
Dissecting the Genetic Diversity of USDA Cowpea Germplasm Collection Using Kompetitive Allele Specific PCR-Single Nucleotide Polymorphism Markers

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and DNA Isolation
2.2. SNP Selection, Primer Design and Genotyping
2.3. Genetic Diversity and Population Structure
3. Results
3.1. Validation of KASP-SNP Markers
3.2. Genetic Diversity of V. unguiculata
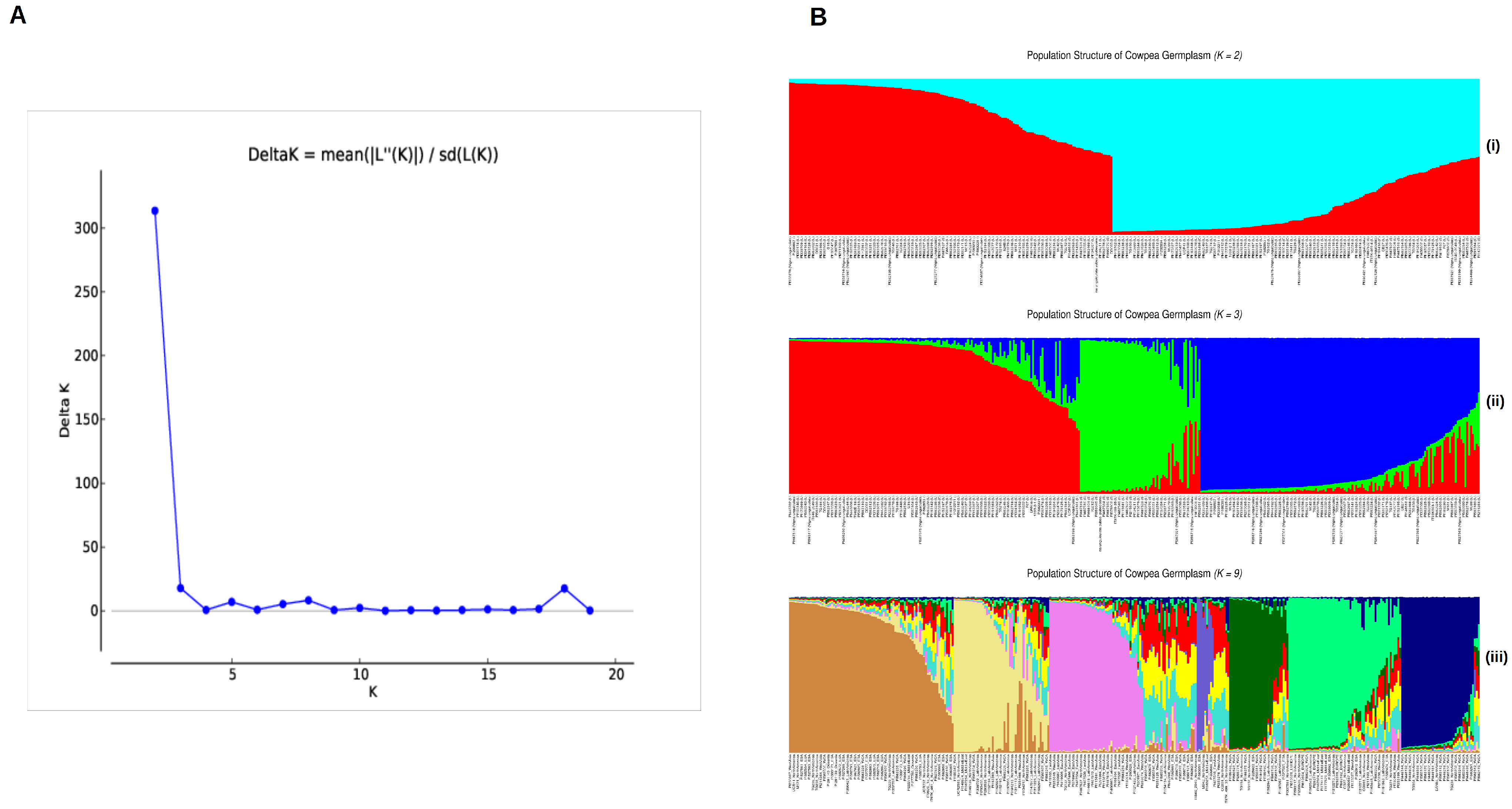
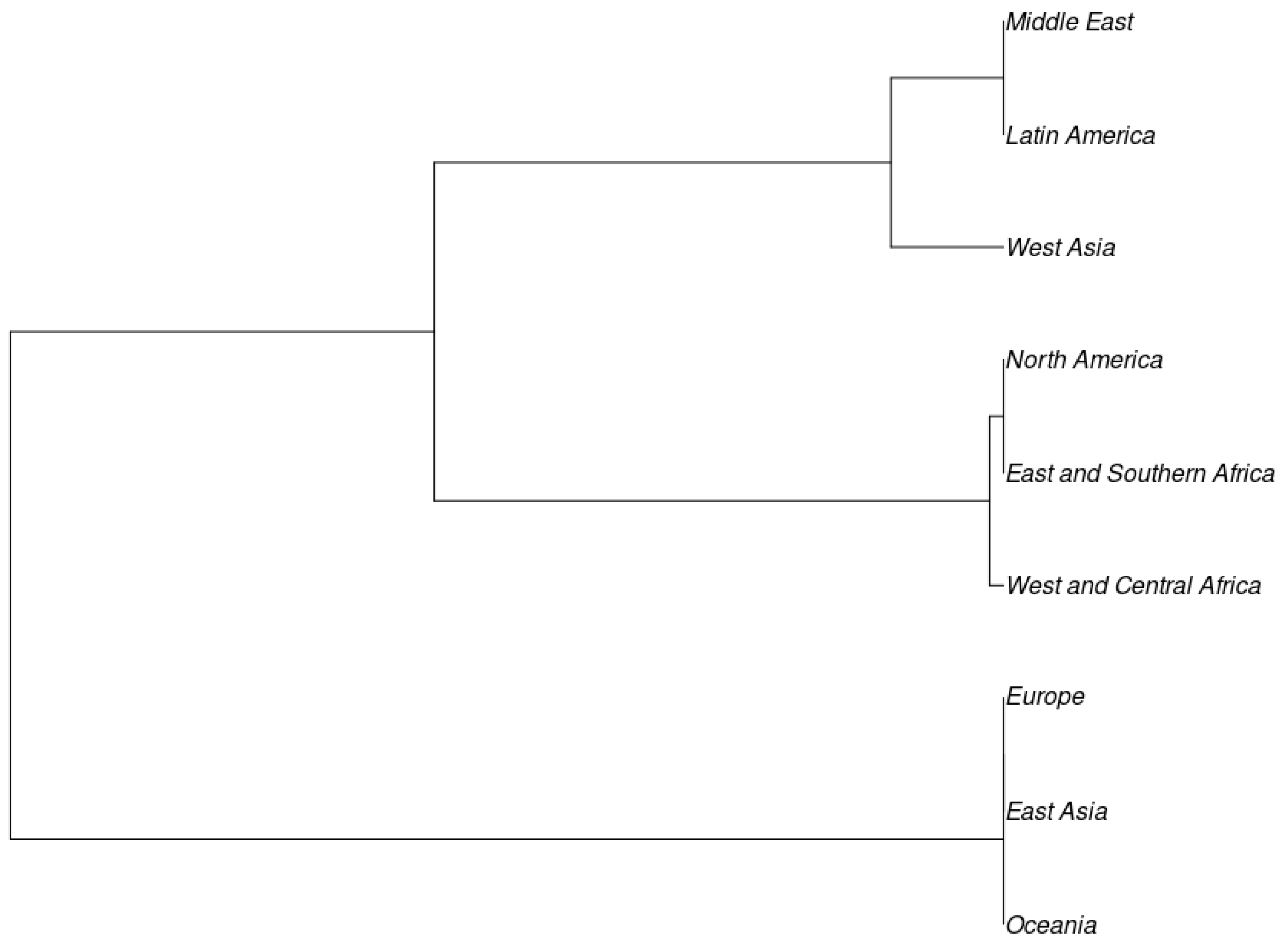
3.3. Population Structure in the Global Cowpea Germplasm

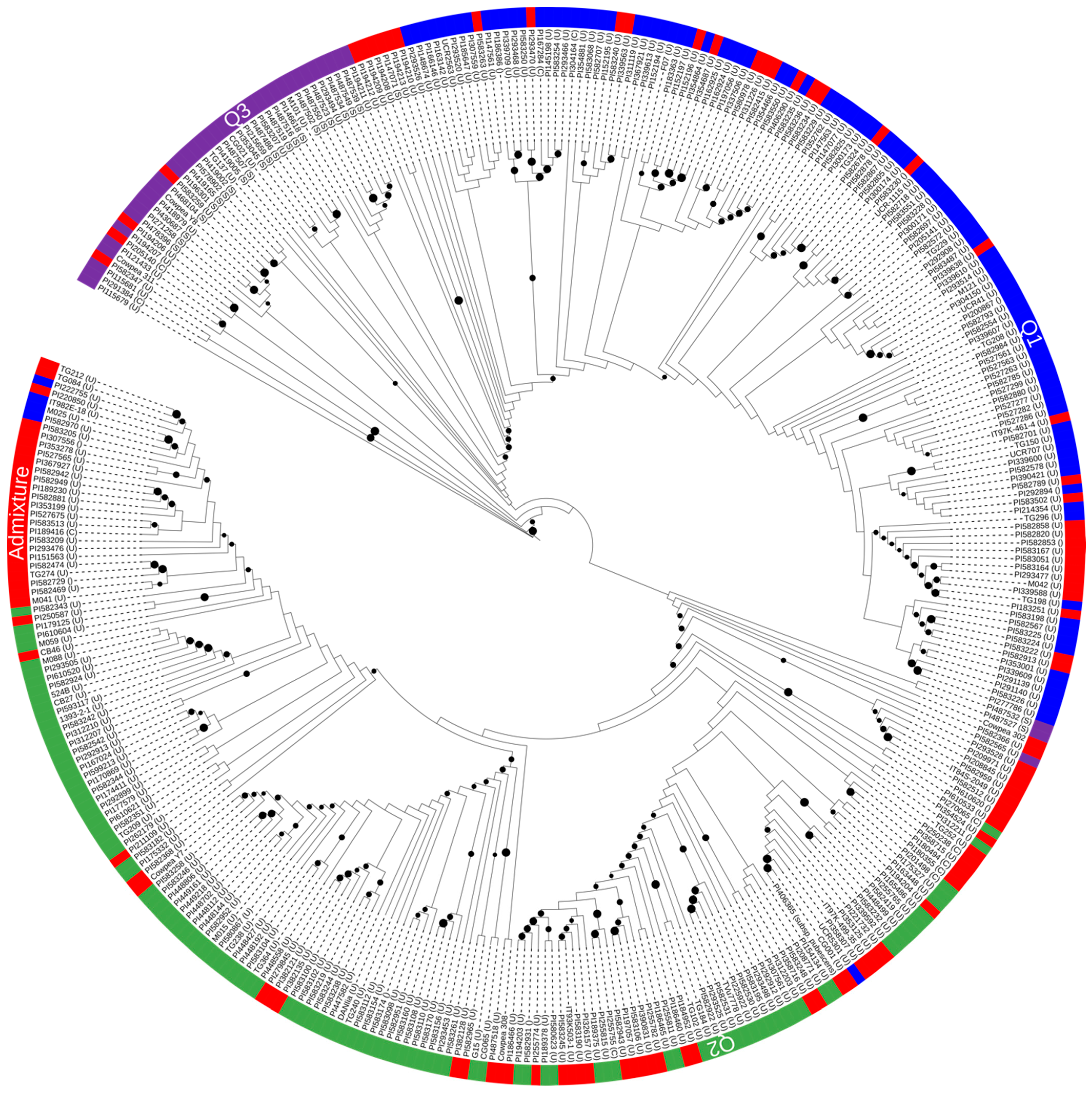

4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singh, R.J.; Jauhar, P.P. (Eds.) Genetic Resources, Chromosome Engineering, and Crop Improvement; CRC Press: Boca Raton, FL, USA, 2005; Volume 1, ISBN 978-0-203-48928-4. [Google Scholar]
- Sun, F.; Pan, K.; Olatunji, O.A.; Li, Z.; Chen, W.; Zhang, A.; Song, D.; Sun, X.; Huang, D.; Tan, X. Specific Legumes Allay Drought Effects on Soil Microbial Food Web Activities of the Focal Species in Agroecosystem. Plant Soil. 2019, 437, 455–471. [Google Scholar] [CrossRef]
- Horn, L.N.; Nghituwamata, S.N.; Isabella, U. Cowpea Production Challenges and Contribution to Livelihood in Sub-Saharan Region. Agric. Sci. 2022, 13, 25–32. [Google Scholar] [CrossRef]
- Boukar, O.; Belko, N.; Chamarthi, S.; Togola, A.; Batieno, J.; Owusu, E.; Haruna, M.; Diallo, S.; Umar, M.L.; Olufajo, O.; et al. Cowpea (Vigna unguiculata): Genetics, Genomics and Breeding. Plant Breed. 2019, 138, 415–424. [Google Scholar] [CrossRef]
- Fatokun, C.; Girma, G.; Abberton, M.; Gedil, M.; Unachukwu, N.; Oyatomi, O.; Yusuf, M.; Rabbi, I.; Boukar, O. Genetic Diversity and Population Structure of a Mini-Core Subset from the World Cowpea (Vigna unguiculata (L.) Walp.) Germplasm Collection. Sci. Rep. 2018, 8, 16035. [Google Scholar] [CrossRef]
- Zafeiriou, I.; Sakellariou, M.; Mylona, P.V. Seed Phenotyping and Genetic Diversity Assessment of Cowpea (Vigna unguiculata) Germplasm Collection. Agronomy 2023, 13, 274. [Google Scholar] [CrossRef]
- Close, T.; Oyatomi, O.; Guo, Y.-N.; Paliwal, R.; Muñoz-Amatriaín, M.; Roberts, P.; Abberton, M.; Marimagne, T.; Boukar, O.; Fatokun, C.; et al. SNP Genotypes of the International Institute of Tropical Agriculture Cowpea Core. 2023. Available online: https://datadryad.org/stash/datase (accessed on 13 February 2024).
- Liang, Q.; Muñoz-Amatriaín, M.; Shu, S.; Lo, S.; Wu, X.; Carlson, J.W.; Davidson, P.; Goodstein, D.M.; Phillips, J.; Janis, N.M.; et al. A View of the Pan-genome of Domesticated Cowpea (Vigna unguiculata [L.] Walp.). Plant Genome 2023, e20319. [Google Scholar] [CrossRef] [PubMed]
