
Principles in the Management of Glioblastoma
by
 ,
,
Domingos Roda
1,†,
Pedro Veiga
2,†,
Joana Barbosa Melo
2,3,4,
Isabel Marques Carreira
2,3,4,* and
Ilda Patrícia Ribeiro
2,3,4 1
Algarve Radiation Oncology Unit—Joaquim Chaves Saúde (JCS), 8000-316 Faro, Portugal
2
Institute of Cellular and Molecular Biology, Cytogenetics and Genomics Laboratory, Faculty of Medicine, University of Coimbra, 3000-548 Coimbra, Portugal
3
Coimbra Institute for Clinical and Biomedical Research (iCBR) and Center of Investigation on Environment Genetics and Oncobiology (CIMAGO), Faculty of Medicine, University of Coimbra, 3000-548 Coimbra, Portugal
4
Center for Innovative Biomedicine and Biotechnology (CIBB) and Clinical Academic Center of Coimbra (CACC), University of Coimbra, 3000-548 Coimbra, Portugal
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Genes 2024, 15(4), 501; https://doi.org/10.3390/genes15040501
Submission received: 15 March 2024
/
Revised: 11 April 2024
/
Accepted: 15 April 2024
/
Published: 17 April 2024
(This article belongs to the Section Human Genomics and Genetic Diseases)
Abstract
:Glioblastoma, the most aggressive and common malignant primary brain tumour, is characterized by infiltrative growth, abundant vascularization, and aggressive clinical evolution. Patients with glioblastoma often face poor prognoses, with a median survival of approximately 15 months. Technological progress and the subsequent improvement in understanding the pathophysiology of these tumours have not translated into significant achievements in therapies or survival outcomes for patients. Progress in molecular profiling has yielded new omics data for a more refined classification of glioblastoma. Several typical genetic and epigenetic alterations in glioblastoma include mutations in genes regulating receptor tyrosine kinase (RTK)/rat sarcoma (RAS)/phosphoinositide 3-kinase (PI3K), p53, and retinoblastoma protein (RB) signalling, as well as mutation of isocitrate dehydrogenase (IDH), methylation of O6-methylguanine-DNA methyltransferase (MGMT), amplification of epidermal growth factor receptor vIII, and codeletion of 1p/19q. Certain microRNAs, such as miR-10b and miR-21, have also been identified as prognostic biomarkers. Effective treatment options for glioblastoma are limited. Surgery, radiotherapy, and alkylating agent chemotherapy remain the primary pillars of treatment. Only promoter methylation of the gene MGMT predicts the benefit from alkylating chemotherapy with temozolomide and it guides the choice of first-line treatment in elderly patients. Several targeted strategies based on tumour-intrinsic dominant signalling pathways and antigenic tumour profiles are under investigation in clinical trials. This review explores the potential genetic and epigenetic biomarkers that could be deployed as analytical tools in the diagnosis and prognostication of glioblastoma. Recent clinical advancements in treating glioblastoma are also discussed, along with the potential of liquid biopsies to advance personalized medicine in the field of glioblastoma, highlighting the challenges and promises for the future.
1. Introduction
Glioblastoma originates from astrocytic glial cells [1] and is a higher-grade malignant glioma (grade 4) according to the World Health Organization (WHO) classification. This highly aggressive cancer has a poor prognosis, with a survival rate of only 15 months after diagnosis [2]. The overall 5-year relative survival rate is one of the lowest among all cancer types (4–5%), and in the last three decades, improvements in survival rates for glioblastoma patients have been very limited [2]. The current standard of care treatment includes surgical resection followed by radiotherapy and chemotherapy.
Glioblastoma is characterized by its highly invasive and therapy-resistant nature. Clinical trials testing new drugs have been on the rise, mostly those involving immunotherapy and targeted therapies [3]. Recently, glioblastoma has been classified into three subtypes based on transcriptome analysis of tumours: proneural, classical, and mesenchymal [4,5]. The mesenchymal subtype of glioblastoma is associated with the worst prognosis, highlighting the importance of these molecular data in advancing personalized treatment. However, translating the growing knowledge about glioblastoma biology into clinical practice and achieving significant improvements in therapies or patient outcomes remains challenging.
In this review, we have discussed recent advances in the management of glioblastoma, as well as the current challenges and future directions for research.
2. Epidemiology
Glioblastoma has a global incidence of less than 10 cases per 100,000 persons and its prevalence is even lower in the paediatric population, although this rate varies worldwide [6]. It accounts for approximately 16% of all central nervous system tumours and constitutes 54% of all gliomas [2].
Regarding age and gender, glioblastoma can occur at any age but tends to affect older adults. Its incidence increases with age, reaching a peak at 75–84 years, with the median age at diagnosis being 64 years old [2]. Glioblastoma is more common in males than in females [2,6]. In terms of race and ethnicity, there is a higher incidence of glioblastoma and poorest survival in Caucasians when compared to Asians and Black individuals [6,7]. While most glioblastomas occur without any apparent family history, there are certain genetic conditions that have been associated with an increased risk of developing primary brain tumours, such as Li–Fraumeni, Turcot, BRCA syndrome and neurofibromatosis type 1, among others [8].
Exogenous factors may also play a role in the origin of glioblastoma and other primary central nervous system tumours. Currently, several studies are attempting to establish an association between the development and progression of the disease and environmental factors, such as exposure to radiation and toxic elements, dietary habits, lifestyle, mobile phone use, or exposure to electromagnetic waves [9,10,11]. Certain studies have provided evidence that exposure to radiation and toxic agents, such as pesticides, can increase susceptibility to the development of this disease [9,12]. The correlation between disease development and factors such as lifestyle, mobile phone use and dietary habits remains inconclusive [13,14,15].
3. Classification of Glioblastoma
The fourth WHO classification of gliomas from 2016 is based on the degree of malignancy, as determined by histopathological criteria, in which four types of this neoplasm have been distinguished [16]:
- −
- Glioblastoma, isocitrate dehydrogenase (IDH) wildtype (90% of cases), developing de novo at about 60 years of age;
- −
- Glioblastoma, IDH-mutant (10% of cases), secondary glioblastoma that usually develops in younger patients with gliomas of higher differentiation (WHO grades I–III); it carries a significantly better prognosis than wildtype IDH [17];
- −
- Glioblastoma not otherwise specified (NOS), the IDH mutation status could not be determined due to a lack of histological or molecular material for testing;
- −
- Not-elsewhere-classified (NEC) glioblastoma, fourth category distinguished in recent years.
