Characterization of the Common Japonica-Originated Genomic Regions in the High-Yielding Varieties Developed from Inter-Subspecific Crosses in Temperate Rice (Oryza sativa L.)

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant DNA Materials
2.2. Whole Genome Sequencing and DNA Variation
2.3. SNP Allele Calling
2.4. SNP Marker Development for the Fluidigm Platform
2.5. DNA Extraction and Fluidigm Genotyping
2.6. Data Analysis and QTL Comparison
3. Results
3.1. Whole Genome Sequencing and SNP Calling
3.2. Evaluation of Japonica-Type SNP Value
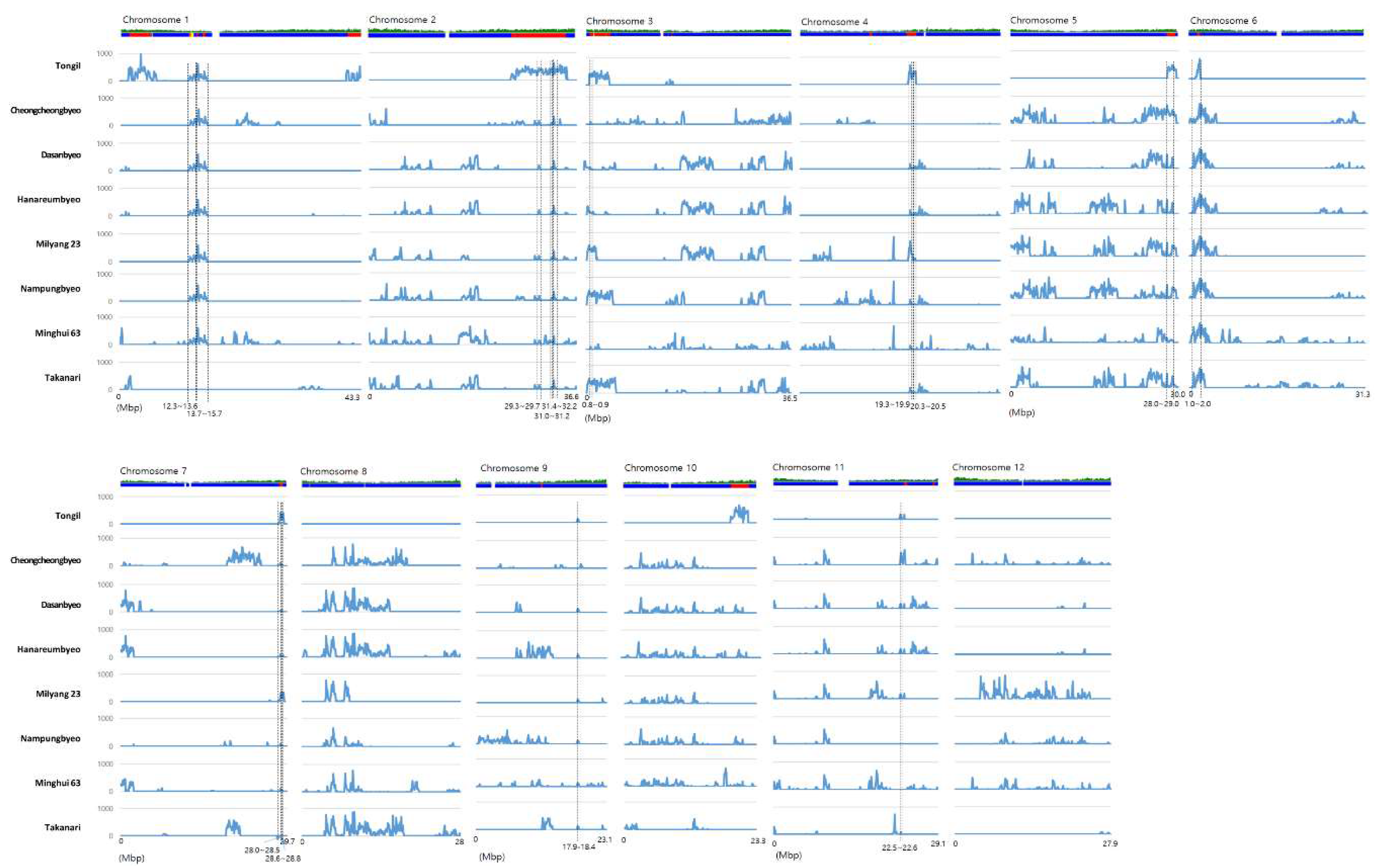
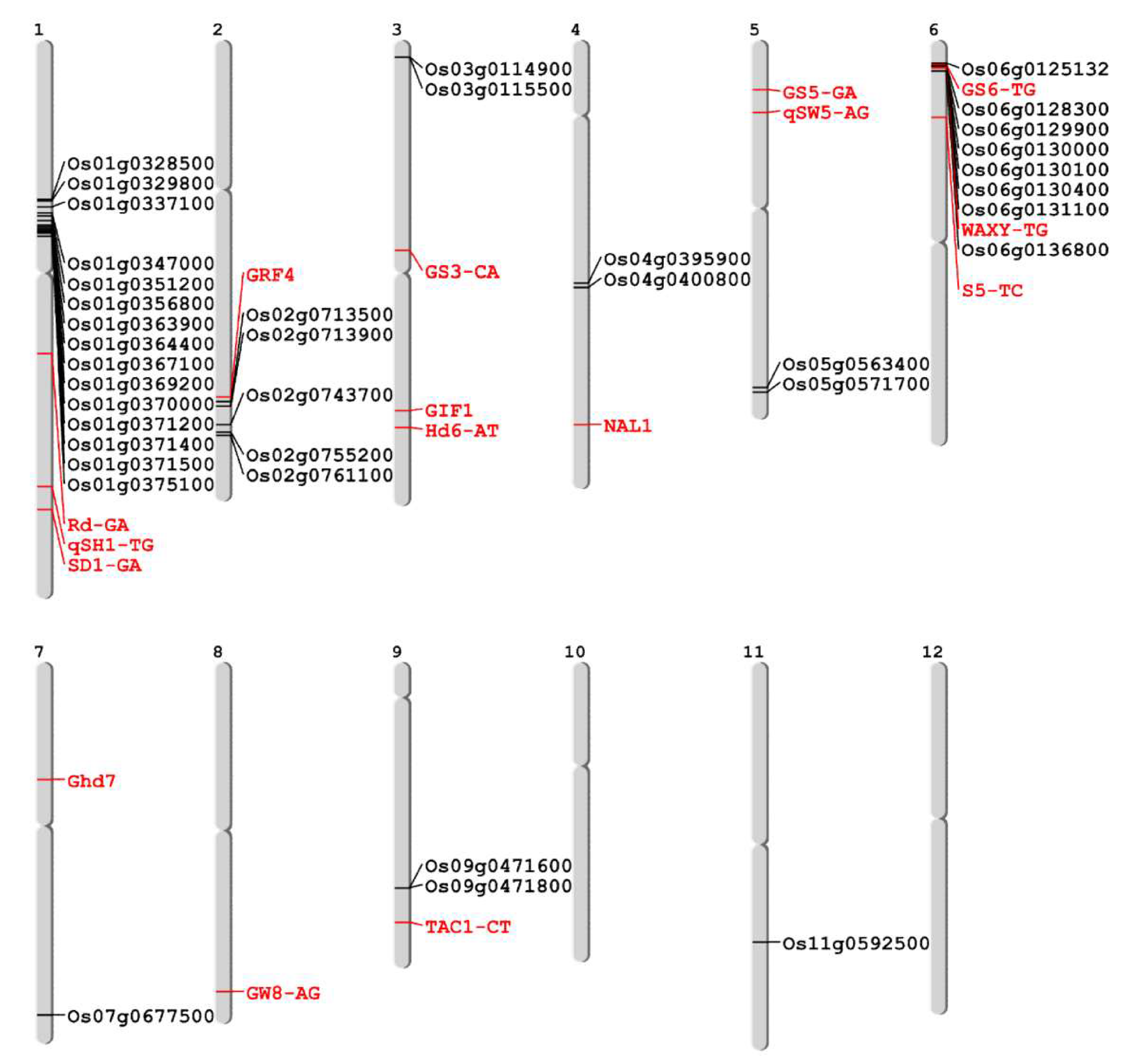
3.3. QTL Comparison and Representative Gene Selection in Common japonica-Originated Genomic Regions
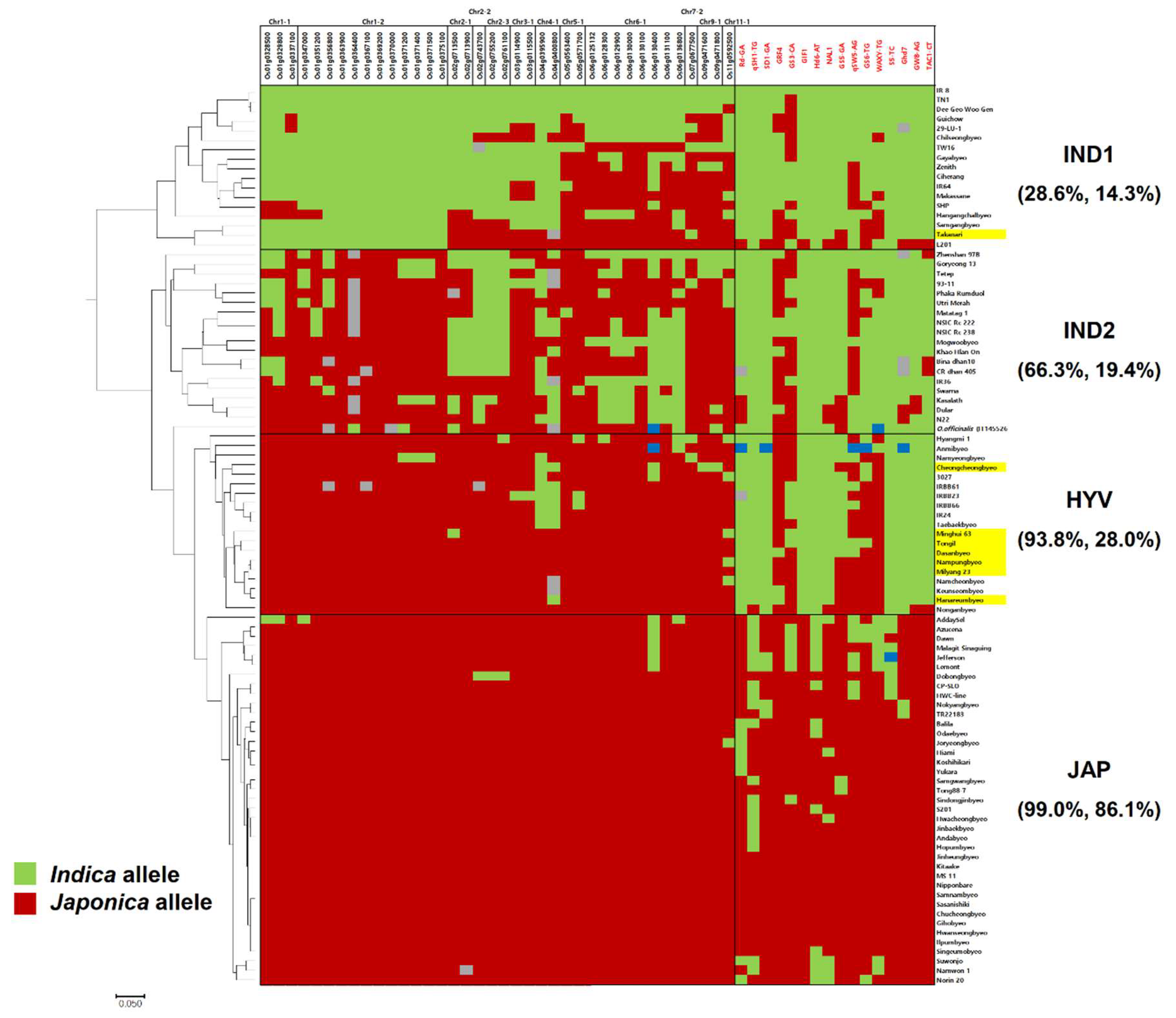
3.4. SNP Marker Development and Genotyping Using Fluidigm Platform
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Oka, H.I. Origin of Cultivated Rice; Japan Scientific Societies Press: Tokyo, Japan, 1988. [Google Scholar]
- Chung, G.S.; Heu, M.H. Improvement of Tongil-Type Rice Cultivars from Indica/Japonica Hybridization in Korea. In Rice; Bajaj, Y.P.S., Ed.; Springer: Berlin/Heidelberg, Germany, 1991; pp. 105–112. [Google Scholar] [CrossRef]
- Kim, B.; Kim, D.-G.; Lee, G.; Seo, J.; Choi, I.-Y.; Choi, B.-S.; Yang, T.-J.; Kim, K.; Lee, J.; Chin, J.; et al. Defining the genome structure of ‘Tongil’ rice, an important cultivar in the Korean “Green Revolution”. Rice 2014, 7, 22. [Google Scholar] [CrossRef]
- Takai, T.; Arai-Sanoh, Y.; Iwasawa, N.; Hayashi, T.; Yoshinaga, S.; Kondo, M. Comparative Mapping Suggests Repeated Selection of the Same Quantitative Trait Locus for High Leaf Photosynthesis Rate in Rice High-Yield Breeding Programs. Crop Sci. 2012, 52, 2649–2658. [Google Scholar] [CrossRef]
- Takai, T.; Ikka, T.; Kondo, K.; Nonoue, Y.; Ono, N.; Arai-Sanoh, Y.; Yoshinaga, S.; Nakano, H.; Yano, M.; Kondo, M.; et al. Genetic mechanisms underlying yield potential in the rice high-yielding cultivar Takanari, based on reciprocal chromosome segment substitution lines. BMC Plant Biol. 2014, 14, 295. [Google Scholar] [CrossRef]