- Fatokun, C.A.; Menancio-Hautea, D.I.; Danesh, D.; Young, N.D. Evidence for Orthologous Seed Weight Genes in Cowpea and Mung Bean Based on RFLP Mapping. Genetics 1992, 132, 841–846. [Google Scholar] [CrossRef]
- Boukar, O.; Kong, L.; Singh, B.B.; Murdock, L.; Ohm, H.W. AFLP and AFLP-Derived SCAR Markers Associated with Striga Gesnerioides Resistance in Cowpea. Crop Sci. 2004, 44, 1259–1264. [Google Scholar] [CrossRef]
- Fang, J.; Chao, C.-C.T.; Roberts, P.A.; Ehlers, J.D. Genetic Diversity of Cowpea [Vigna unguiculata (L.) Walp.] in Four West African and USA Breeding Programs as Determined by AFLP Analysis. Genet. Resour. Crop Evol. 2007, 54, 1197–1209. [Google Scholar] [CrossRef]
- Diouf, D.; Hilu, K.W. Microsatellites and RAPD Markers to Study Genetic Relationships Among Cowpea Breeding Lines and Local Varieties in Senegal. Genet. Resour. Crop Evol. 2005, 52, 1057–1067. [Google Scholar] [CrossRef]
- Ba, F.S.; Pasquet, R.S.; Gepts, P. Genetic Diversity in Cowpea [Vigna unguiculata (L.) Walp.] as Revealed by RAPD Markers. Genet. Resour. Crop Evol. 2004, 51, 539–550. [Google Scholar] [CrossRef]
- Zhang, W.T.; Yang, J.K.; Yuan, T.Y.; Zhou, J.C. Genetic Diversity and Phylogeny of Indigenous Rhizobia from Cowpea [Vigna unguiculata (L.) Walp.]. Biol. Fertil. Soils 2007, 44, 201–210. [Google Scholar] [CrossRef]
- Dagnon, Y.D.; Palanga, K.K.; Bammite, D.; Bodian, A.; Akabassi, G.C.; Foncéka, D.; Tozo, K. Genetic Diversity and Population Structure of Cowpea [Vigna unguiculata (L.) Walp.] Accessions from Togo Using SSR Markers. PLoS ONE 2022, 17, e0252362. [Google Scholar] [CrossRef] [PubMed]
- Gajera, H.P.; Domadiya, R.K.; Patel, S.V.; Golakiya, B.A. Appraisal of RAPD and ISSR Markers for Genetic Diversity Analysis among Cowpea (Vigna unguiculata L.) Genotypes. J. Crop Sci. Biotechnol. 2014, 17, 79–88. [Google Scholar] [CrossRef]
- Egbadzor, K.F.; Ofori, K.; Yeboah, M.; Aboagye, L.M.; Opoku-Agyeman, M.O.; Danquah, E.Y.; Offei, S.K. Diversity in 113 Cowpea [Vigna unguiculata (L.) Walp] Accessions Assessed with 458 SNP Markers. SpringerPlus 2014, 3, 541. [Google Scholar] [CrossRef]
- Seo, E.; Kim, K.; Jun, T.-H.; Choi, J.; Kim, S.-H.; Muñoz-Amatriaín, M.; Sun, H.; Ha, B.-K. Population Structure and Genetic Diversity in Korean Cowpea Germplasm Based on SNP Markers. Plants 2020, 9, 1190. [Google Scholar] [CrossRef]
- Kalia, R.K.; Rai, M.K.; Kalia, S.; Singh, R.; Dhawan, A.K. Microsatellite Markers: An Overview of the Recent Progress in Plants. Euphytica 2011, 177, 309–334. [Google Scholar] [CrossRef]