For NEC, the necessary decision to classify the tumour had been made, but the results did not allow assignment of the tumour to any of the above categories of the 2016 WHO classification. This may occur if there are differences among the clinical, histological, immunohistological and genetic characteristics of the tumour. It is also possible that there are unknown combinations of characteristics of glioblastoma subunits that have not yet been classified by the WHO division [6,16]. Their consecutive classifications are strongly related to histological features and clinical outcome, together with the relatively recent incorporation of molecular characteristics. The 5th edition of the World Health Organization (WHO) classification of tumours of the central nervous system (CNS), published at the end of 2021, has introduced new taxonomy and nomenclature of a great number of tumours including gliomas [18].
During the last decade, there has been a paradigm shift in CNS tumour diagnostics as advances in molecular genetics have revealed alterations in these tumours. In the 2016 WHO classification, molecular changes have been introduced into the diagnostic workup of some tumours and a comprehensive multi-layered diagnosis has been established that includes histopathological and molecular information [16,19]. The WHO 2021 version (WHO CNS5) considers molecular genetics and clinical relevance to a greater extent, so the latest version includes elements of both histopathology and molecular genetics, resulting in a somewhat hybrid taxonomy [20,21]. In this setting, the classifications include three genetic variables (TERT promoter mutation, EGFR gene amplification, +7/−10 chromosome copy number variations) as criteria to diagnose glioblastoma (grade 4), IDH-wildtype [20,22].
In this classification, to make the diagnosis of a glioblastoma the following are required [20]:
- adult patient
- diffuse astrocytic tumour
- IDH-wildtype
- and at least one of the following:
- ○
- necrosis
- ○
- microvascular proliferation
- ○
- TERT promoter mutation
- ○
- EGFR gene amplification
- ○
- combined gain of whole chromosome 7 and loss of chromosome 10 [+7/−10].
MGMT (O6-methylguanine-DNA methyltransferase) promoter methylation status is an essential part of molecular diagnostics for all high-grade gliomas (grade 3 and 4). MGMT promoter methylation is associated with better survival outcomes in patients with high-grade glioma and is a predictive factor for response to treatment with alkylating chemotherapy, as pointed out by major clinical guidelines in association with the classification [22].
4. Molecular Pathogenesis of Glioblastoma
4.1. Cell Signalling Pathways in Glioblastoma
The molecular pathogenesis of glioblastoma is complex and involves multiple alterations in various genes, cell signalling pathways, and genome regulatory elements. Several candidate genes in glioblastoma have been described that may influence the development and progression of the disease—CDKN2A, TP53, EGFR, PTEN, NF1, CDK4, RB1, IDH1, PIK3CA, and PIK3R1. These genes are implicated in various cell signalling pathways such as the PI3K/AKT/mTOR pathway, RAS/RAF/MAPK pathway, and p16INK4a/CDK4/Rb pathway [23].
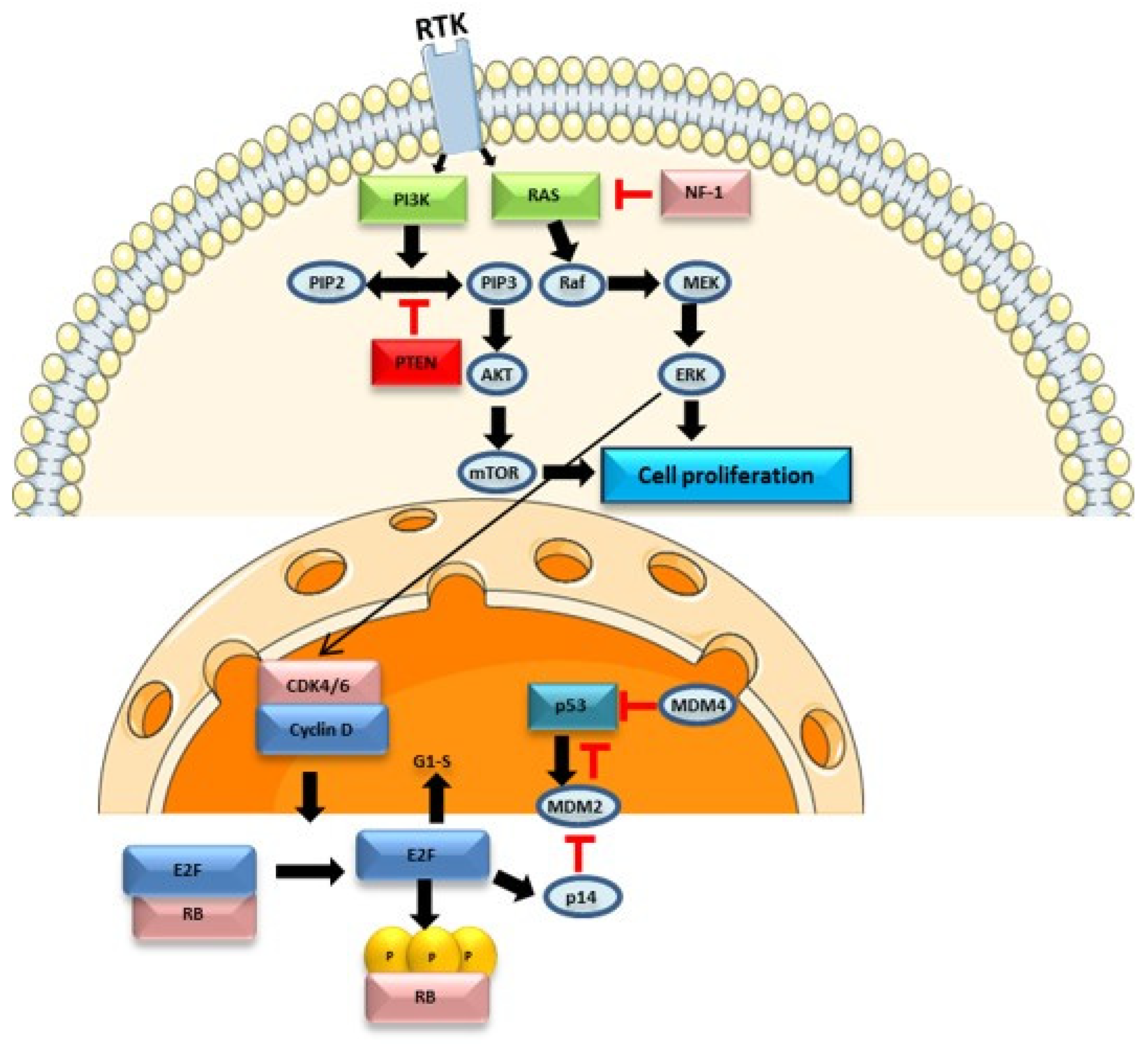
The PI3K/AKT/mTOR pathway involves receptor tyrosine kinases (RTKs) such as EGFR and a tumour suppressor protein, PTEN, which acts as an antagonist of PI3K, contributing to the downregulation of this pathway and inhibiting cell proliferation. In the absence of PTEN, PI3K phosphorylates phosphatidylinositol-4,5-bisphosphate (PIP2) into phosphatidylinositol-3,4,5-trisphosphate (PIP3), leading to the activation of the AKT protein. Both AKT and mammalian targets of rapamycin (mTOR) are serine/threonine-specific protein kinases (STKs) that play key roles in cell proliferation. This pathway is dysregulated in 80% of glioblastomas [24].