- Xie, F.; Zhang, J. Shanyou 63: An elite mega rice hybrid in China. Rice 2018, 11, 17. [Google Scholar] [CrossRef]
- Zhu, L.; Lu, C.; Li, P.; Shen, L.; Xu, Y.; He, P.; Chen, Y. Using doubled haploid populations of rice for quantitative trait locus mapping. In Rice Genetics III. Proceedings of the Third International Rice Genetics Symposium, Manila, Philippines, 16–20 October 1995; Khush, G.S., Ed.; International Rice Research Institute: Los Baños, Philippines, 1996; pp. 631–636. [Google Scholar]
- Xie, W.; Wang, G.; Yuan, M.; Yao, W.; Lyu, K.; Zhao, H.; Yang, M.; Li, P.; Zhang, X.; Yuan, J.; et al. Breeding signatures of rice improvement revealed by a genomic variation map from a large germplasm collection. Proc. Natl. Acad. Sci. USA 2015, 112, E5411–E5419. [Google Scholar] [CrossRef]
- Sasaki, T. The map-based sequence of the rice genome. Nature 2005, 436, 793–800. [Google Scholar] [CrossRef]
- Yu, J.; Hu, S.; Wang, J.; Wong, G.K.-S.; Li, S.; Liu, B.; Deng, Y.; Dai, L.; Zhou, Y.; Zhang, X.; et al. A Draft Sequence of the Rice Genome (Oryza sativa L. ssp. indica). Science 2002, 296, 79–92. [Google Scholar] [CrossRef]
- Schatz, M.C.; Maron, L.G.; Stein, J.C.; Wences, A.H.; Gurtowski, J.; Biggers, E.; Lee, H.; Kramer, M.; Antoniou, E.; Ghiban, E.; et al. Whole genome de novo assemblies of three divergent strains of rice, Oryza sativa, document novel gene space of aus and indica. Genome Biol. 2014, 15, 1–16. [Google Scholar] [CrossRef]
- Sakai, H.; Kanamori, H.; Arai-Kichise, Y.; Shibata-Hatta, M.; Ebana, K.; Oono, Y.; Kurita, K.; Fujisawa, H.; Katagiri, S.; Mukai, Y.; et al. Construction of Pseudomolecule Sequences of the aus Rice Cultivar Kasalath for Comparative Genomics of Asian Cultivated Rice. DNA Res. 2014. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, L.-L.; Xing, F.; Kudrna, D.A.; Yao, W.; Copetti, D.; Mu, T.; Li, W.; Song, J.-M.; Xie, W.; et al. Extensive sequence divergence between the reference genomes of two elite indica rice varieties Zhenshan 97 and Minghui 63. Proc. Natl. Acad. Sci. USA 2016, 113, E5163–E5171. [Google Scholar] [CrossRef]