- Mammadov, J.; Aggarwal, R.; Buyyarapu, R.; Kumpatla, S. SNP Markers and Their Impact on Plant Breeding. Int. J. Plant Genom. 2012, 2012, 728398. [Google Scholar] [CrossRef]
- Huynh, B.; Close, T.J.; Roberts, P.A.; Hu, Z.; Wanamaker, S.; Lucas, M.R.; Chiulele, R.; Cissé, N.; David, A.; Hearne, S.; et al. Gene Pools and the Genetic Architecture of Domesticated Cowpea. Plant Genome 2013, 6. [Google Scholar] [CrossRef]
- Xiong, H.; Shi, A.; Mou, B.; Qin, J.; Motes, D.; Lu, W.; Ma, J.; Weng, Y.; Yang, W.; Wu, D. Genetic Diversity and Population Structure of Cowpea (Vigna unguiculata L. Walp). PLoS ONE 2016, 11, e0160941. [Google Scholar] [CrossRef]
- He, C.; Holme, J.; Anthony, J. SNP Genotyping: The KASP Assay. In Crop Breeding; Fleury, D., Whitford, R., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2014; Volume 1145, pp. 75–86. ISBN 978-1-4939-0445-7. [Google Scholar]
- Saxena, R.K.; Varma Penmetsa, R.; Upadhyaya, H.D.; Kumar, A.; Carrasquilla-Garcia, N.; Schlueter, J.A.; Farmer, A.; Whaley, A.M.; Sarma, B.K.; May, G.D.; et al. Large-Scale Development of Cost-Effective Single-Nucleotide Polymorphism Marker Assays for Genetic Mapping in Pigeonpea and Comparative Mapping in Legumes. DNA Res. 2012, 19, 449–461. [Google Scholar] [CrossRef]
- Steele, K.A.; Quinton-Tulloch, M.J.; Amgai, R.B.; Dhakal, R.; Khatiwada, S.P.; Vyas, D.; Heine, M.; Witcombe, J.R. Accelerating Public Sector Rice Breeding with High-Density KASP Markers Derived from Whole Genome Sequencing of Indica Rice. Mol. Breeding 2018, 38, 38. [Google Scholar] [CrossRef]
- Chandra, S.; Singh, D.; Pathak, J.; Kumari, S.; Kumar, M.; Poddar, R.; Balyan, H.S.; Prabhu, K.V.; Gupta, P.K.; Mukhopadhyay, K. SNP Discovery from Next-Generation Transcriptome Sequencing Data and Their Validation Using KASP Assay in Wheat (Triticum aestivum L.). Mol. Breed. 2017, 37, 92. [Google Scholar] [CrossRef]
- Cheng, P.; Gedling, C.R.; Patil, G.; Vuong, T.D.; Shannon, J.G.; Dorrance, A.E.; Nguyen, H.T. Genetic Mapping and Haplotype Analysis of a Locus for Quantitative Resistance to Fusarium Graminearum in Soybean Accession PI 567516C. Theor. Appl. Genet. 2017, 130, 999–1010. [Google Scholar] [CrossRef]
- Ertiro, B.T.; Ogugo, V.; Worku, M.; Das, B.; Olsen, M.; Labuschagne, M.; Semagn, K. Comparison of Kompetitive Allele Specific PCR (KASP) and Genotyping by Sequencing (GBS) for Quality Control Analysis in Maize. BMC Genomics 2015, 16, 908. [Google Scholar] [CrossRef]
- Ongom, P.O.; Fatokun, C.; Togola, A.; Salvo, S.; Oyebode, O.G.; Ahmad, M.S.; Jockson, I.D.; Bala, G.; Boukar, O. Molecular Fingerprinting and Hybridity Authentication in Cowpea Using Single Nucleotide Polymorphism Based Kompetitive Allele-Specific PCR Assay. Front. Plant Sci. 2021, 12, 734117. [Google Scholar] [CrossRef]