The RAS/RAF/MAPK pathway (Figure 1) is involved in the regulation of apoptosis, proliferation, cell differentiation, and development. In this signalling pathway, multiple isoforms of RAS (H-RAS, N-RAS, K-RAS) and RAF play distinct roles and exhibit oncogenic potential. RAS activation is controlled by binding to guanosine triphosphate (GTP) and its inhibition occurs by binding to guanosine diphosphate (GDP). RAS activation leads to RAF kinase activation, regulating downstream signalling pathways such as the mitogen-activated protein kinase (MAPK) pathway [25,26]. The BRAF protein is a serine/threonine kinase belonging to the RAF family, whose alteration is also associated with brain tumours. The BRAFV600E mutation is particularly relevant, leading to the activation of the RAS/RAF/MAPK pathway, promoting cell proliferation and survival, and inhibiting apoptosis. However, this missense alteration is rare in glioblastoma, detected in only 1–2% of cases [27,28]. This signalling pathway also interacts with other pathways, namely the PI3K/AKT/mTOR pathway and the p53 protein, contributing to the normal functioning of cell processes. Activation of this pathway can also lead to activation of hypoxia-inducible factor-1α (HIF-1α) and vascular endothelial growth factor receptor (VEGF), promoting tumorigenesis and angiogenesis [29]. Dysregulation of this pathway may contribute to increased cell proliferation, as seen in various glioblastoma cases with RAS overexpression. Additionally, growth factors such as EGFR, PDGFR, and other RTKs are often overexpressed, further contributing to glioblastoma pathogenesis. In this context, this signalling pathway may constitute a potential therapeutic target [25].
The Rb protein, encoded by the RB1 gene (retinoblastoma 1), controls the cell cycle between the transition of the G1 to S phase. In normal cells, cyclin D1 activates CDK4, phosphorylating the Rb protein, releasing the E2F1 transcription factor, and contributing to cell cycle progression (Figure 1). CDK4 activity is suppressed by INK4 family proteins. Approximately 78% of glioblastoma show alterations in the p16INK4a/CDK-4/Rb signalling pathway [24]. The RB1 gene is typically downregulated in glioblastoma, contributing to cell cycle progression.
Cell cycle checkpoints are crucial for activating DNA repair pathways in case of DNA damage. In certain cases, cyclin-dependent kinase inhibitors may be activated by the p53 protein, encoded by the TP53 gene, blocking the cell cycle and activating apoptosis pathways, preventing tumour development. However, 87% of glioblastoma patients have mutations in the TP53 gene, an important tumour suppressor gene [23,24].
Cell signalling pathways are interconnected, acting in different cellular processes. Therefore, alterations in genes related to one pathway may have implications in others, contributing to the complexity of glioblastoma pathogenesis [29]. Currently, there is also evidence that certain cell pathways are regulated by epigenetic mechanisms, which may contribute to disease progression and therapy resistance [30].
4.2. Epigenetic Mechanisms
Epigenetic modifications, such as DNA methylation, histone modifications, chromatin remodelling, and non-coding RNAs, play a significant role in the development and progression of cancer [30]. The analysis of epigenetic modifications in glioblastoma allows for better stratification of patients and can serve as therapeutic targets [31]. Promoter methylation of the MGMT gene, an important repair gene, appears to be associated with a favourable prognosis and a better response to temozolomide (TMZ) treatment [32].
Another relevant mechanism is histone modification, where changes in epigenetic regulatory genes, such as HDACs (histone deacetylases), histone demethylases and methyltransferases contribute to glioblastoma. Targeting histone acetylation as a therapeutic strategy has gained attention in the field of glioblastoma research. Histone deacetylase (HDAC) inhibitors have been investigated as potential treatments. By inhibiting HDACs, these drugs aim to increase histone acetylation and promote a more open chromatin structure, potentially reactivating tumour suppressor genes and inhibiting oncogenes [30].
Chromatin remodelling is closely associated with histone modifications. Genes involved in chromatin remodelling are frequently mutated or dysregulated in glioblastomas [33]. Ganguly et al., 2018, demonstrated that the protein BRG1, the catalytic subunit of the SWI/SNF chromatin remodelling complex, contributes to maintaining glioma-initiating cells (GlCs) in their stem-like state, promoting the development of the tumour and contributing to their heterogeneity. Furthermore, inhibition of BRG1 sensitized GlCs to chemotherapy and TMZ, suggesting that this protein could be a novel therapeutic target in glioblastoma [34]. However, chromatin remodelling complexes are complicated structures that still require more knowledge of their regulatory mechanisms and their implications in the genesis of glioblastoma.
