forks instead of in general HR-mediated repair of DNA double-strand breaks.
6. Helicase-Mediated Regulation of Recombination at Sites of Replication
A recombination-based process called ‘break-induced recombination’ can restart replication forks that collapse at obstacles such as a single-strand breaks or DNA cross-links [125]. Replication forks do not immediately collapse on encountering obstacles, however, and can instead pause for a period of several hours, allowing a chance for any obstacles to be removed and for replication to continue [6]. Helicase activity contributes to stability of stalled replication forks, thereby reducing replication fork collapse and break-induced recombination (Figure 5). Electron microscopy has shown that stalled replication forks frequently become reversed to assume a ‘chicken foot structure’, which becomes loaded with RAD51 to promote stabilization. SMARCAL1 acts as an annealing helicase to bring single-stranded DNA molecules at the replication fork together, to promote reversal of stalled replication forks, and help protect them from endonucleolytic degradation [121,122,126]. The ZRANB3 helicase can also reverse stalled replication forks, and acts alongside SMARCAL1 to protect forks in the presence of oncogene-induced replication stress [127]. Another annealing helicase, TWINKLE, is required for replication of mitochondrial DNA [128]. TWINKLE activity is proposed to contribute to a form of recombination-mediated replication initiation, similar to replication by bacteriophage T4, which has been observed in mitochondria [129]. The exact mechanism of helicase-assisted replication that is carried out by TWINKLE is not fully understood, but it is clearly of substantial importance, because mutations in TWINKLE are associated with the severe genetic diseases, progressive external ophthalmoplegia, and infantile-onset spinocerebellar ataxia [130].
Two helicases of the Fanconi Anemia gene family, FANCJ and FANCM, are important for fork stability. FANCJ associates with TOPBP1 at sites of replication stress to properly signal the stress response and to limit fork reversal at these sites [131,132]. FANCM is able to migrate replication forks, and is required for fork stability during replication stress [124,133]. Mph1, the yeast homolog of FANCM, can also reverse replication forks, and this activity is regulated in part by the SMC5-6 complex [134,135,136]. The HELQ helicase is required for normal DNA replication, and interacts with the recombination mediators, RAD51, and RAD51 paralogs of the BCDX complex [98]. In HELQ-deficient cells, RAD51 accumulates normally at replication forks after induction of replication stress, but remains bound there, indicating a defect in replication-associated recombination. This inability to resolve stalled replication forks is linked to attrition of germ cells and tumor susceptibility of HELQ-deficient mice.
7. Conclusions
Although it is clear that helicases play essential roles in HR, understanding the importance of specific helicases in different recombination processes will require substantial further research. For example, as discussed above, BLM helicase has been reported to function in DNA end resection, displacement of RAD51 from nucleoprotein filaments, disassembly of D-loops, and dissolution of Holliday junctions. These activities represent both pro- and anti-recombinogenic functions, and it is not clear which are most important for maintenance of genomic integrity by BLM. Likewise, many different helicases have been reported to have branch migration activity in eukaryotic cells, and it is not clear to what extent they act redundantly, or whether they are regulated for specific purposes. Solving these questions can be challenging, because it is not always easy to test whether the biochemical activities of helicase enzymes in vitro are also relevant in cells. Future work to clarify the roles of helicases in recombination will give us a better understanding of how HR operates, and potentially open the way to pharmacological intervention to manipulate helicase activities and achieve useful therapeutic outcomes [137].
Author Contributions
E.H. and S.F.B. contributed equally to writing of the manuscript and design of the figures. All authors have read and agreed to the published version of the manuscript.
Funding
Work in the Bunting lab is supported by grant R01CA90858, the Robert Wood Johnson Foundation award RWJF-74763, and by funding provided by the Rutgers Cancer Institute of New Jersey through Cancer Center Support grant NCI-CCSG P30CA072720.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jasin, M.; Rothstein, R. Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 2013, 5, a012740. [Google Scholar] [CrossRef] [PubMed]
- Awate, S.; Brosh, R.M., Jr. Interactive Roles of DNA Helicases and Translocases with the Single-Stranded DNA Binding Protein RPA in Nucleic Acid Metabolism. Int. J. Mol. Sci. 2017, 18, 1233. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Gaines, W.A.; Kwon, Y.; Sung, P. Regulation of DNA pairing in homologous recombination. Cold Spring Harb. Perspect. Biol. 2014, 6, a017954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ait Saada, A.; Lambert, S.A.E.; Carr, A.M. Preserving replication fork integrity and competence via the homologous recombination pathway. DNA Repair 2018, 71, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.M.; Ko, J.H.; Hu, L.; Kim, S.A.; Bishop, A.J.; Vijg, J.; Montagna, C.; Hasty, P. RAD51 mutants cause replication defects and chromosomal instability. Mol. Cell. Biol. 2012, 32, 3663–3680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petermann, E.; Orta, M.L.; Issaeva, N.; Schultz, N.; Helleday, T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell 2010, 37, 492–502. [Google Scholar] [CrossRef] [Green Version]