- Du, H.; Yu, Y.; Ma, Y.; Gao, Q.; Cao, Y.; Chen, Z.; Ma, B.; Qi, M.; Li, Y.; Zhao, X.; et al. Sequencing and de novo assembly of a near complete indica rice genome. Nat. Commun. 2017, 8, 15324. [Google Scholar] [CrossRef]
- Subbaiyan, G.K.; Waters, D.L.; Katiyar, S.K.; Sadananda, A.R.; Vaddadi, S.; Henry, R.J. Genome-wide DNA polymorphisms in elite indica rice inbreds discovered by whole-genome sequencing. Plant Biotechnol. J. 2012, 10, 623–634. [Google Scholar] [CrossRef]
- Feltus, F.A.; Wan, J.; Schulze, S.R.; Estill, J.C.; Jiang, N.; Paterson, A.H. An SNP resource for rice genetics and breeding based on subspecies indica and japonica genome alignments. Genome Res. 2004, 14, 1812–1819. [Google Scholar] [CrossRef]
- Sun, C.; Hu, Z.; Zheng, T.; Lu, K.; Zhao, Y.; Wang, W.; Shi, J.; Wang, C.; Lu, J.; Zhang, D.; et al. RPAN: Rice pan-genome browser for approximately 3000 rice genomes. Nucleic Acids Res. 2017, 45, 597–605. [Google Scholar] [CrossRef]
- Abbai, R.; Singh, V.K.; Nachimuthu, V.V.; Sinha, P.; Selvaraj, R.; Vipparla, A.K.; Singh, A.K.; Singh, U.M.; Varshney, R.K.; Kumar, A. Haplotype analysis of key genes governing grain yield and quality traits across 3K RG panel reveals scope for the development of tailor-made rice with enhanced genetic gains. Plant Biotechnol. J. 2019, 17, 1612–1622. [Google Scholar] [CrossRef]
- Carpentier, M.C.; Manfroi, E.; Wei, F.J.; Wu, H.P.; Lasserre, E.; Llauro, C.; Debladis, E.; Akakpo, R.; Hsing, Y.I.; Panaud, O. Retrotranspositional landscape of Asian rice revealed by 3000 genomes. Nat. Commun. 2019, 10, 24. [Google Scholar] [CrossRef]
- Fuentes, R.R.; Chebotarov, D.; Duitama, J.; Smith, S.; De la Hoz, J.F.; Mohiyuddin, M.; Wing, R.A.; McNally, K.L.; Tatarinova, T.; Grigoriev, A.; et al. Structural variants in 3000 rice genomes. Genome Res. 2019, 29, 870–880. [Google Scholar] [CrossRef]