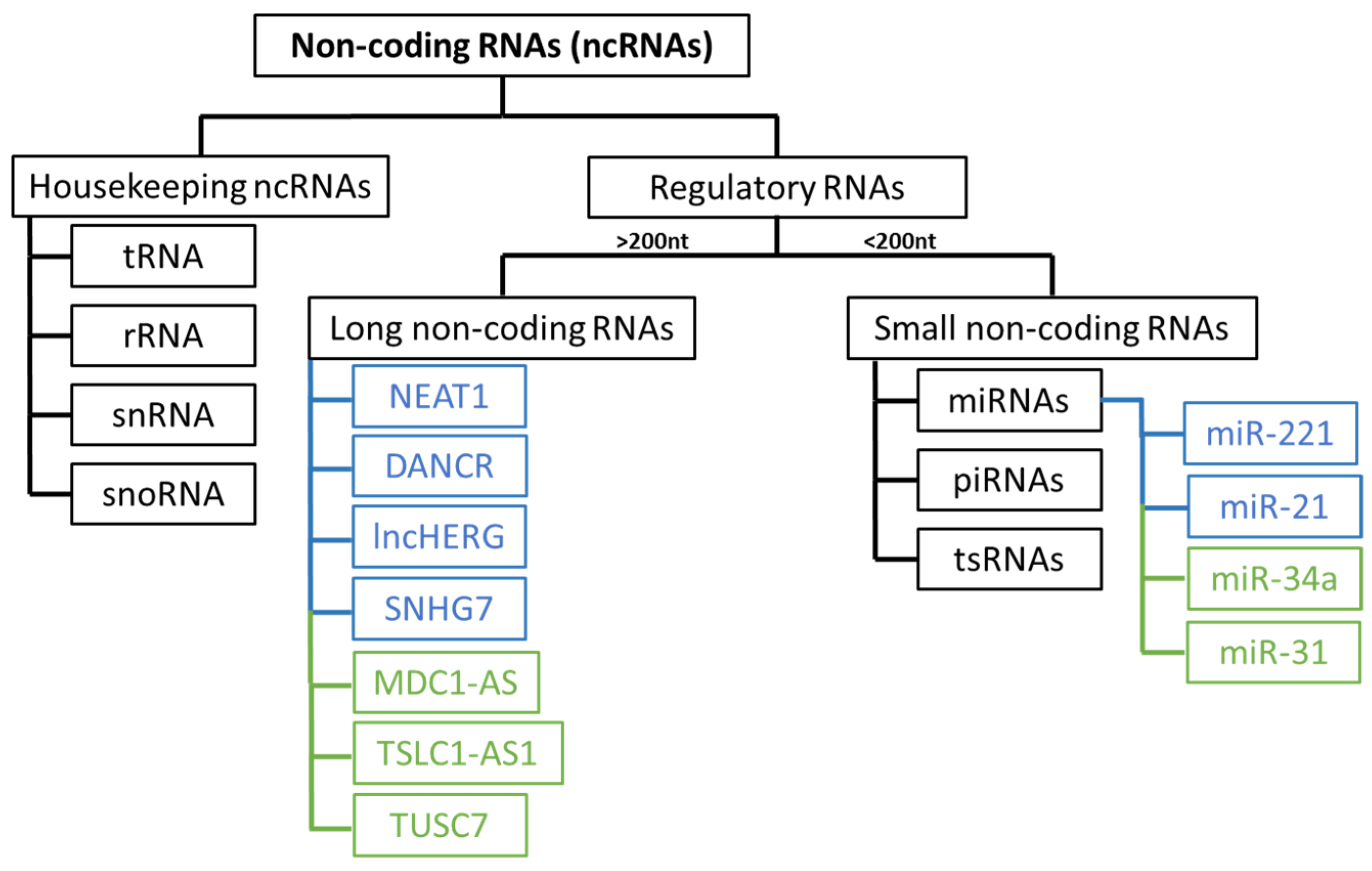
Recently, there has been a growing focus on understanding the role of non-coding RNAs (ncRNAs) in the development and progression of glioblastoma. These RNA molecules are divided into several groups (Figure 2) and play an important role in regulating gene expression [35,36].
Dysregulation of microRNAs is common in various types of cancer, including glioblastoma [37]. The loss of tumour suppressor miRNAs, which typically regulate mRNAs that may have oncogenic potential, such as miR-31, which inhibits CDKN2A/B and miR-34a, affecting EGFR protein levels, contributes to cell proliferation, apoptosis resistance, and subsequent disease progression [36]. The overexpression of other miRNAs, such as miR-221 and miR-21 (oncomiRs), facilitates cell growth and proliferation in glioblastoma by interacting with various genes involved in key signalling pathways [37].
Long non-coding RNAs (lncRNAs) are frequently dysregulated in various types of cancers, including glioblastoma, and this dysregulation can alter the activity of oncogene promoters, facilitating interactions with transcription factors and contributing to cell proliferation and disease progression [35,36,38]. Numerous oncogenic lncRNAs associated with glioblastoma, such as NEAT1, DANCR, lncHERG, SNHG7, MNX1-AS1, MCM3AP-AS1, and LINC01446, contribute to invasion, migration, chemoresistance, apoptosis resistance, and cell proliferation [39]. Certain lncRNAs have tumour suppressor functions and are typically downregulated, such as MDC1-AS, TSLC1-AS1, ADAMTS9-AS2, and TUSC7 [40].
From a therapeutic point of view, epigenetic modifications can be associated with treatment resistance and can contribute to tumour development. Therefore, targeting epigenetic regulators as a therapeutic strategy can offer advantages for the clinical management of the disease [38]. Moreover, understanding the interactions between the cell signalling pathways involved in glioblastoma’s pathogenesis and epigenetics offers promising insights into potential therapeutic targets and personalized treatments, providing better outcomes for the patients.
4.3. Multi-Omics Approach
An approach that involves the integration of genomics, transcriptomics, proteomics, and other omics data allows researchers to gain a comprehensive understanding of the underlying molecular mechanisms in glioblastoma development. This integration of diverse omics data facilitates the identification of genetic variants, gene expression changes, and protein alterations, contributing to the stratification of patients based on molecular alterations present in the tumour. Additionally, it potentially allows for the discovery of novel therapeutic targets [41]. With the technological progress in the molecular characterization of cancer, we may be a step closer to glioblastoma personalized medicine, offering individualized diagnosis and targeted therapies, which will contribute to better management of the disease.