- Ellis, N.A.; Groden, J.; Ye, T.Z.; Straughen, J.; Lennon, D.J.; Ciocci, S.; Proytcheva, M.; German, J. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell 1995, 83, 655–666. [Google Scholar] [CrossRef] [Green Version]
- Gray, M.D.; Shen, J.C.; Kamath-Loeb, A.S.; Blank, A.; Sopher, B.L.; Martin, G.M.; Oshima, J.; Loeb, L.A. The Werner syndrome protein is a DNA helicase. Nat. Genet. 1997, 17, 100–103. [Google Scholar] [CrossRef]
- Karow, J.K.; Chakraverty, R.K.; Hickson, I.D. The Bloom’s syndrome gene product is a 3′-5′ DNA helicase. J. Biol. Chem. 1997, 272, 30611–30614. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.E.; Oshima, J.; Fu, Y.H.; Wijsman, E.M.; Hisama, F.; Alisch, R.; Matthews, S.; Nakura, J.; Miki, T.; Ouais, S.; et al. Positional cloning of the Werner’s syndrome gene. Science 1996, 272, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Jimeno, S.; Camarillo, R.; Mejias-Navarro, F.; Fernandez-Avila, M.J.; Soria-Bretones, I.; Prados-Carvajal, R.; Huertas, P. The Helicase PIF1 Facilitates Resection over Sequences Prone to Forming G4 Structures. Cell Rep. 2018, 25, 3543. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Shamanna, R.A.; Keijzers, G.; Anand, R.; Rasmussen, L.J.; Cejka, P.; Croteau, D.L.; Bohr, V.A. RECQL4 Promotes DNA End Resection in Repair of DNA Double-Strand Breaks. Cell Rep. 2016, 16, 161–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamanna, R.A.; Singh, D.K.; Lu, H.; Mirey, G.; Keijzers, G.; Salles, B.; Croteau, D.L.; Bohr, V.A. RECQ helicase RECQL4 participates in non-homologous end joining and interacts with the Ku complex. Carcinogenesis 2014, 35, 2415–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukowy, Z.; Harrigan, J.A.; Ramsden, D.A.; Tudek, B.; Bohr, V.A.; Stevnsner, T. WRN Exonuclease activity is blocked by specific oxidatively induced base lesions positioned in either DNA strand. Nucleic Acids Res. 2008, 36, 4975–4987. [Google Scholar] [CrossRef] [Green Version]
- Cooper, M.P.; Machwe, A.; Orren, D.K.; Brosh, R.M.; Ramsden, D.; Bohr, V.A. Ku complex interacts with and stimulates the Werner protein. Genes Dev. 2000, 14, 907–912. [Google Scholar]
- Orren, D.K.; Machwe, A.; Karmakar, P.; Piotrowski, J.; Cooper, M.P.; Bohr, V.A. A functional interaction of Ku with Werner exonuclease facilitates digestion of damaged DNA. Nucleic Acids Res. 2001, 29, 1926–1934. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.C.; Loeb, L.A. Werner syndrome exonuclease catalyzes structure-dependent degradation of DNA. Nucleic Acids Res. 2000, 28, 3260–3268. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, N.; Shiratori, M.; Goto, M.; Furuichi, Y. Werner syndrome helicase contains a 5′-->3′ exonuclease activity that digests DNA and RNA strands in DNA/DNA and RNA/DNA duplexes dependent on unwinding. Nucleic Acids Res. 1999, 27, 2361–2368. [Google Scholar] [CrossRef] [Green Version]
- Keka, I.S.; Mohiuddin; Maede, Y.; Rahman, M.M.; Sakuma, T.; Honma, M.; Yamamoto, T.; Takeda, S.; Sasanuma, H. Smarcal1 promotes double-strand-break repair by nonhomologous end-joining. Nucleic Acids Res. 2015, 43, 6359–6372. [Google Scholar] [CrossRef] [Green Version]
- Cejka, P. DNA End Resection: Nucleases Team Up with the Right Partners to Initiate Homologous Recombination. J. Biol. Chem. 2015, 290, 22931–22938. [Google Scholar] [CrossRef] [Green Version]
- Gravel, S.; Chapman, J.R.; Magill, C.; Jackson, S.P. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008, 22, 2767–2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mimitou, E.P.; Symington, L.S. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 2008, 455, 770–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011, 25, 350–362. [Google Scholar] [CrossRef] [Green Version]
- Nimonkar, A.V.; Ozsoy, A.Z.; Genschel, J.; Modrich, P.; Kowalczykowski, S.C. Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc. Natl. Acad. Sci. USA 2008, 105, 16906–16911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Chung, W.H.; Shim, E.Y.; Lee, S.E.; Ira, G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 2008, 134, 981–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Lisby, M.; Symington, L.S. RPA coordinates DNA end resection and prevents formation of DNA hairpins. Mol. Cell 2013, 50, 589–600. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, M.; Sommers, J.A.; Morris, C.; Brosh, R.M., Jr. Delineation of WRN helicase function with EXO1 in the replicational stress response. DNA Repair (Amst) 2010, 9, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Doherty, K.M.; Sharma, S.; Uzdilla, L.A.; Wilson, T.M.; Cui, S.; Vindigni, A.; Brosh, R.M., Jr. RECQ1 helicase interacts with human mismatch repair factors that regulate genetic recombination. J. Biol. Chem. 2005, 280, 28085–28094. [Google Scholar] [CrossRef] [Green Version]
- Palermo, V.; Rinalducci, S.; Sanchez, M.; Grillini, F.; Sommers, J.A.; Brosh, R.M., Jr.; Zolla, L.; Franchitto, A.; Pichierri, P. CDK1 phosphorylates WRN at collapsed replication forks. Nat. Commun. 2016, 7, 12880. [Google Scholar] [CrossRef]
- Sturzenegger, A.; Burdova, K.; Kanagaraj, R.; Levikova, M.; Pinto, C.; Cejka, P.; Janscak, P. DNA2 cooperates with the WRN and BLM RecQ helicases to mediate long-range DNA end resection in human cells. J. Biol. Chem. 2014, 289, 27314–27326. [Google Scholar] [CrossRef] [Green Version]