- Huang, J.; Li, J.; Zhou, J.; Wang, L.; Yang, S.; Hurst, L.D.; Li, W.H.; Tian, D. Identifying a large number of high-yield genes in rice by pedigree analysis, whole-genome sequencing, and CRISPR-Cas9 gene knockout. Proc. Natl. Acad. Sci. USA 2018, 115, E7559–E7567. [Google Scholar] [CrossRef]
- Adachi, S.; Yamamoto, T.; Nakae, T.; Yamashita, M.; Uchida, M.; Karimata, R.; Ichihara, N.; Soda, K.; Ochiai, T.; Ao, R.; et al. Genetic architecture of leaf photosynthesis in rice revealed by different types of reciprocal mapping populations. J. Exp. Bot. 2019, 70, 5131–5144. [Google Scholar] [CrossRef]
- Takai, T.; Adachi, S.; Taguchi-Shiobara, F.; Sanoh-Arai, Y.; Iwasawa, N.; Yoshinaga, S.; Hirose, S.; Taniguchi, Y.; Yamanouchi, U.; Wu, J.; et al. A natural variant of NAL1, selected in high-yield rice breeding programs, pleiotropically increases photosynthesis rate. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef]
- Kawahara, Y.; de la Bastide, M.; Hamilton, J.P.; Kanamori, H.; McCombie, W.R.; Ouyang, S.; Schwartz, D.C.; Tanaka, T.; Wu, J.; Zhou, S.; et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 2013, 6, 4. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Barnett, D.W.; Garrison, E.K.; Quinlan, A.R.; Stromberg, M.P.; Marth, G.T. BamTools: A C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 2011, 27, 1691–1692. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Huang, X.; Wei, X.; Sang, T.; Zhao, Q.; Feng, Q.; Zhao, Y.; Li, C.; Zhu, C.; Lu, T.; Zhang, Z.; et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 2010, 42, 961–967. [Google Scholar] [CrossRef]
- Seo, J.; Lee, G.; Jin, Z.; Kim, B.; Chin, J.H.; Koh, H.-J. Development and application of indica–japonica SNP assays using the Fluidigm platform for rice genetic analysis and molecular breeding. Mol. Breed. 2020, 40, 39. [Google Scholar] [CrossRef]
- D3 Assay Design-Fluidigm. Available online: https://d3.fluidigm.com (accessed on 25 September 2019).