5. Prognostic Biomarkers
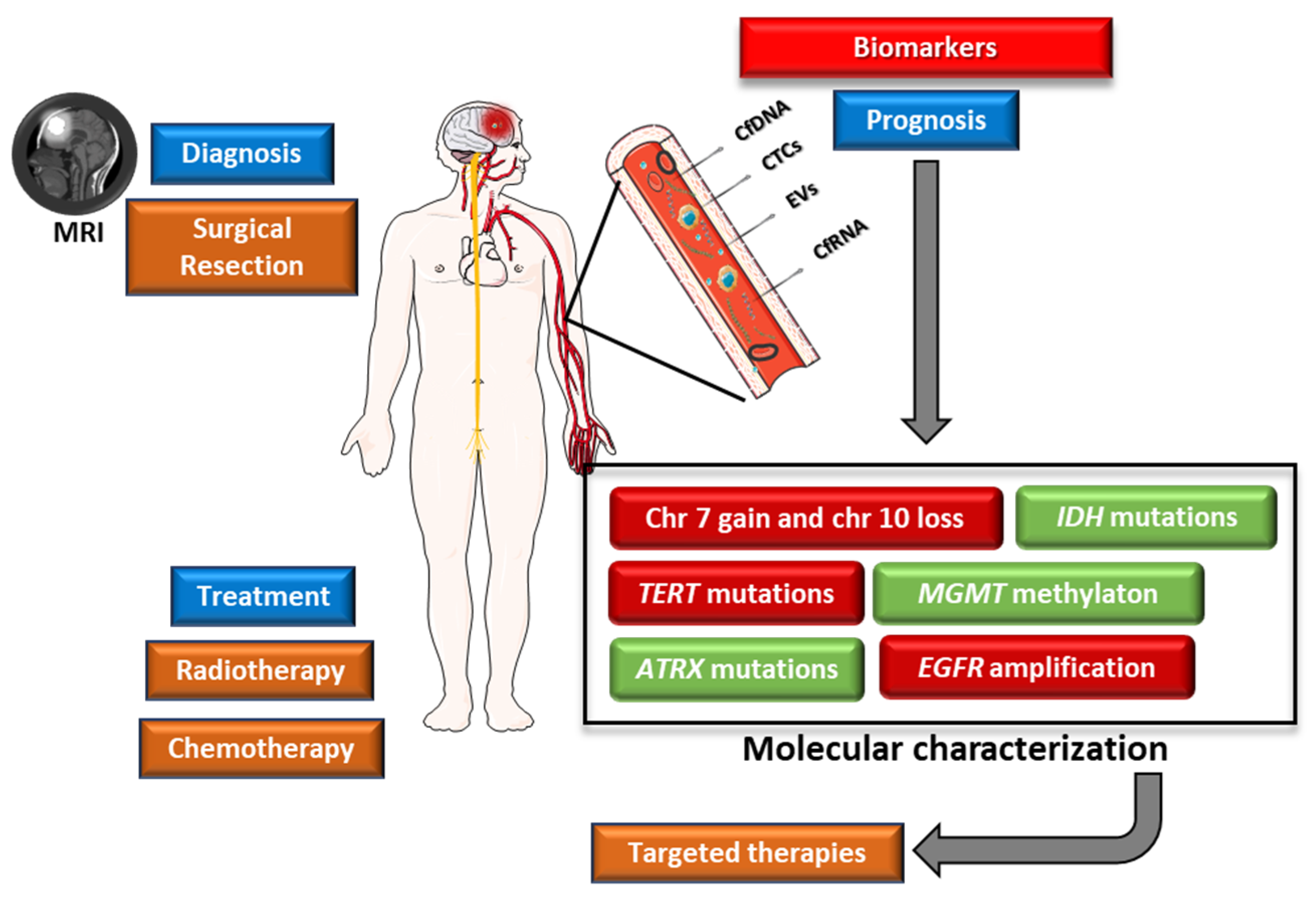
The invasive nature of glioblastoma is a critical factor in its aggressive growth and progression. It is therefore essential to identify recurring cytogenetic, genomic, and epigenomic changes that contribute to the development of this cancer. These changes have the potential to serve as genetic biomarkers, offering insights to improve patient care and treatment outcomes and therefore may influence the survival rates of this highly aggressive disease (Figure 3) [32]. Several genetic alterations are used as potential prognostic biomarkers in glioblastoma (Table 1).
5.1. IDH1 and IDH2 Mutational Status
Isocitrate dehydrogenase 1 (IDH1) and isocitrate dehydrogenase 2 (IDH2) are enzymes that play crucial roles in cell metabolism, particularly in the Krebs cycle. IDH1 is primarily located in the cytoplasm and peroxisomes of cells. Its main function is to catalyse the oxidative decarboxylation of isocitrate to produce α-ketoglutarate [42]. This enzyme also plays a role in maintaining the balance of NADP+ and NADPH, protecting cells against oxidative stress. IDH2 has a similar function as IDH1 [43].
Mutations in IDH1 or IDH2 are positive prognostic factors, improving the prognosis of the patients when compared to IDH-wildtype glioblastomas. IDH mutations lead to a gain-of-function mutation where the enzyme produces the oncometabolite D-2-hydroxyglutarate (D2HG) instead of the normal product, α-ketoglutarate (αKG) [44]. This metabolite inhibits αKG-dependent dioxygenases that are involved in the regulation of epigenetics and differentiation, which is thought to induce epigenetic dysfunction and therefore slows down tumour growth by inhibiting normal cell differentiation. Elevated levels of D2HG are also reported to induce DNA hypermethylation which may reduce tumour cell proliferation since hypermethylation is associated with gene silencing [32].
5.2. Alterations in ATRX (ATRX Chromatin Remodeler)
The ATRX gene provides instructions for making a protein involved in gene regulation by a process known as chromatin remodelling. ATRX is frequently mutated in astrocytomas IDH-mutant. In terms of prognosis, in patients diagnosed with glioblastoma IDH-wildtype, ATRX alterations were associated with favourable outcomes [32].
5.3. Alterations in TERT (Telomerase Reverse Transcriptase)
The TERT gene encodes a subunit of telomerase, which is responsible for maintaining telomeres. Mutations in the promoter of this gene are associated with an increase in telomerase activity, contributing to cell immortalization [45]. Arita et al., 2013 concluded that in 98% of the tumours analysed, a mutation in the promoter of the TERT gene was present concomitantly with 1p/19q loss and IDH1/2 mutations [46]. Mutations in the TERT gene are associated with a worse prognosis, particularly in IDH-wildtype gliomas [32].