- Wu, X.; Wang, B.; Wu, S.; Li, S.; Zhang, Y.; Wang, Y.; Li, Y.; Wang, J.; Wu, X.; Lu, Z.; et al. Development of a Core Set of Single Nucleotide Polymorphism Markers for Genetic Diversity Analysis and Cultivar Fingerprinting in Cowpea. Legume Sci. 2021, 3, e93. [Google Scholar] [CrossRef]
- Hazra, P.; Chattopadhaya, A.; Dasgupta, T.; Kar, N.; Das, P.; Som, M. Breeding Strategy for Improving Plant Type, Pod Yield and Protein Content in Vegetable Cowpea (Vigna unguiculata). In Proceedings of the I International Conference on Indigenous Vegetables and Legumes, Prospectus for Fighting Poverty, Hunger and Malnutrition, Hyderabad, India, 12–15 December 2006; pp. 275–280. [Google Scholar]
- Wu, X.; Cortés, A.J.; Blair, M.W. Genetic Differentiation of Grain, Fodder and Pod Vegetable Type Cowpeas (Vigna unguiculata L.) Identified through Single Nucleotide Polymorphisms from Genotyping-by-Sequencing. Mol. Hortic. 2022, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Boukar, O.; Abberton, M.; Oyatomi, O.; Togola, A.; Tripathi, L.; Fatokun, C. Introgression Breeding in Cowpea [Vigna unguiculata (L.) Walp.]. Front. Plant Sci. 2020, 11, 567425. [Google Scholar] [CrossRef] [PubMed]
- Vega-Vela, N.E.; Sánchez, M.I.C. Isolation of High-Quality DNA in 16 Aromatic and Medicinal Colombian Species Using Silica-Based Extraction Columns. Agron. Colomb. 2011, 29, 349–357. [Google Scholar]
- Muchero, W.; Diop, N.N.; Bhat, P.R.; Fenton, R.D.; Wanamaker, S.; Pottorff, M.; Hearne, S.; Cisse, N.; Fatokun, C.; Ehlers, J.D.; et al. A Consensus Genetic Map of Cowpea [Vigna unguiculata (L.) Walp.] and Synteny Based on EST-Derived SNPs. Proc. Natl. Acad. Sci. USA 2009, 106, 18159–18164. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Muse, S.V. PowerMarker: An Integrated Analysis Environment for Genetic Marker Analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Lischer, H.E.L. Arlequin Suite Ver 3.5: A New Series of Programs to Perform Population Genetics Analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the Number of Clusters of Individuals Using the Software Structure: A Simulation Study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A Website and Program for Visualizing STRUCTURE Output and Implementing the Evanno Method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A Cluster Matching and Permutation Program for Dealing with Label Switching and Multimodality in Analysis of Population Structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef]