- Tomimatsu, N.; Mukherjee, B.; Deland, K.; Kurimasa, A.; Bolderson, E.; Khanna, K.K.; Burma, S. Exo1 plays a major role in DNA end resection in humans and influences double-strand break repair and damage signaling decisions. DNA Repair (Amst) 2012, 11, 441–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, A.T.; Kim, T.; Wagner, J.E.; Conti, B.A.; Lach, F.P.; Huang, A.L.; Molina, H.; Sanborn, E.M.; Zierhut, H.; Cornes, B.K.; et al. A Dominant Mutation in Human RAD51 Reveals Its Function in DNA Interstrand Crosslink Repair Independent of Homologous Recombination. Mol. Cell 2015, 59, 478–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levikova, M.; Pinto, C.; Cejka, P. The motor activity of DNA2 functions as an ssDNA translocase to promote DNA end resection. Genes Dev. 2017, 31, 493–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.S.; Daley, J.M.; Pham, N.T.; Niu, H.; Xue, X.; Ira, G.; Sung, P. A novel role of the Dna2 translocase function in DNA break resection. Genes Dev. 2017, 31, 503–510. [Google Scholar] [CrossRef] [Green Version]
- Tkac, J.; Xu, G.; Adhikary, H.; Young, J.T.F.; Gallo, D.; Escribano-Diaz, C.; Krietsch, J.; Orthwein, A.; Munro, M.; Sol, W.; et al. HELB Is a Feedback Inhibitor of DNA End Resection. Mol. Cell 2016, 61, 405–418. [Google Scholar] [CrossRef] [Green Version]
- Costelloe, T.; Louge, R.; Tomimatsu, N.; Mukherjee, B.; Martini, E.; Khadaroo, B.; Dubois, K.; Wiegant, W.W.; Thierry, A.; Burma, S.; et al. The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature 2012, 489, 581–584. [Google Scholar] [CrossRef] [Green Version]
- Awad, S.; Ryan, D.; Prochasson, P.; Owen-Hughes, T.; Hassan, A.H. The Snf2 homolog Fun30 acts as a homodimeric ATP-dependent chromatin-remodeling enzyme. J. Biol. Chem. 2010, 285, 9477–9484. [Google Scholar] [CrossRef] [Green Version]
- Rowbotham, S.P.; Barki, L.; Neves-Costa, A.; Santos, F.; Dean, W.; Hawkes, N.; Choudhary, P.; Will, W.R.; Webster, J.; Oxley, D.; et al. Maintenance of silent chromatin through replication requires SWI/SNF-like chromatin remodeler SMARCAD1. Mol. Cell 2011, 42, 285–296. [Google Scholar] [CrossRef]
- Suhasini, A.N.; Rawtani, N.A.; Wu, Y.; Sommers, J.A.; Sharma, S.; Mosedale, G.; North, P.S.; Cantor, S.B.; Hickson, I.D.; Brosh, R.M., Jr. Interaction between the helicases genetically linked to Fanconi anemia group J and Bloom’s syndrome. EMBO J. 2011, 30, 692–705. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Peng, M.; Guillemette, S.; Quan, S.; Maniatis, S.; Wu, Y.; Venkatesh, A.; Shaffer, S.A.; Brosh, R.M., Jr.; Cantor, S.B. FANCJ/BACH1 acetylation at lysine 1249 regulates the DNA damage response. PLoS Genet. 2012, 8, e1002786. [Google Scholar] [CrossRef]
- Nath, S.; Nagaraju, G. FANCJ helicase promotes DNA end resection by facilitating CtIP recruitment to DNA double-strand breaks. PLoS Genet. 2020, 16, e1008701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.Y.; Im, J.S.; Shibata, E.; Park, J.; Handa, N.; Kowalczykowski, S.C.; Dutta, A. MCM8-9 complex promotes resection of double-strand break ends by MRE11-RAD50-NBS1 complex. Nat. Commun. 2015, 6, 7744. [Google Scholar] [CrossRef] [PubMed]
- Lutzmann, M.; Grey, C.; Traver, S.; Ganier, O.; Maya-Mendoza, A.; Ranisavljevic, N.; Bernex, F.; Nishiyama, A.; Montel, N.; Gavois, E.; et al. MCM8- and MCM9-deficient mice reveal gametogenesis defects and genome instability due to impaired homologous recombination. Mol. Cell 2012, 47, 523–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, K.; Ishiai, M.; Horikawa, K.; Fukagawa, T.; Takata, M.; Takisawa, H.; Kanemaki, M.T. Mcm8 and Mcm9 form a complex that functions in homologous recombination repair induced by DNA interstrand crosslinks. Mol. Cell 2012, 47, 511–522. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Long, D.T.; Lee, K.Y.; Abbas, T.; Shibata, E.; Negishi, M.; Luo, Y.; Schimenti, J.C.; Gambus, A.; Walter, J.C.; et al. The MCM8-MCM9 complex promotes RAD51 recruitment at DNA damage sites to facilitate homologous recombination. Mol. Cell. Biol. 2013, 33, 1632–1644. [Google Scholar] [CrossRef]
- Sullivan, M.R.; Bernstein, K.A. RAD-ical New Insights into RAD51 Regulation. Genes (Basel) 2018, 9, 629. [Google Scholar] [CrossRef] [Green Version]
- Veaute, X.; Delmas, S.; Selva, M.; Jeusset, J.; Le Cam, E.; Matic, I.; Fabre, F.; Petit, M.A. UvrD helicase, unlike Rep helicase, dismantles RecA nucleoprotein filaments in Escherichia coli. EMBO J. 2005, 24, 180–189. [Google Scholar] [CrossRef] [Green Version]
- Krejci, L.; Van Komen, S.; Li, Y.; Villemain, J.; Reddy, M.S.; Klein, H.; Ellenberger, T.; Sung, P. DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature 2003, 423, 305–309. [Google Scholar] [CrossRef]
- Veaute, X.; Jeusset, J.; Soustelle, C.; Kowalczykowski, S.C.; Le Cam, E.; Fabre, F. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature 2003, 423, 309–312. [Google Scholar] [CrossRef]
- Papouli, E.; Chen, S.; Davies, A.A.; Huttner, D.; Krejci, L.; Sung, P.; Ulrich, H.D. Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol. Cell 2005, 19, 123–133. [Google Scholar] [CrossRef]
- Pfander, B.; Moldovan, G.L.; Sacher, M.; Hoege, C.; Jentsch, S. SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature 2005, 436, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Chanet, R.; Heude, M.; Adjiri, A.; Maloisel, L.; Fabre, F. Semidominant mutations in the yeast Rad51 protein and their relationships with the Srs2 helicase. Mol. Cell. Biol. 1996, 16, 4782–4789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moldovan, G.L.; Dejsuphong, D.; Petalcorin, M.I.; Hofmann, K.; Takeda, S.; Boulton, S.J.; D’Andrea, A.D. Inhibition of homologous recombination by the PCNA-interacting protein PARI. Mol. Cell 2012, 45, 75–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommers, J.A.; Rawtani, N.; Gupta, R.; Bugreev, D.V.; Mazin, A.V.; Cantor, S.B.; Brosh, R.M., Jr. FANCJ uses its motor ATPase to destabilize protein-DNA complexes, unwind triplexes, and inhibit RAD51 strand exchange. J. Biol. Chem. 2009, 284, 7505–7517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, A.; Osman, F.; Folkyte, V.; Sofueva, S.; Whitby, M.C. Fbh1 limits Rad51-dependent recombination at blocked replication forks. Mol. Cell. Biol. 2009, 29, 4742–4756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osman, F.; Dixon, J.; Barr, A.R.; Whitby, M.C. The F-Box DNA helicase Fbh1 prevents Rhp51-dependent recombination without mediator proteins. Mol. Cell. Biol. 2005, 25, 8084–8096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fugger, K.; Mistrik, M.; Danielsen, J.R.; Dinant, C.; Falck, J.; Bartek, J.; Lukas, J.; Mailand, N. Human Fbh1 helicase contributes to genome maintenance via pro- and anti-recombinase activities. J. Cell Biol. 2009, 186, 655–663. [Google Scholar] [CrossRef]