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4326. [Google Scholar] [CrossRef]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef]
- Cavalli-Sforza, L.L.; Edwards, A.W.F. Phylogenetic analysis. Models and estimation procedures. Am. J. Hum. Genet. 1967, 19, 233–257. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Yonemaru, J.-i.; Yamamoto, T.; Fukuoka, S.; Uga, Y.; Hori, K.; Yano, M. Q-TARO: QTL Annotation Rice Online Database. Rice 2010, 3, 194–203. [Google Scholar] [CrossRef]
- Seo, J.; Lee, S.-M.; Han, J.-H.; Shin, N.-H.; Koh, H.-J.; Chin, J.H. Identification of Yield and Yield-Related Quantitative Trait Loci for the Field High Temperature Condition in Backcross Populations of Rice (Oryza sativa L.). Plant Breed. Biotechnol. 2019, 7, 415–426. [Google Scholar] [CrossRef]
- Ha, U.-G.; Song, Y.-C.; Yeo, U.-S.; Cho, J.-H.; Hwang, H.-G.; Kim, Y.-D.; Cho, Y.-H.; Yang, S.-J.; Lee, J.-H.; Oh, B.-G.; et al. A New High Yielding Rice Variety with Multi-Disease Resistance, ‘Keunseom’. Korean J. Breed. Sci. 2011, 43, 576–580. [Google Scholar]
- Huang, N.; Angeles, E.R.; Domingo, J.; Magpantay, G.; Singh, S.; Zhang, G.; Kumaravadivel, N.; Bennett, J.; Khush, G.S. Pyramiding of bacterial blight resistance genes in rice: Marker-assisted selection using RFLP and PCR. Theor. Appl. Genet. 1997, 95, 313–320. [Google Scholar] [CrossRef]
- Khush, G.; Virk, P. IR Varieties and Their Impact, 1st ed.; International Rice Research Institute: Los Baños, Philippines, 2005; pp. 46–48. [Google Scholar]
- Kim, S.-J.; Jeong, D.-H.; An, G.; Kim, S.-R. Characterization of a drought-responsive gene, OsTPS1, identified by the T-DNA Gene-Trap system in rice. J. Plant Biol. 2005, 48, 371–379. [Google Scholar] [CrossRef]
- Li, H.W.; Zang, B.S.; Deng, X.W.; Wang, X.P. Overexpression of the trehalose-6-phosphate synthase gene OsTPS1 enhances abiotic stress tolerance in rice. Planta 2011, 234, 1007–1018. [Google Scholar] [CrossRef]
- Li, C.; Wang, Y.; Liu, L.; Hu, Y.; Zhang, F.; Mergen, S.; Wang, G.; Schlappi, M.R.; Chu, C. A rice plastidial nucleotide sugar epimerase is involved in galactolipid biosynthesis and improves photosynthetic efficiency. PLoS Genet. 2011, 7, e1002196. [Google Scholar] [CrossRef]
- Sreenivasulu, N.; Butardo, V.M., Jr.; Misra, G.; Cuevas, R.P.; Anacleto, R.; Kavi Kishor, P.B. Designing climate-resilient rice with ideal grain quality suited for high-temperature stress. J. Exp. Bot. 2015, 66, 1737–1748. [Google Scholar] [CrossRef]
- Olsen, K.M.; Caicedo, A.L.; Polato, N.; McClung, A.; McCouch, S.; Purugganan, M.D. Selection Under Domestication: Evidence for a Sweep in the Rice Waxy Genomic Region. Genetics 2006, 173, 975–983. [Google Scholar] [CrossRef]
- Ji, H.; Ahn, E.; Seo, B.; Kang, H.; Choi, I.; Kim, K. Genome-wide detection of SNPs between two Korean Tongil-type rice varieties. Korean J. Breed. Sci. 2016, 48, 460–469. [Google Scholar] [CrossRef]
- Kim, H.-Y.; Yang, C.-I.; Choi, Y.-H.; Won, Y.-J.; Lee, Y.-T. Changes of Seed Viability and Physico-Chemical Properties of Milled Rice with Different Ecotypes and Storage Duration. Korean J. Crop Sci. 2007, 52, 375–379. [Google Scholar]
- Kwak, J.; Yoon, M.R.; Lee, J.S.; Lee, J.H.; Ko, S.; Tai, T.H.; Won, Y.J. Morphological and starch characteristics of the Japonica rice mutant variety Seolgaeng for dry-milled flour. Food Sci. Biotechnol. 2017, 26, 43–48. [Google Scholar] [CrossRef]
- Matsushima, R.; Maekawa, M.; Kusano, M.; Tomita, K.; Kondo, H.; Nishimura, H.; Crofts, N.; Fujita, N.; Sakamoto, W. Amyloplast Membrane Protein SUBSTANDARD STARCH GRAIN6 Controls Starch Grain Size in Rice Endosperm. Plant Physiol. 2016, 170, 1445–1459. [Google Scholar] [CrossRef]
- Fekih, R.; Tamiru, M.; Kanzaki, H.; Abe, A.; Yoshida, K.; Kanzaki, E.; Saitoh, H.; Takagi, H.; Natsume, S.; Undan, J.R.; et al. The rice (Oryza sativa L.) LESION MIMIC RESEMBLING, which encodes an AAA-type ATPase, is implicated in defense response. Mol. Genet. Genom. 2015, 290, 611–622. [Google Scholar] [CrossRef]