- Rosenberg, N.A. Distruct: A Program for the Graphical Display of Population Structure: PROGRAM NOTE. Mol. Ecol. Notes 2003, 4, 137–138. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Fatokun, C.; Singh, B.B. Interspecific Hybridization between Vigna pubescens and V. unquiculata (L.) Walp through Embryo Rescue. Plant Cell Tissue Organ Cult. 1987, 9, 229–233. [Google Scholar] [CrossRef]
- Ogunkanmi, L.; Fakoya, A.; Adekoya, K. Breeding for Long Pod Trait in Cowpea (Vigna unguiculata L. Walp). J. Sci. Res. Dev. 2013, 14, 9–14. [Google Scholar]
- Muchero, W.; Roberts, P.A.; Diop, N.N.; Drabo, I.; Cisse, N.; Close, T.J.; Muranaka, S.; Boukar, O.; Ehlers, J.D. Genetic Architecture of Delayed Senescence, Biomass, and Grain Yield under Drought Stress in Cowpea. PLoS ONE 2013, 8, e70041. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Islam, A.S.M.F.; Limpot, N.; Mackasmiel, L.; Mierzwa, J.; Cortés, A.J.; Blair, M.W. Genome-Wide SNP Identification and Association Mapping for Seed Mineral Concentration in Mung Bean (Vigna radiata L.). Front. Genet. 2020, 11, 656. [Google Scholar] [CrossRef] [PubMed]
- Ravelombola, W.; Qin, J.; Shi, A.; Lu, W.; Weng, Y.; Xiong, H.; Yang, W.; Bhattarai, G.; Mahamane, S.; Payne, W.A.; et al. Association Mapping Revealed SNP Markers for Adaptation to Low Phosphorus Conditions and Rock Phosphate Response in USDA Cowpea (Vigna unguiculata (L.) Walp.) Germplasm. Euphytica 2017, 213, 183. [Google Scholar] [CrossRef]
- Xu, P.; Wu, X.; Wang, B.; Liu, Y.; Ehlers, J.D.; Close, T.J.; Roberts, P.A.; Diop, N.-N.; Qin, D.; Hu, T.; et al. A SNP and SSR Based Genetic Map of Asparagus Bean (Vigna. unguiculata Ssp. Sesquipedialis) and Comparison with the Broader Species. PLoS ONE 2011, 6, e15952. [Google Scholar] [CrossRef]
- Herniter, I.A.; Muñoz-Amatriaín, M.; Close, T.J. Genetic, Textual, and Archeological Evidence of the Historical Global Spread of Cowpea (Vigna unguiculata [L.] Walp.). Legume Sci. 2020, 2, e57. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| SNP_ID | p | q | MAF | He | Ho | GD | PIC | Missing | X2 | pval |
|---|---|---|---|---|---|---|---|---|---|---|
| 9815_2051 | 0.3 | 0.7 | 0.3 | 0.42 | 0.57 | 0.42 | 0.33 | 0.01 | 46.34 | 0.00 |
| 9400_502 | 0.41 | 0.59 | 0.41 | 0.48 | 0.76 | 0.48 | 0.37 | 0 | 114.58 | 0.00 |
| 9263_376 | 0.17 | 0.83 | 0.17 | 0.29 | 0.32 | 0.29 | 0.25 | 0.01 | 4.85 | 0.03 |
| 8677_1492 | 0.1 | 0.9 | 0.1 | 0.17 | 0.17 | 0.17 | 0.16 | 0.02 | 0.14 | 0.71 |
| 8645_1960 | 0.31 | 0.69 | 0.31 | 0.43 | 0.58 | 0.43 | 0.34 | 0.01 | 47.58 | 0.00 |