- Kohzaki, M.; Hatanaka, A.; Sonoda, E.; Yamazoe, M.; Kikuchi, K.; Vu Trung, N.; Szuts, D.; Sale, J.E.; Shinagawa, H.; Watanabe, M.; et al. Cooperative roles of vertebrate Fbh1 and Blm DNA helicases in avoidance of crossovers during recombination initiated by replication fork collapse. Mol. Cell. Biol. 2007, 27, 2812–2820. [Google Scholar] [CrossRef] [Green Version]
- Simandlova, J.; Zagelbaum, J.; Payne, M.J.; Chu, W.K.; Shevelev, I.; Hanada, K.; Chatterjee, S.; Reid, D.A.; Liu, Y.; Janscak, P.; et al. FBH1 helicase disrupts RAD51 filaments in vitro and modulates homologous recombination in mammalian cells. J. Biol. Chem. 2013, 288, 34168–34180. [Google Scholar] [CrossRef] [Green Version]
- Chu, W.K.; Payne, M.J.; Beli, P.; Hanada, K.; Choudhary, C.; Hickson, I.D. FBH1 influences DNA replication fork stability and homologous recombination through ubiquitylation of RAD51. Nat. Commun. 2015, 6, 5931. [Google Scholar] [CrossRef] [Green Version]
- Crickard, J.B.; Xue, C.; Wang, W.; Kwon, Y.; Sung, P.; Greene, E.C. The RecQ helicase Sgs1 drives ATP-dependent disruption of Rad51 filaments. Nucleic Acids Res. 2019, 47, 4694–4706. [Google Scholar] [CrossRef] [PubMed]
- Bugreev, D.V.; Yu, X.; Egelman, E.H.; Mazin, A.V. Novel pro- and anti-recombination activities of the Bloom’s syndrome helicase. Genes Dev. 2007, 21, 3085–3094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, D.S.; Misenko, S.M.; Her, J.; Bunting, S.F. BLM helicase regulates DNA repair by counteracting RAD51 loading at DNA double-strand break sites. J. Cell Biol. 2017, 216, 3521–3534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Raynard, S.; Sehorn, M.G.; Lu, X.; Bussen, W.; Zheng, L.; Stark, J.M.; Barnes, E.L.; Chi, P.; Janscak, P.; et al. RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes Dev. 2007, 21, 3073–3084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwendener, S.; Raynard, S.; Paliwal, S.; Cheng, A.; Kanagaraj, R.; Shevelev, I.; Stark, J.M.; Sung, P.; Janscak, P. Physical interaction of RECQ5 helicase with RAD51 facilitates its anti-recombinase activity. J. Biol. Chem. 2010, 285, 15739–15745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzaki, K.; Kondo, S.; Ishikawa, T.; Shinohara, A. Human RAD51 paralogue SWSAP1 fosters RAD51 filament by regulating the anti-recombinase FIGNL1 AAA+ ATPase. Nat. Commun. 2019, 10, 1407. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Chen, J. FIGNL1-containing protein complex is required for efficient homologous recombination repair. Proc. Natl. Acad. Sci. USA 2013, 110, 10640–10645. [Google Scholar] [CrossRef] [Green Version]
- Black, S.J.; Kashkina, E.; Kent, T.; Pomerantz, R.T. DNA Polymerase theta: A Unique Multifunctional End-Joining Machine. Genes (Basel) 2016, 7, 67. [Google Scholar] [CrossRef] [Green Version]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef] [Green Version]
- Ira, G.; Malkova, A.; Liberi, G.; Foiani, M.; Haber, J.E. Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell 2003, 115, 401–411. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Ede, C.; Wright, W.D.; Gore, S.K.; Jenkins, S.S.; Freudenthal, B.D.; Todd Washington, M.; Veaute, X.; Heyer, W.D. Srs2 promotes synthesis-dependent strand annealing by disrupting DNA polymerase delta-extending D-loops. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Prakash, R.; Satory, D.; Dray, E.; Papusha, A.; Scheller, J.; Kramer, W.; Krejci, L.; Klein, H.; Haber, J.E.; Sung, P.; et al. Yeast Mph1 helicase dissociates Rad51-made D-loops: Implications for crossover control in mitotic recombination. Genes Dev. 2009, 23, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Barber, L.J.; Youds, J.L.; Ward, J.D.; McIlwraith, M.J.; O’Neil, N.J.; Petalcorin, M.I.; Martin, J.S.; Collis, S.J.; Cantor, S.B.; Auclair, M.; et al. RTEL1 maintains genomic stability by suppressing homologous recombination. Cell 2008, 135, 261–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, H.; Schertzer, M.; Wu, X.; Gertsenstein, M.; Selig, S.; Kammori, M.; Pourvali, R.; Poon, S.; Vulto, I.; Chavez, E.; et al. Regulation of murine telomere length by Rtel: An essential gene encoding a helicase-like protein. Cell 2004, 117, 873–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkovics, P.; Dome, L.; Juhasz, S.; Altmannova, V.; Sebesta, M.; Pacesa, M.; Fugger, K.; Sorensen, C.S.; Lee, M.Y.; Haracska, L.; et al. The PCNA-associated protein PARI negatively regulates homologous recombination via the inhibition of DNA repair synthesis. Nucleic Acids Res. 2016, 44, 3176–3189. [Google Scholar] [CrossRef] [Green Version]
- Meetei, A.R.; Medhurst, A.L.; Ling, C.; Xue, Y.; Singh, T.R.; Bier, P.; Steltenpool, J.; Stone, S.; Dokal, I.; Mathew, C.G.; et al. A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat. Genet. 2005, 37, 958–963. [Google Scholar] [CrossRef] [Green Version]
- Mosedale, G.; Niedzwiedz, W.; Alpi, A.; Perrina, F.; Pereira-Leal, J.B.; Johnson, M.; Langevin, F.; Pace, P.; Patel, K.J. The vertebrate Hef ortholog is a component of the Fanconi anemia tumor-suppressor pathway. Nat. Struct. Mol. Biol. 2005, 12, 763–771. [Google Scholar] [CrossRef]