5.4. Alterations in CDKN2A (Cyclin Dependent Kinase Inhibitor 2A)
The CDKN2A gene encodes a protein of the INK4 family, which acts as a cell cycle inhibitor, controlling cell cycle progression. The main function of this protein is to inhibit the action of cyclin-dependent kinases 4 and 6 (CDK4/6) by preventing the phosphorylation of retinoblastoma, blocking the entry of cells to the S phase [47]. Homozygous deletion of this gene is associated with a poor prognosis [32].
5.5. 1p/19q Codeletion
The 1p/19q codeletion represents a translocation t(1;19)(q10;p10), and in addition to its prognostic significance, it is also utilized for the classification of the tumour [20,32]. Regarding its value as a prognostic biomarker, this alteration is associated with a favourable prognosis and chemosensitivity in lower-grade gliomas [32]. Clark et al., state that the 1p/19q codeletion has no prognostic impact on gliomas classified as glioblastomas [48]. In addition, this alteration is uncommon in glioblastomas. However, Mizoguchi et al., studied a small group of glioblastoma patients with 1p/19q deletion who had a more favourable clinical outcome [49].
Data regarding the impact of this alteration in glioblastoma remains limited, and thus, further studies are necessary to understand the significance of this alteration in the management of this disease.
5.6. Chromosome 7 Gain and Chromosome 10 Loss
Gain of chromosome 7 and loss of chromosome 10 are common alterations in glioblastoma and are typically associated with a poor prognosis [32]. These alterations can involve the entire chromosome or be partial and are considered a molecular marker of IDH-wildtype glioblastomas [50]. Chromosome 10 monosomy is associated with the loss of an important tumour suppressor gene—PTEN (phosphatase and tensin homolog). This gene encodes a phosphatase protein that plays a crucial role in regulating cell growth and division by interacting with phosphatidylinositol 3,4,5-triphosphate (PIP3), thus blocking the PI3K/AKT pathway and inhibiting cell proliferation [51].
On the other hand, trisomy of chromosome 7 is associated with the amplification of oncogenes, particularly the EGFR (epidermal growth factor receptor) gene, contributing to increased cell proliferation and disease progression [32].
5.7. EGFR Mutations
The EGFR gene encodes for a transmembrane receptor with tyrosine kinase activity involved in various cell signalling pathways, such as PI3K/AKT/mTOR and RAS/RAF/MAPK, participating in multiple cellular processes, including cell proliferation, apoptosis, differentiation, cell growth, and migration [52]. Overexpression of this growth factor due to mutations or amplifications of this gene is often detected in glioblastoma [32,52]. Most patients with EGFR amplification have a deletion of exons 2–7 (EGFRvIII). This variant is usually expressed by extrachromosomal DNA fragments called “double minutes” [53].
Amplification of this gene is associated with a poor prognosis as the tumour has an increased capacity for proliferation, cell migration, neovascularization, and resistance to chemotherapy [52,54]. However, despite numerous studies reporting this negative association [32,55,56,57], Ohgaki et al., 2004 reported that in a cohort of 715 glioblastoma patients, the presence of EGFR gene amplification did not affect overall survival [58]. Faulkner et al., 2015 also concluded that overexpression of this gene is not a predictive biomarker of overall survival based on data from a cohort of 51 individuals, 49% of whom had alterations in the EGFR gene [59]. Therefore, there is a need for further studies to better understand the impact of this alteration on the management of this disease.
In addition to its function as a potential predictive biomarker, EGFR gene amplification is also used for glioblastoma classification [20].
5.8. MGMT Promoter Methylation
MGMT (O6-methylguanine-DNA methyltransferase) encodes a protein involved in DNA repair, specifically responsible for removing alkyl groups from the O6 position of guanine. Temozolomide is an alkylating agent that induces DNA damage by adding a methyl group to the N7 and O6 positions of guanine and the N3 position of adenines. This alteration leads to activation of the MMR (mismatch repair) pathway during DNA replication, culminating in double-strand breaks and ultimately leading to apoptosis [60]. The MGMT protein prevents this by removing and transferring the methyl group, thus inhibiting the cytotoxic action of TMZ. Consequently, promoter methylation of the MGMT gene is associated with a favourable prognosis and a better response to TMZ treatment [32]. Several studies confirmed this association and reported a higher overall survival and better response to treatment compared to patients without methylation of the MGMT gene [61,62,63,64,65]. For patients in this situation, O6-benzylguanine (O6-BG) can be used for MGMT inactivation. This O6-meG analogue passes through the blood–brain barrier so it can be used as a potential treatment for glioblastoma, sensitizing cells to TMZ [66].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Diagnostic, prognostic and predictive biomarkers in glioblastoma.
| Biomarker | Classification | Effect | References |
|---|---|---|---|
| IDH1 and IDH2 | Prognostic | Mutations in IDH1 or IDH2 are positive prognostic factors | [44] |
| ATRX | Prognostic | ATRX alterations are associated with a better prognosis | [32] |
| TERT | Diagnostic and prognostic | Mutations in the TERT gene are associated with a worse prognosis | [32] |
| CDKN2A | Prognostic | Homozygous deletion of this gene is associated with a poor prognosis | [32] |
| 1p/19q Codeletion | Prognostic | 1p/19q deletion is associated with a more favourable clinical outcome, although this alteration is uncommon in glioblastoma | [49] |
| 7+/10− | Diagnostic and prognostic | Gain of chromosome 7 and loss of chromosome 10 are common alterations in glioblastoma and are typically associated with a poor prognosis | [32] |
| EGFR | Diagnostic, prognostic and predictive | Amplification of this gene is associated with a poor prognosis and resistance to therapy | [52,54] |
| MGMT | Prognostic and predictive | Promoter methylation of the MGMT gene is associated with a favourable prognosis and a better response to treatment | [32] |
It is important to acknowledge that even though some of these biomarkers have a positive impact on the patient’s prognosis, they are just one aspect of the complex molecular and genetic landscape of glioblastoma.