| 8276_369 | 0.44 | 0.56 | 0.44 | 0.49 | 0.83 | 0.49 | 0.37 | 0 | 172.67 | 0.00 |
| 8408_1086 | 0.42 | 0.58 | 0.42 | 0.49 | 0.8 | 0.49 | 0.37 | 0 | 150.60 | 0.00 |
| 7953_664 | 0.29 | 0.71 | 0.29 | 0.41 | 0.55 | 0.41 | 0.33 | 0.1 | 38.37 | 0.00 |
| 8044_1006 | 0.27 | 0.73 | 0.27 | 0.39 | 0.49 | 0.39 | 0.32 | 0.01 | 23.99 | 0.00 |
| 8118_1675 | 0.79 | 0.21 | 0.21 | 0.34 | 0.46 | 0.34 | 0.28 | 0.02 | 32.84 | 0.00 |
| 8119_299 | 0.69 | 0.31 | 0.31 | 0.43 | 0.65 | 0.43 | 0.34 | 0.01 | 89.75 | 0.00 |
| 7383_1042 | 0.38 | 0.62 | 0.38 | 0.47 | 0.7 | 0.47 | 0.36 | 0.01 | 89.01 | 0.00 |
| 7392_569 | 0.38 | 0.62 | 0.38 | 0.47 | 0.7 | 0.47 | 0.36 | 0 | 90.41 | 0.00 |
| 7436_791 | 0.43 | 0.57 | 0.43 | 0.49 | 0.8 | 0.49 | 0.37 | 0 | 151.83 | 0.00 |
| 7627_617 | 0.43 | 0.57 | 0.43 | 0.49 | 0.81 | 0.49 | 0.37 | 0.01 | 151.77 | 0.00 |
| 7281_555 | 0.41 | 0.59 | 0.41 | 0.48 | 0.78 | 0.48 | 0.37 | 0.03 | 127.76 | 0.00 |
| 7034_542 | 0.08 | 0.92 | 0.08 | 0.15 | 0.16 | 0.15 | 0.14 | 0.01 | 0.15 | 0.70 |
| 7087_1100 | 0.15 | 0.85 | 0.15 | 0.26 | 0.27 | 0.26 | 0.22 | 0.01 | 1.83 | 0.18 |
| 5993_278 | 0.09 | 0.91 | 0.09 | 0.16 | 0.18 | 0.16 | 0.15 | 0.01 | 3.49 | 0.06 |
| 6796_936 | 0.08 | 0.92 | 0.08 | 0.15 | 0.16 | 0.15 | 0.14 | 0.01 | 2.83 | 0.09 |
| 699_429 | 0.92 | 0.08 | 0.08 | 0.14 | 0.15 | 0.14 | 0.13 | 0.05 | 0.67 | 0.41 |
| 673_473 | 0.57 | 0.43 | 0.43 | 0.49 | 0.84 | 0.49 | 0.37 | 0.03 | 183.69 | 0.00 |
| 5295_403 | 0.06 | 0.94 | 0.06 | 0.11 | 0.12 | 0.11 | 0.1 | 0.09 | 1.28 | 0.26 |
| 4238_636 | 0.43 | 0.57 | 0.43 | 0.49 | 0.82 | 0.49 | 0.37 | 0 | 165.95 | 0.00 |
| 4533_841 | 0.14 | 0.86 | 0.14 | 0.25 | 0.28 | 0.25 | 0.22 | 0.07 | 7.18 | 0.01 |
| 4558_472 | 0.24 | 0.76 | 0.24 | 0.37 | 0.47 | 0.37 | 0.3 | 0.04 | 27.25 | 0.00 |
| 5026_672 | 0.15 | 0.85 | 0.15 | 0.26 | 0.3 | 0.26 | 0.23 | 0.03 | 6.91 | 0.01 |
| 5074_629 | 0.36 | 0.64 | 0.36 | 0.46 | 0.68 | 0.46 | 0.36 | 0.18 | 69.84 | 0.00 |
| 5268_412 | 0.07 | 0.93 | 0.07 | 0.14 | 0.13 | 0.14 | 0.13 | 0.01 | 0.00 | 0.96 |
| 311_1536 | 0.11 | 0.89 | 0.11 | 0.19 | 0.2 | 0.19 | 0.17 | 0.09 | 1.02 | 0.31 |
| 313_221 | 0.08 | 0.92 | 0.08 | 0.16 | 0.16 | 0.16 | 0.14 | 0.02 | 0.16 | 0.69 |
| 3571_469 | 0.13 | 0.87 | 0.13 | 0.22 | 0.25 | 0.22 | 0.2 | 0.02 | 3.39 | 0.07 |
| 3701_796 | 0.15 | 0.85 | 0.15 | 0.25 | 0.28 | 0.25 | 0.22 | 0.12 | 3.13 | 0.08 |
| 3900_562 | 0.33 | 0.67 | 0.33 | 0.44 | 0.63 | 0.44 | 0.34 | 0.01 | 67.56 | 0.00 |
| 3939_496 | 0.16 | 0.84 | 0.16 | 0.26 | 0.3 | 0.26 | 0.23 | 0.04 | 5.25 | 0.02 |
| 4131_472 | 0.62 | 0.38 | 0.38 | 0.47 | 0.73 | 0.47 | 0.36 | 0.08 | 107.52 | 0.00 |
| 17107_475 | 0.28 | 0.72 | 0.28 | 0.41 | 0.54 | 0.41 | 0.32 | 0.18 | 33.06 | 0.00 |