- Rosado, I.V.; Niedzwiedz, W.; Alpi, A.F.; Patel, K.J. The Walker B motif in avian FANCM is required to limit sister chromatid exchanges but is dispensable for DNA crosslink repair. Nucleic Acids Res. 2009, 37, 4360–4370. [Google Scholar] [CrossRef]
- Fasching, C.L.; Cejka, P.; Kowalczykowski, S.C.; Heyer, W.D. Top3-Rmi1 dissolve Rad51-mediated D loops by a topoisomerase-based mechanism. Mol. Cell 2015, 57, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Van Brabant, A.J.; Ye, T.; Sanz, M.; German, I.J.; Ellis, N.A.; Holloman, W.K. Binding and melting of D-loops by the Bloom syndrome helicase. Biochemistry 2000, 39, 14617–14625. [Google Scholar] [CrossRef] [PubMed]
- Prince, P.R.; Emond, M.J.; Monnat, R.J., Jr. Loss of Werner syndrome protein function promotes aberrant mitotic recombination. Genes Dev. 2001, 15, 933–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saintigny, Y.; Makienko, K.; Swanson, C.; Emond, M.J.; Monnat, R.J., Jr. Homologous recombination resolution defect in werner syndrome. Mol. Cell. Biol. 2002, 22, 6971–6978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constantinou, A.; Tarsounas, M.; Karow, J.K.; Brosh, R.M.; Bohr, V.A.; Hickson, I.D.; West, S.C. Werner’s syndrome protein (WRN) migrates Holliday junctions and co-localizes with RPA upon replication arrest. EMBO Rep. 2000, 1, 80–84. [Google Scholar] [CrossRef]
- Opresko, P.L.; Sowd, G.; Wang, H. The Werner syndrome helicase/exonuclease processes mobile D-loops through branch migration and degradation. PLoS ONE 2009, 4, e4825. [Google Scholar] [CrossRef] [Green Version]
- Orren, D.K.; Theodore, S.; Machwe, A. The Werner syndrome helicase/exonuclease (WRN) disrupts and degrades D-loops in vitro. Biochemistry 2002, 41, 13483–13488. [Google Scholar] [CrossRef]
- Bugreev, D.V.; Brosh, R.M., Jr.; Mazin, A.V. RECQ1 possesses DNA branch migration activity. J. Biol. Chem. 2008, 283, 20231–20242. [Google Scholar] [CrossRef] [Green Version]
- Allers, T.; Lichten, M. Differential timing and control of noncrossover and crossover recombination during meiosis. Cell 2001, 106, 47–57. [Google Scholar] [CrossRef] [Green Version]
- LaRocque, J.R.; Stark, J.M.; Oh, J.; Bojilova, E.; Yusa, K.; Horie, K.; Takeda, J.; Jasin, M. Interhomolog recombination and loss of heterozygosity in wild-type and Bloom syndrome helicase (BLM)-deficient mammalian cells. Proc. Natl. Acad. Sci. USA 2011, 108, 11971–11976. [Google Scholar] [CrossRef] [Green Version]
- Hustedt, N.; Saito, Y.; Zimmermann, M.; Alvarez-Quilon, A.; Setiaputra, D.; Adam, S.; McEwan, A.; Yuan, J.Y.; Olivieri, M.; Zhao, Y.; et al. Control of homologous recombination by the HROB-MCM8-MCM9 pathway. Genes Dev. 2019, 33, 1397–1415. [Google Scholar] [CrossRef]
- Griffin, W.C.; Trakselis, M.A. The MCM8/9 complex: A recent recruit to the roster of helicases involved in genome maintenance. DNA Repair (Amst) 2019, 76, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Natsume, T.; Nishimura, K.; Minocherhomji, S.; Bhowmick, R.; Hickson, I.D.; Kanemaki, M.T. Acute inactivation of the replicative helicase in human cells triggers MCM8-9-dependent DNA synthesis. Genes Dev. 2017, 31, 816–829. [Google Scholar] [CrossRef] [Green Version]
- Hartford, S.A.; Luo, Y.; Southard, T.L.; Min, I.M.; Lis, J.T.; Schimenti, J.C. Minichromosome maintenance helicase paralog MCM9 is dispensible for DNA replication but functions in germ-line stem cells and tumor suppression. Proc. Natl. Acad. Sci. USA 2011, 108, 17702–17707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutzmann, M.; Bernex, F.; da Costa de Jesus, C.; Hodroj, D.; Marty, C.; Plo, I.; Vainchenker, W.; Tosolini, M.; Forichon, L.; Bret, C.; et al. MCM8- and MCM9 Deficiencies Cause Lifelong Increased Hematopoietic DNA Damage Driving p53-Dependent Myeloid Tumors. Cell Rep. 2019, 28, 2851–2865. [Google Scholar] [CrossRef] [PubMed]
- Luebben, S.W.; Kawabata, T.; Akre, M.K.; Lee, W.L.; Johnson, C.S.; O’Sullivan, M.G.; Shima, N. Helq acts in parallel to Fancc to suppress replication-associated genome instability. Nucleic Acids Res. 2013, 41, 10283–10297. [Google Scholar] [CrossRef]
- Muzzini, D.M.; Plevani, P.; Boulton, S.J.; Cassata, G.; Marini, F. Caenorhabditis elegans POLQ-1 and HEL-308 function in two distinct DNA interstrand cross-link repair pathways. DNA Repair (Amst) 2008, 7, 941–950. [Google Scholar] [CrossRef]
- Takata, K.; Reh, S.; Tomida, J.; Person, M.D.; Wood, R.D. Human DNA helicase HELQ participates in DNA interstrand crosslink tolerance with ATR and RAD51 paralogs. Nat. Commun. 2013, 4, 2338. [Google Scholar] [CrossRef] [Green Version]
- Adelman, C.A.; Lolo, R.L.; Birkbak, N.J.; Murina, O.; Matsuzaki, K.; Horejsi, Z.; Parmar, K.; Borel, V.; Skehel, J.M.; Stamp, G.; et al. HELQ promotes RAD51 paralogue-dependent repair to avert germ cell loss and tumorigenesis. Nature 2013, 502, 381–384. [Google Scholar] [CrossRef] [Green Version]
- Ward, J.D.; Muzzini, D.M.; Petalcorin, M.I.; Martinez-Perez, E.; Martin, J.S.; Plevani, P.; Cassata, G.; Marini, F.; Boulton, S.J. Overlapping mechanisms promote postsynaptic RAD-51 filament disassembly during meiotic double-strand break repair. Mol. Cell 2010, 37, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Dyer, M.A.; Qadeer, Z.A.; Valle-Garcia, D.; Bernstein, E. ATRX and DAXX: Mechanisms and Mutations. Cold Spring Harb Perspect Med 2017, 7. [Google Scholar] [CrossRef]