- Ouyang, S.Q.; Liu, Y.F.; Liu, P.; Lei, G.; He, S.J.; Ma, B.; Zhang, W.K.; Zhang, J.S.; Chen, S.Y. Receptor-like kinase OsSIK1 improves drought and salt stress tolerance in rice (Oryza sativa) plants. Plant J. 2010, 62, 316–329. [Google Scholar] [CrossRef]
- Dalrymple, D.G. Development of High-Yielding Rice Varieties. In Development and Spread of High-Yielding Rice Varieties in Developing Countries, 2nd ed.; Bureau for Science and Technology Agency for International Development: Washington, DC, USA, 1986; pp. 15–35. [Google Scholar]
- Zeng, D.; Tian, Z.; Rao, Y.; Dong, G.; Yang, Y.; Huang, L.; Leng, Y.; Xu, J.; Sun, C.; Zhang, G.; et al. Rational design of high-yield and superior-quality rice. Nat. Plants 2017, 3, 17031. [Google Scholar] [CrossRef]
- Li, L.; Mao, D.; Prasad, M. Deployment of cold tolerance loci from Oryza sativa ssp. Japonica cv. ‘Nipponbare’ in a high-yielding Indica rice Cultivar ‘93-11’. Plant Breed. 2018, 137, 553–560. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Type | Variety Name | Yield (bp) | Read | N (%) | GC (%) | Q30 (%) | Depth (X) | Sequencing Platform |
|---|---|---|---|---|---|---|---|---|
| HYV | Cheongcheongbyeo | 9,991,040,272 | 66,464,246 | 0.07 | 43.83 | 83.37 | 21.76 | Illumina NextSeq 500 |
| HYV | Dasanbyeo | 5,677,243,407 | 93,838,734 | 1.05 | 39.75 | 69.57 | 10.32 | Illumina HiSeq 1000 |
| HYV | Hanareumbyeo | 8,701,463,207 | 57,884,660 | 0.07 | 43.74 | 81.74 | 18.58 | Illumina NextSeq 500 |
| HYV | Milyang 23 | 8,909,120,495 | 147,258,190 | 0.31 | 41.08 | 76.44 | 17.79 | Illumina HiSeq 1000 |
| HYV | Minghui 63 | 10,346,919,646 | 68,794,138 | 0.07 | 43.93 | 83.18 | 22.48 | Illumina NextSeq 500 |
| HYV | Nampungbyeo | 10,366,586,498 | 68,923,446 | 0.07 | 43.88 | 83.52 | 22.62 | Illumina NextSeq 500 |
| HYV | Takanari | 9,372,371,998 | 62,353,794 | 0.08 | 43.38 | 83.10 | 20.35 | Illumina NextSeq 500 |
| HYV | Tongil | 13,362,670,165 | 264,607,330 | 0.16 | 42.57 | 70.41 | 24.58 | Illumina HiSeq 1000 |
| Japonica | Nipponbare | 22,212,867,380 | 439,858,760 | 0.19 | 42.10 | 89.12 | 51.72 | Illumina HiSeq 1000 |
| Japonica | Yukara | 9,155,887,048 | 151,336,976 | 0.28 | 41.13 | 77.37 | 18.51 | Illumina HiSeq 1000 |
| Indica | IR 8 | 8,287,794,812 | 136,988,344 | 0.40 | 41.14 | 78.94 | 17.09 | Illumina HiSeq 1000 |
| Indica | TN 1 | 8,337,247,875 | 137,805,750 | 0.31 | 41.51 | 77.25 | 16.83 | Illumina HiSeq 1000 |
| Varieties | Non-Synonymous | Synonymous | Intron | 5′ UTR | 3′ UTR | Intergenic | Total |
|---|---|---|---|---|---|---|---|
| Cheongcheongbyeo | 19,193 | 17,439 | 21,671 | 7076 | 29,403 | 1,019,466 | 1,114,248 |
| Dasanbyeo | 18,534 | 16,944 | 21,247 | 7006 | 28,598 | 975,433 | 1,067,762 |
| Hanareumbyeo | 18,059 | 16,492 | 20,657 | 6825 | 27,766 | 937,990 | 1,027,789 |
| Milyang 23 | 17,836 | 16,414 | 20,110 | 6628 | 27,271 | 933,778 | 1,022,037 |
| Minghui 63 | 20,117 | 18,601 | 22,351 | 7350 | 30,731 | 1,042,153 | 1,141,303 |
| Nampungbyeo | 19,592 | 17,960 | 21,588 | 7195 | 29,286 | 1,005,553 | 1,101,174 |
| Takanari | 18,631 | 16,936 | 20,881 | 6887 | 28,157 | 966,035 | 1,057,527 |
| Tongil | 18,427 | 16,837 | 20,816 | 6750 | 27,021 | 952,421 | 1,042,272 |
| IR 8 | 20,578 | 18,834 | 22,847 | 7494 | 31,152 | 1,061,383 | 1,162,288 |
| TN1 | 20,171 | 18,179 | 22,095 | 7614 | 30,286 | 1,041,363 | 1,139,708 |
| Region | Narrowed Range (Mb) | Size (Mb) | Tongil | Cheongcheongbyeo | Dasanbyeo | Hanareumbyeo | Milyang 23 | Nampungbyeo | Minghui 63 | Takanari | Type |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Chr1-1 | 12.3 ~ 13.6 | 1.3 | 23.3 | 23.3 | 23.3 | 23.3 | 23.3 | 23.3 | 23.3 | 0.0 | All (ex. Takanari) |
| Chr1-2 | 13.7 ~ 15.7 | 2 | 31.4 | 31.4 | 31.4 | 31.4 | 31.4 | 31.5 | 31.5 | 0.0 | All (ex. Takanari) |