| 2680_1095 | 0.2 | 0.8 | 0.2 | 0.32 | 0.39 | 0.32 | 0.27 | 0.07 | 13.53 | 0.00 |
| 2682_1169 | 0.21 | 0.79 | 0.21 | 0.33 | 0.41 | 0.33 | 0.27 | 0.05 | 20.91 | 0.00 |
| 2728_121 | 0.27 | 0.73 | 0.27 | 0.4 | 0.53 | 0.4 | 0.32 | 0.15 | 33.58 | 0.00 |
| 2829_305 | 0.25 | 0.75 | 0.25 | 0.38 | 0.48 | 0.38 | 0.31 | 0.09 | 24.62 | 0.00 |
| 14619_471 | 0.36 | 0.64 | 0.36 | 0.46 | 0.69 | 0.46 | 0.35 | 0.09 | 79.71 | 0.00 |
| 15183_436 | 0.24 | 0.76 | 0.24 | 0.36 | 0.47 | 0.36 | 0.3 | 0.14 | 24.62 | 0.00 |
| 15288_527 | 0.49 | 0.51 | 0.49 | 0.5 | 0.95 | 0.5 | 0.37 | 0.09 | 269.96 | 0.00 |
| 15534_890 | 0.28 | 0.72 | 0.28 | 0.4 | 0.54 | 0.4 | 0.32 | 0.07 | 37.32 | 0.00 |
| 15779_1173 | 0.34 | 0.66 | 0.34 | 0.45 | 0.66 | 0.45 | 0.35 | 0.04 | 77.40 | 0.00 |
| 16011_113 | 0.37 | 0.63 | 0.37 | 0.47 | 0.71 | 0.47 | 0.36 | 0.13 | 90.72 | 0.00 |
| 16043_314 | 0.23 | 0.77 | 0.23 | 0.36 | 0.45 | 0.36 | 0.29 | 0.05 | 22.39 | 0.00 |
| 13842_975 | 0.19 | 0.81 | 0.19 | 0.31 | 0.38 | 0.31 | 0.26 | 0.03 | 14.88 | 0.00 |
| 139_439 | 0.43 | 0.57 | 0.43 | 0.49 | 0.83 | 0.49 | 0.37 | 0.05 | 172.65 | 0.00 |
| 13987_174 | 0.08 | 0.92 | 0.08 | 0.15 | 0.16 | 0.15 | 0.14 | 0.05 | 0.11 | 0.74 |
| 14110_2536 | 0.38 | 0.62 | 0.38 | 0.47 | 0.72 | 0.47 | 0.36 | 0.05 | 102.41 | 0.00 |
| 1441_128 | 0.92 | 0.08 | 0.08 | 0.15 | 0.19 | 0.15 | 0.14 | 0.05 | 3.77 | 0.05 |
| 1202_1215 | 0.4 | 0.6 | 0.4 | 0.48 | 0.79 | 0.48 | 0.37 | 0.12 | 131.03 | 0.00 |
| 12041_453 | 0.31 | 0.69 | 0.31 | 0.42 | 0.59 | 0.42 | 0.33 | 0.01 | 54.81 | 0.00 |
| 1257_517 | 0.29 | 0.71 | 0.29 | 0.41 | 0.56 | 0.41 | 0.33 | 0.04 | 44.28 | 0.00 |
| 12793_473 | 0.3 | 0.7 | 0.3 | 0.42 | 0.59 | 0.42 | 0.33 | 0.17 | 46.41 | 0.00 |
| 12933_387 | 0.21 | 0.79 | 0.21 | 0.34 | 0.42 | 0.34 | 0.28 | 0.01 | 24.04 | 0.00 |
| 13034_542 | 0.14 | 0.86 | 0.14 | 0.24 | 0.26 | 0.24 | 0.21 | 0.07 | 2.45 | 0.12 |
| 13252_298 | 0.55 | 0.45 | 0.45 | 0.5 | 0.88 | 0.5 | 0.37 | 0 | 224.36 | 0.00 |
| 13786_529 | 0.29 | 0.71 | 0.29 | 0.41 | 0.54 | 0.41 | 0.33 | 0.03 | 36.26 | 0.00 |
| 11303_254 | 0.18 | 0.82 | 0.18 | 0.3 | 0.37 | 0.3 | 0.26 | 0.04 | 17.98 | 0.00 |
| 11470_272 | 0.35 | 0.65 | 0.35 | 0.45 | 0.68 | 0.45 | 0.35 | 0.06 | 85.18 | 0.00 |
| 11515_820 | 0.08 | 0.92 | 0.08 | 0.14 | 0.15 | 0.14 | 0.13 | 0.07 | 0.57 | 0.45 |
| 11585_1881 | 0.24 | 0.76 | 0.24 | 0.37 | 0.47 | 0.37 | 0.3 | 0.02 | 27.39 | 0.00 |
| 11683_874 | 0.14 | 0.86 | 0.14 | 0.25 | 0.28 | 0.25 | 0.22 | 0.05 | 7.37 | 0.01 |
| 11737_146 | 0.4 | 0.6 | 0.4 | 0.48 | 0.76 | 0.48 | 0.36 | 0.03 | 121.93 | 0.00 |
| 11783_1366 | 0.38 | 0.62 | 0.38 | 0.47 | 0.75 | 0.47 | 0.36 | 0.09 | 115.93 | 0.00 |