- Mazin, A.V.; Mazina, O.M.; Bugreev, D.V.; Rossi, M.J. Rad54, the motor of homologous recombination. DNA Repair (Amst) 2010, 9, 286–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petukhova, G.; Stratton, S.; Sung, P. Catalysis of homologous DNA pairing by yeast Rad51 and Rad54 proteins. Nature 1998, 393, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Mazin, A.V.; Alexeev, A.A.; Kowalczykowski, S.C. A novel function of Rad54 protein. Stabilization of the Rad51 nucleoprotein filament. J. Biol. Chem. 2003, 278, 14029–14036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solinger, J.A.; Kiianitsa, K.; Heyer, W.D. Rad54, a Swi2/Snf2-like recombinational repair protein, disassembles Rad51:dsDNA filaments. Mol. Cell 2002, 10, 1175–1188. [Google Scholar] [CrossRef]
- Bugreev, D.V.; Mazina, O.M.; Mazin, A.V. Rad54 protein promotes branch migration of Holliday junctions. Nature 2006, 442, 590–593. [Google Scholar] [CrossRef] [PubMed]
- Mazina, O.M.; Mazin, A.V. Human Rad54 protein stimulates human Mus81-Eme1 endonuclease. Proc. Natl. Acad. Sci. USA 2008, 105, 18249–18254. [Google Scholar] [CrossRef] [Green Version]
- Lacoste, C.; Leheup, B.; Agouti, I.; Mowat, D.; Giuliano, F.; Badens, C. Mutations of codon 2085 in the helicase domain of ATRX are recurrent and cause ATRX syndrome. Clin. Genet. 2014, 86, 502–503. [Google Scholar] [CrossRef]
- Juhasz, S.; Elbakry, A.; Mathes, A.; Lobrich, M. ATRX Promotes DNA Repair Synthesis and Sister Chromatid Exchange during Homologous Recombination. Mol. Cell 2018, 71, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Mazina, O.M.; Mazin, A.V.; Nakagawa, T.; Kolodner, R.D.; Kowalczykowski, S.C. Saccharomyces cerevisiae Mer3 helicase stimulates 3′-5′ heteroduplex extension by Rad51; implications for crossover control in meiotic recombination. Cell 2004, 117, 47–56. [Google Scholar] [CrossRef] [Green Version]
- Guiraldelli, M.F.; Eyster, C.; Wilkerson, J.L.; Dresser, M.E.; Pezza, R.J. Mouse HFM1/Mer3 is required for crossover formation and complete synapsis of homologous chromosomes during meiosis. PLoS Genet. 2013, 9, e1003383. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, H.D.; West, S.C. Holliday junction resolvases. Cold Spring Harb. Perspect. Biol. 2014, 6, a023192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, R.J.; Keck, J.L.; Wang, J.C. Binding specificity determines polarity of DNA unwinding by the Sgs1 protein of S. cerevisiae. J. Mol. Biol. 1999, 289, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Karow, J.K.; Constantinou, A.; Li, J.L.; West, S.C.; Hickson, I.D. The Bloom’s syndrome gene product promotes branch migration of holliday junctions. Proc. Natl. Acad. Sci. USA 2000, 97, 6504–6508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raynard, S.; Bussen, W.; Sung, P. A double Holliday junction dissolvasome comprising BLM, topoisomerase IIIalpha, and BLAP75. J. Biol. Chem. 2006, 281, 13861–13864. [Google Scholar] [CrossRef] [Green Version]
- Singh, T.R.; Ali, A.M.; Busygina, V.; Raynard, S.; Fan, Q.; Du, C.H.; Andreassen, P.R.; Sung, P.; Meetei, A.R. BLAP18/RMI2, a novel OB-fold-containing protein, is an essential component of the Bloom helicase-double Holliday junction dissolvasome. Genes Dev. 2008, 22, 2856–2868. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Bachrati, C.Z.; Ou, J.; Xu, C.; Yin, J.; Chang, M.; Wang, W.; Li, L.; Brown, G.W.; Hickson, I.D. BLAP75/RMI1 promotes the BLM-dependent dissolution of homologous recombination intermediates. Proc. Natl. Acad. Sci. USA 2006, 103, 4068–4073. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Hickson, I.D. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature 2003, 426, 870–874. [Google Scholar] [CrossRef]
- Chaganti, R.S.; Schonberg, S.; German, J. A manyfold increase in sister chromatid exchanges in Bloom’s syndrome lymphocytes. Proc. Natl. Acad. Sci. USA 1974, 71, 4508–4512. [Google Scholar] [CrossRef] [Green Version]
- Jessop, L.; Rockmill, B.; Roeder, G.S.; Lichten, M. Meiotic chromosome synapsis-promoting proteins antagonize the anti-crossover activity of sgs1. PLoS Genet. 2006, 2, e155. [Google Scholar] [CrossRef]
- Wang, W.; Seki, M.; Narita, Y.; Nakagawa, T.; Yoshimura, A.; Otsuki, M.; Kawabe, Y.; Tada, S.; Yagi, H.; Ishii, Y.; et al. Functional relation among RecQ family helicases RecQL1, RecQL5, and BLM in cell growth and sister chromatid exchange formation. Mol. Cell. Biol. 2003, 23, 3527–3535. [Google Scholar] [CrossRef] [Green Version]
- Betous, R.; Couch, F.B.; Mason, A.C.; Eichman, B.F.; Manosas, M.; Cortez, D. Substrate-selective repair and restart of replication forks by DNA translocases. Cell Rep. 2013, 3, 1958–1969. [Google Scholar] [CrossRef] [Green Version]
- Betous, R.; Mason, A.C.; Rambo, R.P.; Bansbach, C.E.; Badu-Nkansah, A.; Sirbu, B.M.; Eichman, B.F.; Cortez, D. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012, 26, 151–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccia, A.; Nimonkar, A.V.; Hu, Y.; Hajdu, I.; Achar, Y.J.; Izhar, L.; Petit, S.A.; Adamson, B.; Yoon, J.C.; Kowalczykowski, S.C.; et al. Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol. Cell 2012, 47, 396–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gari, K.; Decaillet, C.; Stasiak, A.Z.; Stasiak, A.; Constantinou, A. The Fanconi anemia protein FANCM can promote branch migration of Holliday junctions and replication forks. Mol. Cell 2008, 29, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D. Replication-Coupled DNA Repair. Mol. Cell 2019, 74, 866–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Ghosal, G.; Chen, J. The annealing helicase HARP protects stalled replication forks. Genes Dev. 2009, 23, 2394–2399. [Google Scholar] [CrossRef] [Green Version]
- Puccetti, M.V.; Adams, C.M.; Kushinsky, S.; Eischen, C.M. Smarcal1 and Zranb3 Protect Replication Forks from Myc-Induced DNA Replication Stress. Cancer Res. 2019, 79, 1612–1623. [Google Scholar] [CrossRef]