5.9. Liquid Biopsy
Liquid biopsies allow the detection of nucleic acids (DNA or RNA), circulating tumour cells (CTCs), and extracellular vesicles (EVs), from blood samples, cerebrospinal fluid (CSF), urine, or other body fluid. In the context of glioblastoma, the most relevant are CSF [67], blood [68], and, although with limited evidence, urine [69]. Currently, glioblastoma diagnosis relies on imaging and histopathological analysis, requiring surgical intervention for tissue sampling and subsequent molecular analysis [32].
The principal benefit of this non-invasive approach lies in its capacity for early detection of the disease, tracking the possibility of relapse after treatment, and providing a means for molecular tumour profiling through a simple blood or other body fluid sample. Liquid biopsy also allows the patient to be followed throughout treatment, without the need for multiple surgical interventions during the course of the disease.
Glioblastoma is characterized by high heterogeneity, leading to diverse cell populations with various molecular alterations. Therefore, a tissue biopsy may not provide an accurate representation of the tumour [70]. The main objective of liquid biopsies is to detect biomarkers with implications for disease management, treatment, prognosis, and monitoring [71]. Despite advances, liquid biopsies also have limitations, particularly in tumour representation, as some alterations may remain undetected due to the low concentrations of relevant biomarkers in biological fluids. Therefore, larger cohort studies are needed to determine the sensitivity and effectiveness of liquid biopsies in glioblastoma molecular characterization.
Cerebrospinal fluid is in close contact with the brain and spinal cord, making it a receptor for various cell-secreted products. Also, it is not separated from the tumour by the blood–brain barrier (BBB) so it may have a greater representation of the tumour cells than plasma samples. Compared to blood samples, CSF is obtained through an invasive method, making it challenging to repeat at key timepoints during patient follow-up. Several studies [67,72,73,74,75] demonstrated the possibility of detecting ctDNA in CSF, making it a strong candidate for its use in liquid biopsies, with the potential to significantly impact tumour characterization, diagnosis, prognosis, and disease management [67]. Pan et al., 2019 showed that the molecular profile obtained by sequencing ctDNA from CSF coincided with the tumour profile and, in some cases, ctDNA analysis detected genetic alterations that had not been found in the tumour [74]. This can be explained due to the limitations in the collection of the tissue and tumour heterogeneity.
The analysis of biomarkers present in plasma has been studied in the oncological context, namely in neurological tumours, as it allows their molecular study and patient follow-up without the use of multiple surgical interventions [71]. In contrast to CSF, blood is subject to the BBB. Therefore, for a liquid biopsy using a plasma sample to be representative of the tumour, the materials secreted by tumour cells must be able to cross this barrier [76]. The integrity of the BBB changes during the development of a brain tumour, becoming more permeable and forming a brain–tumour barrier (BTB). This alteration is attributed to the downregulation of tight junctions, primarily due to the overexpression of vascular endothelial growth factor (VEGF), a mitogen associated with angiogenesis [77]. Thus, the overexpression of this growth factor can constitute a prognostic biomarker and be used as a target for the development of targeted therapies [78]. While the BBB presents a challenge for liquid biopsies, numerous studies have demonstrated potential for the detection of circulating tumour DNA (ctDNA) in blood samples. Some of these studies report detection rates ranging from 27% to 55% using next-generation sequencing (NGS) techniques [79,80,81]. This detection rate is consistent with findings by Bettegowda et al., 2014, where ctDNA was detected in less than 50% of patients with primary brain tumours, compared to 75% of patients with other types of cancers [82]. However, some of these studies have limitations regarding the number of patients. Although the use of plasma as a biofluid for liquid biopsies seems promising, it is essential to consider that the molecular profile of the tumour may be underrepresented, because the molecular alterations in tissue may not entirely reflect those detected in plasma due to the BBB. So, the results obtained should be interpreted cautiously and integrated with data obtained from complementary methods.
Regarding the use of urine samples in liquid biopsies for molecular characterization of glioblastoma, there are relatively few conclusive studies. Some report that it is possible to detect ctDNA in urine but have some limitations in the size of the cohort, which does not allow the clinical impact to be demonstrated [69]. Moreover, it is important to note that urine is produced by the kidneys, and numerous components might not be as effectively filtered into urine compared to blood. Consequently, urine-based liquid biopsy has received more extensive attention in genitourinary cancers, while it poses a significant challenge in non-urological cancers [83].
5.9.1. Circulating Tumour Cells
Circulating tumour cells (CTCs), first described by Ashworth in 1869 [84], constitute a heterogeneous group of cells originating from the primary tumour or metastatic sites, which dissociate from the tumour and enter the bloodstream as either individual cells or clusters [85,86]. This type of cell contributes to metastasis and its detection can serve as a predictive biomarker and a way of monitoring treatment response [76]. However, the proportion of CTCs in the bloodstream is quite low, especially in the early stages of the disease, posing significant challenges to their isolation and characterization. Consequently, an initial enrichment step is necessary for CTC isolation. Currently, for most tumours, this selection process is based on the expression of the epithelial cell adhesion molecule (EpCAM). Nonetheless, glioblastoma tumour cells exhibit a mesenchymal phenotype and, consequently, lack EpCAM expression [76]. Thus, new methods are needed to isolate this type of cell. The detection of CTCs in cases of glioblastoma is also challenging, as this type of cancer rarely has extracranial metastases, largely due to the low survival rate and the constitution of central nervous system tissues [87]. Several studies demonstrated the possibility of detecting CTCs using different isolation methods, including density-gradient centrifugation, immunomagnetic enrichment, Parsortix microfluidic cassettes, and size-based techniques [88,89,90,91]. Sullivan et al. detected CTCs in 39% of the patients enrolled in their study through a blood-based liquid biopsy. They also concluded that patients in an advanced state of the disease exhibited a higher frequency of CTCs, further emphasizing the utility of CTC analysis in disease monitoring [92]. Despite these challenges, the study of CTCs allows for a more comprehensive molecular analysis, allowing characterization at the DNA, RNA, and protein levels. Therefore, CTCs can serve as a valuable predictive and prognostic biomarker. However, more evidence of their impact on the management of the disease is needed before applying this technology in clinical practice [32]. Furthermore, to date, there are few studies dedicated to CTC analysis in glioblastoma, with most of them enrolling a limited number of patients [88,91,92].