| Chr2-1 | 29.3 ~ 29.7 | 0.4 | 64.1 | 14.1 | 14.1 | 14.1 | 14.1 | 14.1 | 14.4 | 14.1 | All |
| Chr2-2 | 31.0 ~ 31.2 | 0.2 | 46.5 | 1.1 | 1.1 | 1.1 | 1.1 | 1.1 | 2.1 | 1.1 | All |
| Chr2-3 | 31.4 ~ 32.2 | 0.8 | 77.4 | 17.3 | 17.6 | 17.6 | 17.6 | 17.6 | 21.6 | 17.6 | All |
| Chr3-1 | 0.8 ~ 0.9 | 0.1 | 73.9 | 19.3 | 16.6 | 16.6 | 73.9 | 73.9 | 19.2 | 73.9 | All |
| Chr4-1 | 19.3 ~ 19.9 | 0.6 | 75.9 | 0.0 | 8.2 | 8.2 | 77.3 | 8.2 | 7.0 | 8.2 | All (ex. Cheongcheong) |
| Chr4-2 | 20.3 ~ 20.5 | 0.2 | 87.9 | 0.0 | 12.8 | 12.8 | 12.8 | 5.1 | 0.3 | 5.1 | All (ex. Cheongcheong) |
| Chr5-1 | 28.0 ~ 29.0 | 1 | 76.9 | 82.1 | 23.9 | 23.9 | 23.9 | 23.9 | 13.7 | 23.9 | All |
| Chr6-1 | 1.0 2.0 | 1 | 55.2 | 51.7 | 70.1 | 70.1 | 70.1 | 70.1 | 70.1 | 70.1 | All |
| Chr7-1 | 28.5 ~ 28.6 | 0.1 | 32.0 | 6.5 | 5.0 | 6.5 | 4.8 | 6.5 | 6.5 | 0.0 | All (ex. Takanari) |
| Chr7-2 | 28.6 ~ 28.8 | 0.2 | 93.5 | 9.7 | 7.7 | 7.6 | 33.2 | 9.7 | 7.6 | 0.0 | All (ex. Takanari) |
| Chr9-1 | 17.9 ~ 18.4 | 0.5 | 13.9 | 0.7 | 13.9 | 13.9 | 13.9 | 13.9 | 13.9 | 13.9 | All |
| Chr11-1 | 22.5 ~ 22.6 | 0.1 | 25.3 | 65.6 | 25.3 | 25.3 | 25.3 | 0.0 | 0.0 | 12.9 | All (ex. Nampung/Minghui63) |
| Region | Eating Quality | Abiotic Tolerance | Biotic Resistance | Yield-Related | Root | Flowering | Other | Total |
|---|---|---|---|---|---|---|---|---|
| Chr1-1/1-2 | 0 | 4 | 0 | 2 | 0 | 0 | 1 | 7 |
| Chr2-1/2-2/2-3 | 8 | 6 | 3 | 5 | 1 | 1 | 1 | 25 |
| Chr3-1 | 1 | 8 | 0 | 0 | 2 | 1 | 0 | 12 |
| Chr4-1/4-2 | 0 | 2 | 0 | 4 | 1 | 0 | 1 | 8 |
| Chr5-1 | 3 | 2 | 0 | 6 | 2 | 0 | 0 | 13 |
| Chr6-1 | 10 | 1 | 1 | 0 | 1 | 2 | 0 | 15 |
| Chr7-1/7-2 | 0 | 1 | 0 | 3 | 0 | 1 | 0 | 5 |
| Chr9-1 | 0 | 4 | 2 | 7 | 0 | 0 | 0 | 13 |
| Chr11-1 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 3 |
| Total | 23 | 29 | 6 | 28 | 7 | 5 | 3 | 101 |
| Region | Gene (RAP DB) | Alternative Name | Known Function | |
|---|---|---|---|---|
| UniProt | RAP DB | |||
| Chr1-1 | Os01g0328500 | Bucentaur or craniofacial development family protein | ||
| Os01g0329800 | IAI1 | YT521-B-like protein family protein | ||
| Os01g0337100 | OsTPS1 | Similar to Sesquiterpene synthase | ||
| Chr1-2 | Os01g0347000 | OsPHS1b, OsPP4, OsSTA14 | Similar to PROPYZAMIDE-HTPERSENSITIVE 1 | |
| Os01g0348900 | salT(sal1), SalT1, sal1, Sal1, SALT, ML, SalT, OsSalT | Salt stress-induced protein | SalT gene product | |
| Os01g0351200 | PARP2-A | Poly [ADP-ribose] polymerase 2-A | Similar to Poly | |
| Os01g0356800 | OsEnS-6 | Domain of unknown function DUF3406, chloroplast translocase domain containing protein | ||
| Os01g0363900 | OsWAK5 | Similar to HASTY | ||
| Os01g0364400 | OsRLCK35 | Protein kinase, catalytic domain domain containing protein | ||
| Os01g0367100 | PHD1 | Chloroplast-localized UDP-glucose epimerase (UGE), Galactolipid biosynthesis for chloroplast membranes, Photosynthetic capability and carbon assimilate homeostasis (Os01t0367100-01); NAD(P)-binding domain containing protein | ||
| Os01g0369200 | CUL1 | Cullin-like protein | Similar to Cullin-1 | |
| Os01g0370000 | OsOPR9, OsOPR01-2 | Putative 12-oxophytodienoate reductase 9 | NADH:flavin oxidoreductase/NADH oxidase, N-terminal domain containing protein | |
| Os01g0371200 | OsGSTF1, RGSI | Probable glutathione S-transferase GSTF1 | Similar to Glutathione-S-transferase 19E50 | |
| Os01g0371400 | OsGSTF9 | Similar to Glutathione s-transferase gstf2 | ||
| Os01g0371500 | OsGSTF10 | Similar to Glutathione-S-transferase 19E50 | ||
| Os01g0375100 | OsDjC6 | DNAJ heat shock N-terminal domain-containing protein-like | Similar to DnAJ-like protein slr0093 | |
| Chr2-1 | Os02g0713500 | OsFbox108, Os_F0236 | F-box domain, cyclin-like domain containing protein. | |
| Os02g0713900 | HMGR I, Hmg1, HMGR1, HMGR 1, OsHMGR1 | 3-hydroxy-3-methylglutaryl-coenzyme A reductase 1 | Similar to 3-hydroxy-3-methylglutaryl-coenzyme A reductase 1 | |