- Sen, D.; Nandakumar, D.; Tang, G.Q.; Patel, S.S. Human mitochondrial DNA helicase TWINKLE is both an unwinding and annealing helicase. J. Biol. Chem. 2012, 287, 14545–14556. [Google Scholar] [CrossRef] [Green Version]
- Pohjoismaki, J.L.; Goffart, S.; Tyynismaa, H.; Willcox, S.; Ide, T.; Kang, D.; Suomalainen, A.; Karhunen, P.J.; Griffith, J.D.; Holt, I.J.; et al. Human heart mitochondrial DNA is organized in complex catenated networks containing abundant four-way junctions and replication forks. J. Biol. Chem. 2009, 284, 21446–21457. [Google Scholar] [CrossRef] [Green Version]
- Goffart, S.; Cooper, H.M.; Tyynismaa, H.; Wanrooij, S.; Suomalainen, A.; Spelbrink, J.N. Twinkle mutations associated with autosomal dominant progressive external ophthalmoplegia lead to impaired helicase function and in vivo mtDNA replication stalling. Hum. Mol. Genet. 2009, 18, 328–340. [Google Scholar] [CrossRef] [Green Version]
- Gong, Z.; Kim, J.E.; Leung, C.C.; Glover, J.N.; Chen, J. BACH1/FANCJ acts with TopBP1 and participates early in DNA replication checkpoint control. Mol. Cell 2010, 37, 438–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, M.; Cong, K.; Panzarino, N.J.; Nayak, S.; Calvo, J.; Deng, B.; Zhu, L.J.; Morocz, M.; Hegedus, L.; Haracska, L.; et al. Opposing Roles of FANCJ and HLTF Protect Forks and Restrain Replication during Stress. Cell Rep. 2018, 24, 3251–3261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luke-Glaser, S.; Luke, B.; Grossi, S.; Constantinou, A. FANCM regulates DNA chain elongation and is stabilized by S-phase checkpoint signalling. EMBO J. 2010, 29, 795–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.H.; Choi, K.; Szakal, B.; Arenz, J.; Duan, X.; Ye, H.; Branzei, D.; Zhao, X. Interplay between the Smc5/6 complex and the Mph1 helicase in recombinational repair. Proc. Natl. Acad. Sci. USA 2009, 106, 21252–21257. [Google Scholar] [CrossRef] [Green Version]
- Xue, X.; Choi, K.; Bonner, J.; Chiba, T.; Kwon, Y.; Xu, Y.; Sanchez, H.; Wyman, C.; Niu, H.; Zhao, X.; et al. Restriction of replication fork regression activities by a conserved SMC complex. Mol. Cell 2014, 56, 436–445. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.F.; Prakash, R.; Saro, D.; Longerich, S.; Niu, H.; Sung, P. Processing of DNA structures via DNA unwinding and branch migration by the S. cerevisiae Mph1 protein. DNA Repair (Amst) 2011, 10, 1034–1043. [Google Scholar] [CrossRef] [Green Version]
- Datta, A.; Brosh, R.M., Jr. New Insights Into DNA Helicases as Druggable Targets for Cancer Therapy. Front. Mol. Biosci. 2018, 5, 59. [Google Scholar] [CrossRef]
Figure 1.
Helicase Proteins involved in the generation of resected DNA ends during recombination. Multiple helicases, such as Bloom Syndrome helicase (BLM) and Sgs1, promote the formation of 3′ single-stranded DNA overhangs necessary for recombination. Other helicases, such as HelB, limit resection, or promote other pathways for repair. For full details, see text.
Figure 1.
Helicase Proteins involved in the generation of resected DNA ends during recombination. Multiple helicases, such as Bloom Syndrome helicase (BLM) and Sgs1, promote the formation of 3′ single-stranded DNA overhangs necessary for recombination. Other helicases, such as HelB, limit resection, or promote other pathways for repair. For full details, see text.

Figure 2.
Displacement of RAD51 from resected DNA breaks by helicase proteins. The stability of the RAD51 nucleoprotein filament is regulated by several helicases, which can remove RAD51, thereby reducing the efficiency of recombination.
Figure 2.
Displacement of RAD51 from resected DNA breaks by helicase proteins. The stability of the RAD51 nucleoprotein filament is regulated by several helicases, which can remove RAD51, thereby reducing the efficiency of recombination.

Figure 3.
Helicase-mediated unwinding of displacement loop (D-loop) intermediates. Strand invasion of a broken DNA molecule into a homologous duplex creates a D-loop. Template-based repair of sequence at the break site can proceed at the paired 3′ end, using the homologous DNA as a template. This process is inhibited by the action of a number of helicases, such as Srs2 and RTEL1, which exhibit ‘anti-recombinase’ activity by unwinding the D-loop. Other helicases, such as HFM1 and MCM8-9, stabilize the D-loop by supporting DNA polymerase activity, increasing the amount of paired heteroduplex DNA.
Figure 3.
Helicase-mediated unwinding of displacement loop (D-loop) intermediates. Strand invasion of a broken DNA molecule into a homologous duplex creates a D-loop. Template-based repair of sequence at the break site can proceed at the paired 3′ end, using the homologous DNA as a template. This process is inhibited by the action of a number of helicases, such as Srs2 and RTEL1, which exhibit ‘anti-recombinase’ activity by unwinding the D-loop. Other helicases, such as HFM1 and MCM8-9, stabilize the D-loop by supporting DNA polymerase activity, increasing the amount of paired heteroduplex DNA.

Figure 4.
Regulation of Holliday Junction disassembly by helicases. Double-Holliday junctions can be moved into close proximity by the branch migration activity of several helicase molecules. Protein complexes formed by helicases such as BLM and Sgs1 promote dissolution of the hemicatenane intermediate produced by branch migration, leading to non-crossover products of recombination.
Figure 4.
Regulation of Holliday Junction disassembly by helicases. Double-Holliday junctions can be moved into close proximity by the branch migration activity of several helicase molecules. Protein complexes formed by helicases such as BLM and Sgs1 promote dissolution of the hemicatenane intermediate produced by branch migration, leading to non-crossover products of recombination.