| 10466_465 | 0.4 | 0.6 | 0.4 | 0.48 | 0.76 | 0.48 | 0.36 | 0.09 | 118.25 | 0.00 |
| 10738_1400 | 0.27 | 0.73 | 0.27 | 0.39 | 0.51 | 0.39 | 0.31 | 0.07 | 31.52 | 0.00 |
| 11266_52 | 0.42 | 0.58 | 0.42 | 0.49 | 0.79 | 0.49 | 0.37 | 0.11 | 126.74 | 0.00 |
| 894_153 | 0.34 | 0.66 | 0.34 | 0.45 | 0.64 | 0.45 | 0.35 | 0.15 | 54.63 | 0.00 |
| Regions | ||||
| Source of Variation | Sum of Squares | Variance Components | Percentage Variation | FST: 0.01880 |
| Among Groups | 265.90 | 0.24 | 1.88 | |
| Within Groups | 8419.67 | 12.50 | 98.12 | |
| Total | 8685.57 | 12.74 | ||
| Sub-species | ||||
| Source of Variation | Sum of Squares | Variance Components | Percentage Variation | FST: 0.02220 |
| Among Groups | 102.43 | 0.29 | 2.22 | |
| Within Groups | 8164.87 | 12.61 | 97.78 | |
| Total | 8267.30 | 12.90 |
| Regions | Number of Accessions in Each Cluster | Percentage of Accessions in Each Cluster | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Q1 | Q2 | Q3 | Admixture | Total | Q1 | Q2 | Q3 | Admixture | Total | |
| Europe | 0 | 8 | 0 | 2 | 10 | 0 | 2.2 | 0 | 0.6 | 2.8 |
| East Asia | 0 | 0 | 18 | 4 | 22 | 0 | 0 | 5 | 1.1 | 6.1 |
| Oceania | 3 | 0 | 0 | 1 | 4 | 0.8 | 0 | 0 | 0.3 | 1.1 |
| Middle East | 3 | 5 | 0 | 2 | 10 | 0.8 | 1.4 | 0 | 0.6 | 2.8 |
| Latin America | 17 | 10 | 5 | 13 | 45 | 4.7 | 2.8 | 1.4 | 3.6 | 12.5 |
| West and Central Africa | 14 | 47 | 1 | 33 | 95 | 3.9 | 13 | 0.3 | 9.1 | 26.3 |
| East and Southern Africa | 34 | 5 | 0 | 20 | 59 | 9.4 | 1.4 | 0 | 5.5 | 16.3 |
| West Asia | 11 | 6 | 7 | 16 | 40 | 3 | 1.7 | 1.9 | 4.4 | 11.1 |
| North America | 22 | 23 | 8 | 23 | 76 | 6.1 | 6.4 | 2.2 | 6.4 | 21.1 |
| Total | 104 | 104 | 39 | 114 | 361 | 28.8 | 28.8 | 10.8 | 31.6 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Potts, J.; Michael, V.N.; Meru, G.; Wu, X.; Blair, M.W. Dissecting the Genetic Diversity of USDA Cowpea Germplasm Collection Using Kompetitive Allele Specific PCR-Single Nucleotide Polymorphism Markers. Genes 2024, 15, 362. https://doi.org/10.3390/genes15030362
Potts J, Michael VN, Meru G, Wu X, Blair MW. Dissecting the Genetic Diversity of USDA Cowpea Germplasm Collection Using Kompetitive Allele Specific PCR-Single Nucleotide Polymorphism Markers. Genes. 2024; 15(3):362. https://doi.org/10.3390/genes15030362
Chicago/Turabian StylePotts, Jesse, Vincent N. Michael, Geoffrey Meru, Xingbo Wu, and Matthew W. Blair. 2024. "Dissecting the Genetic Diversity of USDA Cowpea Germplasm Collection Using Kompetitive Allele Specific PCR-Single Nucleotide Polymorphism Markers" Genes 15, no. 3: 362. https://doi.org/10.3390/genes15030362