| Chr2-2 | Os02g0743700 | Similar to RING-H2 finger protein ATL1Q | ||
| Chr2-3 | Os02g0755200 | OsHDMA702, HDMA702 | Lysine-specific histone demethylase 1 homolog 1 | Similar to amine oxidase family protein |
| Os02g0761100 | OsCYP40b, OsCYP-8, OsCYP40-2 | Similar to Cyclophilin-40 (Expressed protein) | ||
| Chr3-1 | Os03g0114900 | Mitochondrial import inner membrane translocase subunit Tim17 family protein, expressed | Similar to putaive mitochondrial inner membrane protein | |
| Os03g0115500 | Similar to pyridoxine 5’-phosphate oxidase-related | |||
| Chr4-1 | Os04g0395900 | Polynucleotide adenylyltransferase region domain containing protein | ||
| Os04g0400800 | Heavy metal transport/detoxification protein domain containing protein | |||
| Chr5-1 | Os05g0563400 | OsARF15, ETT1, OsETT1, ARF3b | Auxin response factor 15 | Similar to Auxin response factor 5 |
| Os05g0571700 | OsFbox282, Os_F0643 | Cyclin-like F-box domain containing protein | ||
| Chr6-1 | Os06g0125132 | SDH8B | Succinate dehydrogenase subunit 8B, mitochondrial | Conserved hypothetical protein |
| Os06g0128300 | OsABCB23, OsISC32 | Similar to Mitochondrial half-ABC transporter, Similar to STA1 (STARIK 1); ATPase, coupled to transmembrane movement of substances | ||
| Os06g0129900 | Cytochrome P450 | Cytochrome P450 | Similar to Cytochrome P450 CYPD | |
| Os06g0130000 | LMR | AAA-type ATPase, Defense response, Similar to Tobacco mosaic virus helicase domain-binding protein (Fragment) | ||
| Os06g0130100 | OsSIK1, OsER2, ER2 | Receptor-like kinase (RLK), Drought and salt stress tolerance, Oryza sativa stress-induced protein kinase gene 1 | ||
| Os06g0130400 | OsACS6 | ACC synthase, Protein homologous to aminotransferase, Ethylene biosynthesis, Control of starch grain size in rice endosperm | ||
| Os06g0131100 | OsWD40-124 | Similar to guanine nucleotide-binding protein beta subunit-like protein 1, WD40/YVTN repeat-like domain containing protein | ||
| Os06g0136800 | OsClp9, CLP9 | ATP-dependent Clp protease proteolytic subunit | Peptidase S14, ClpP family protein | |
| Chr7-2 | Os07g0677500 | POX3006, prx114 | Peroxidase | Similar to Peroxidase precursor (EC 1.11.1.7) |
| Chr9-1 | Os09g0471600 | OsWAK84 | EGF-like calcium-binding domain containing protein | |
| Os09g0471800 | OsWAK85, YK10 | Similar to WAK80 - OsWAK receptor-like protein kinase | ||
| Chr11-1 | Os11g0592500 | NB-ARC domain containing protein, expressed | Similar to NB-ARC domain containing protein, expressed | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seo, J.; Lee, S.-M.; Han, J.-H.; Shin, N.-H.; Lee, Y.K.; Kim, B.; Chin, J.H.; Koh, H.-J. Characterization of the Common Japonica-Originated Genomic Regions in the High-Yielding Varieties Developed from Inter-Subspecific Crosses in Temperate Rice (Oryza sativa L.). Genes 2020, 11, 562. https://doi.org/10.3390/genes11050562
Seo J, Lee S-M, Han J-H, Shin N-H, Lee YK, Kim B, Chin JH, Koh H-J. Characterization of the Common Japonica-Originated Genomic Regions in the High-Yielding Varieties Developed from Inter-Subspecific Crosses in Temperate Rice (Oryza sativa L.). Genes. 2020; 11(5):562. https://doi.org/10.3390/genes11050562
Chicago/Turabian StyleSeo, Jeonghwan, So-Myeong Lee, Jae-Hyuk Han, Na-Hyun Shin, Yoon Kyung Lee, Backki Kim, Joong Hyoun Chin, and Hee-Jong Koh. 2020. "Characterization of the Common Japonica-Originated Genomic Regions in the High-Yielding Varieties Developed from Inter-Subspecific Crosses in Temperate Rice (Oryza sativa L.)" Genes 11, no. 5: 562. https://doi.org/10.3390/genes11050562
APA StyleSeo, J., Lee, S.-M., Han, J.-H., Shin, N.-H., Lee, Y. K., Kim, B., Chin, J. H., & Koh, H.-J. (2020). Characterization of the Common Japonica-Originated Genomic Regions in the High-Yielding Varieties Developed from Inter-Subspecific Crosses in Temperate Rice (Oryza sativa L.). Genes, 11(5), 562. https://doi.org/10.3390/genes11050562