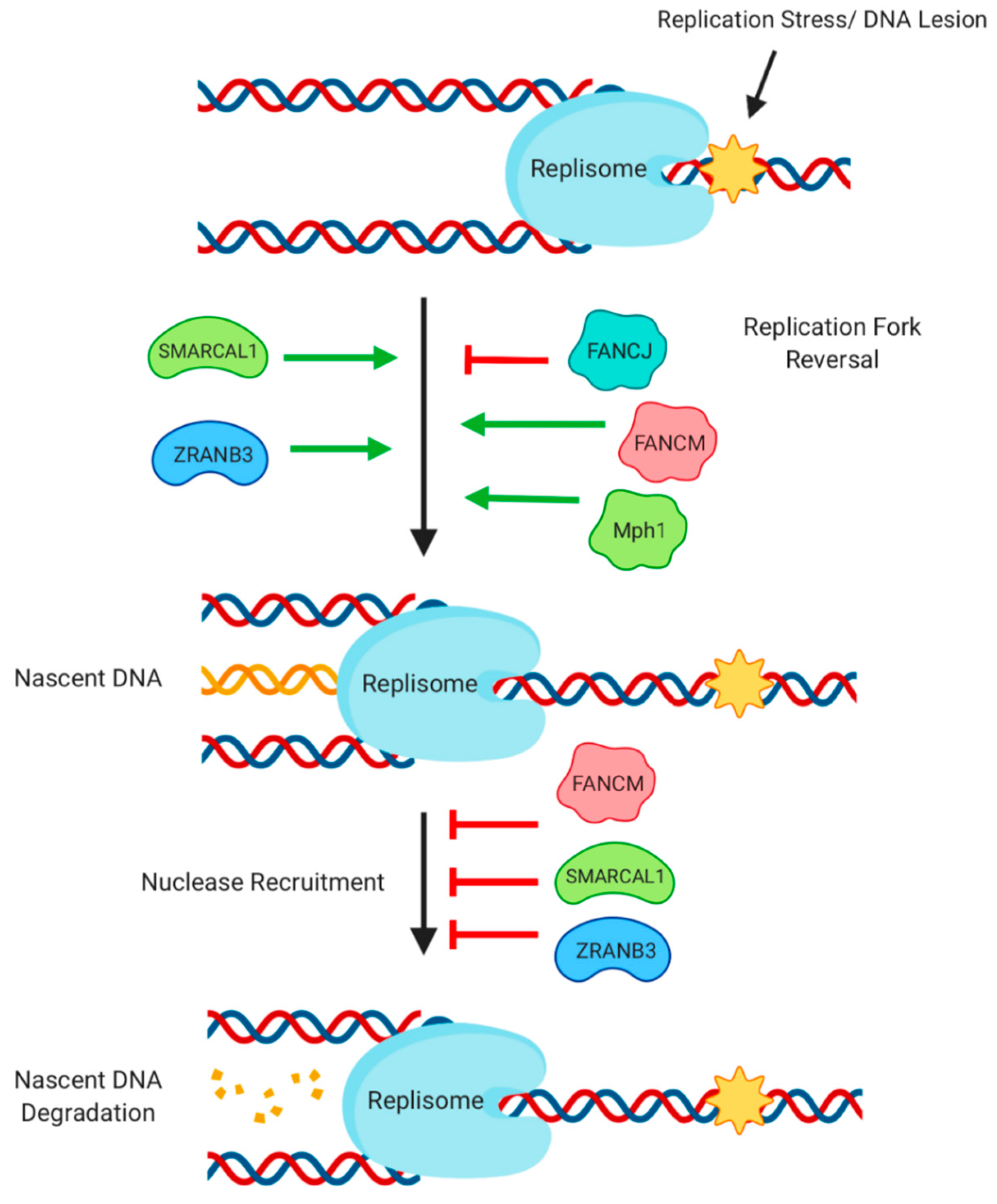
Figure 5.
Activity of helicases during DNA replication and transcription. Replication stress caused by a block in replisome progress at a DNA break or lesion can trigger replication fork reversal, which is regulated by a number of helicases. Newly synthesized DNA at the reversed fork is bound by RAD51, protecting it from nucleolytic degradation. Protection of nascent DNA is supported by the presence of several helicases.
Figure 5.
Activity of helicases during DNA replication and transcription. Replication stress caused by a block in replisome progress at a DNA break or lesion can trigger replication fork reversal, which is regulated by a number of helicases. Newly synthesized DNA at the reversed fork is bound by RAD51, protecting it from nucleolytic degradation. Protection of nascent DNA is supported by the presence of several helicases.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Helicases involved in Homologous Recombination.
| Family | Gene | Key Function | Species |
|---|---|---|---|
| RecQ | RecQ | Promotes End Resection, D-Loop Disruption, Holliday Junction Migration and Dissolution | E. coli |
| Sgs1 | Promotes End Resection, RAD51 Displacement, D-Loop Disruption, Holliday Junction Migration and Dissolution | S. cerevisiae | |
| BLM | Promotes End Resection, RAD51 Displacement, D-Loop Disruption, Holliday Junction Migration and Dissolution | Mammalian | |
| WRN | Regulates End Resection, Migrates Holliday Junctions | Mammalian | |
| RECQL1 | Promotes End Resection, D-Loop Disruption, Migrates Holliday Junctions | Mammalian | |
| RECQL4 | Promotes End Resection in S/G2, Suppresses End Resection in G1 | Mammalian | |
| RECQL5 | RAD51 Displacement | Mammalian | |
| UvrD | UvrD | RecA Displacement, | E. coli |
| SLFN11 | Replication Fork Signaling | Mammalian | |
| Srs2 | RAD51 Displacement, D-loop Disruption, Holliday Junction Migration | S. cerevisiae | |
| FBH1 | RAD51 Displacement and Degradation, Replication Fork Signaling | Mammalian | |
| PARI | RAD51 Displacement, D-loop Disruption | Mammalian | |
| HELB | Suppresses End Resection | Mammalian | |
| Fe-S | FANCJ | Promotes End Resection, RAD51 Disruption, Replication Fork Reversal | Mammalian |
| RTEL1 | D-Loop Disruption | Mammalian | |
| DNA2 | Promotes End Resection. | Mammalian | |
| DEAH Box | FANCM | Replication Fork Reversal, D-Loop Disruption | Mammalian |
| FANCJ | Promotes End Resection, RAD51 Disruption, Replication Fork Reversal | Mammalian | |
| RTEL1 | D-Loop Disruption | Mammalian | |
| Mph1 | Replication Fork Reversal, D-Loop Disruption | S. cerevisiae | |
| POLQ | Mediates alt-NHEJ, Displaces RPA | Mammalian | |
| HELQ | Promotes HR during Replication Stress, Post-Synaptic Recombination Suppression | Mammalian | |
| HFM1 | Meiotic D-Loop Stabilization | Mammalian | |
| SNF2/SWI2-like | RAD54 | RAD51 Displacement, Holliday Junction Migration and Dissolution, Promotes D-Loop Formation | Mammalian |
| Fun30 | Promotes End Resection | S. cerevisiae MammalianMammalian | |
| SMARCAD1 | Promotes End Resection | ||
| SMARCAL1 | Regulates End Resection, Replication Fork Reversal, Holliday Junction Migration, Strand Annealing | ||
| ATRX | Histone H3.3 Replacement, Post-Synaptic Regulation | ||
| MCM | MCM8-9 | Promotes End Resection, D-Loop Disruption | Mammalian |
| AAA ATPase | FIGNL1 | RAD51 Displacement | Mammalian |
| Other | PIF1 | Complex Substrate Unwinding | Mammalian/S. cerevisiae |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Huselid, E.; Bunting, S.F. The Regulation of Homologous Recombination by Helicases. Genes 2020, 11, 498. https://doi.org/10.3390/genes11050498
AMA Style
Huselid E, Bunting SF. The Regulation of Homologous Recombination by Helicases. Genes. 2020; 11(5):498. https://doi.org/10.3390/genes11050498
Chicago/Turabian StyleHuselid, Eric, and Samuel F. Bunting. 2020. "The Regulation of Homologous Recombination by Helicases" Genes 11, no. 5: 498. https://doi.org/10.3390/genes11050498
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.