5.9.2. Cell-Free Nucleic Acids
Liquid biopsy also allows the detection of nucleic acids (DNA/RNA), whose main objective is the detection of point mutations, copy number variations (CNVs) in specific genes, or methylation status, which may have a significant impact on diagnosis, prognosis, and treatment [93]. ctDNA is a component of cell-free DNA (cfDNA) that is released by the tumour into the bloodstream and other body fluids due to cell death and apoptosis. It comprises DNA fragments of approximately 180–200 base pairs and sequencing of these fragments allows the detection of a wide spectrum of changes that may have an impact on the disease [32]. The analysis of ctDNA from CSF appears to have a higher sensitivity, representing the tumour molecular profile in a more reliable way [71]. Regarding the use of blood samples, plasma should be used in ctDNA analysis, since compared to serum it has lower levels of background cfDNA resulting from the cell lysis of lymphocytes [94]. However, ctDNA levels in plasma are also variable among patients, depending on the disease progression, tumour heterogeneity, effects of the treatment and individual patient characteristics. In most cases, low levels of ctDNA are associated with the presence of the BBB [93,94]. Several studies used different techniques for ctDNA detection and analysis, including NGS [95] and PCR-based methods such as digital droplet PCR (ddPCR) [96]. These methods allow the detection of various genetic alterations commonly found in glioblastoma, such as mutations in the EGFR, PTEN, TP53, TERT, and RB1 genes [96,97]. Muralidharan et al., 2021 developed a novel ddPCR assay for the detection of two mutations in the promotor of the TERT gene from cfDNA. They obtained a sensitivity of 62.5% and a specificity of 90% compared to detection in a tissue sample [98]. Through sequencing, Mouliere et al., 2021 detected ctDNA from plasma, CSF, and urine samples of patients with gliomas, demonstrating high sensitivity. Despite the limited number of individuals included, they demonstrated the promising use of liquid biopsies in the management of the disease [69]. However, there is a need for more translational studies to establish liquid biopsy as a routine clinical practice in the management of glioblastoma. In addition to point mutations and CNVs, the analysis of ctDNA also allows the study of methylation. In a study conducted by Dai et al., 2023, a genome-wide analysis of methylation was performed on ctDNA extracted from CSF, demonstrating the possibility of integrating epigenetic studies into the management of this disease [99]. In glioblastoma, the analysis of MGMT methylation patterns significantly impacts prognosis and treatment response.
RNA analysis can also serve as a biomarker for monitoring disease progression and response to treatment. Like cfDNA, cfRNA can also be extracted from CSF, blood, and urine [100]. cfRNA originates from tumour cells and is released into the bloodstream through necrotic or apoptotic cells or a vesicle-free RNA-binding protein-dependent pathway. There are several types of cfRNA, with microRNAs and lncRNAs being the most studied in the context of liquid biopsy [52]. Some microRNAs act as tumour suppressors, including miR-7, miR-34a, miR-128, miR-181a, and miR-181b, which are typically downregulated in glioblastoma. Upregulated microRNAs (oncomiRs) influence the expression of tumour suppressor genes and promote oncogenesis [101]. The most studied oncomiRs in glioblastoma are miR-21, miR-10b, miR-93, miR-221, miR-222, and miR-182 [101,102]. LncRNAs interact with miRNAs, contributing to the regulation of diverse signalling pathways, including NOTCH (neurogenic locus notch homolog protein), MAPKs (mitogen-activated protein kinases), PI3K/AKT/mTOR (phosphoinositide 3-kinase/protein kinase B/mammalian target of rapamycin), Wnt/β-catenin, and BMP (bone morphogenetic protein). Dysregulation of these cell pathways is associated with the development and progression of glioblastoma, and therefore, lncRNAs have potential as predictive biomarkers since they can also influence response to treatment [103].
Wu et al., 2020 reported that LINC00470 promoted cell proliferation, invasion, and resistance to TMZ by competitively interacting with miRNA-134, which targets MYC, negatively affecting its expression [104].
5.9.3. Extracellular Vesicles
In the context of liquid biopsies, extracellular vesicles (EVs) have gained greater focus in recent studies, although they were first reported in 1946 by Chargaff and West [105]. These vesicles are small structures enclosed by a lipid membrane and are secreted by cells. EVs can originate from the endosomal system and are referred to as exosomes or be released from the plasma membrane, known as microvesicles [106,107]. The main interest in the isolation of these structures lies in the informative power they carry since they can transport nucleic acids, lipids, and proteins, acting as vehicles of intercellular communication [103]. EVs can be isolated from blood, CSF, urine, or other body fluids. The use of EVs in liquid biopsies seems promising since they are more stable than CTCs and cfDNA/RNA and are able to pass the BBB, even when it is intact [108].
Tumour cells also release these vesicles, which can contribute to metastasis, angiogenesis, and chemotherapy resistance by affecting other cells within the tumour microenvironment and more distant cells or organs [107]. One of the main challenges in the application of EVs analysis in liquid biopsies is related to the difficulty in isolating these structures that are susceptible to contamination with non-EV proteins, lipoproteins, and high-density lipoproteins (HDL). Currently, there are several methods for the isolation of EVs that can be divided based on their physical and chemical properties. These methods may include centrifugation-based isolation, size, affinity, precipitation, or microfluidic techniques [107,108]. Ma et al., 2022 demonstrated that EVs are released by glioma stem cells and alter the tumour microenvironment, making tumour cells resistant to treatment. However, these results are only based on data obtained from cell lines from three patients [109]. More recently, Tzaridis et al., 2023 investigated differences in the quantity of EVs in the serum of 67 patients with glioblastoma and 22 controls. They found a significant increase in EVs in the patient group, highlighting the possibility of isolating these structures from a blood liquid biopsy. Nevertheless, the authors also reinforce the limitations regarding the number of patients and heterogeneity within the cohort itself, which does not allow definitive conclusions to be drawn [110]. On the other hand, Garcia et al., 2019, concluded that exosome concentrations were higher in patients compared to controls and also demonstrated the possibility of isolating exosomes from plasma samples. This study also had a limited number of participants (19 patients and 19 controls), emphasizing the inherent limitations that are common to several studies [111].
While the use of EVs as predictive biomarkers in glioblastoma appears promising, further research involving larger cohorts is needed to obtain more data concerning clinical applicability and disease management. Standardization of laboratory practices in EVs isolation is also required to overcome methodological challenges.
6. Diagnosis
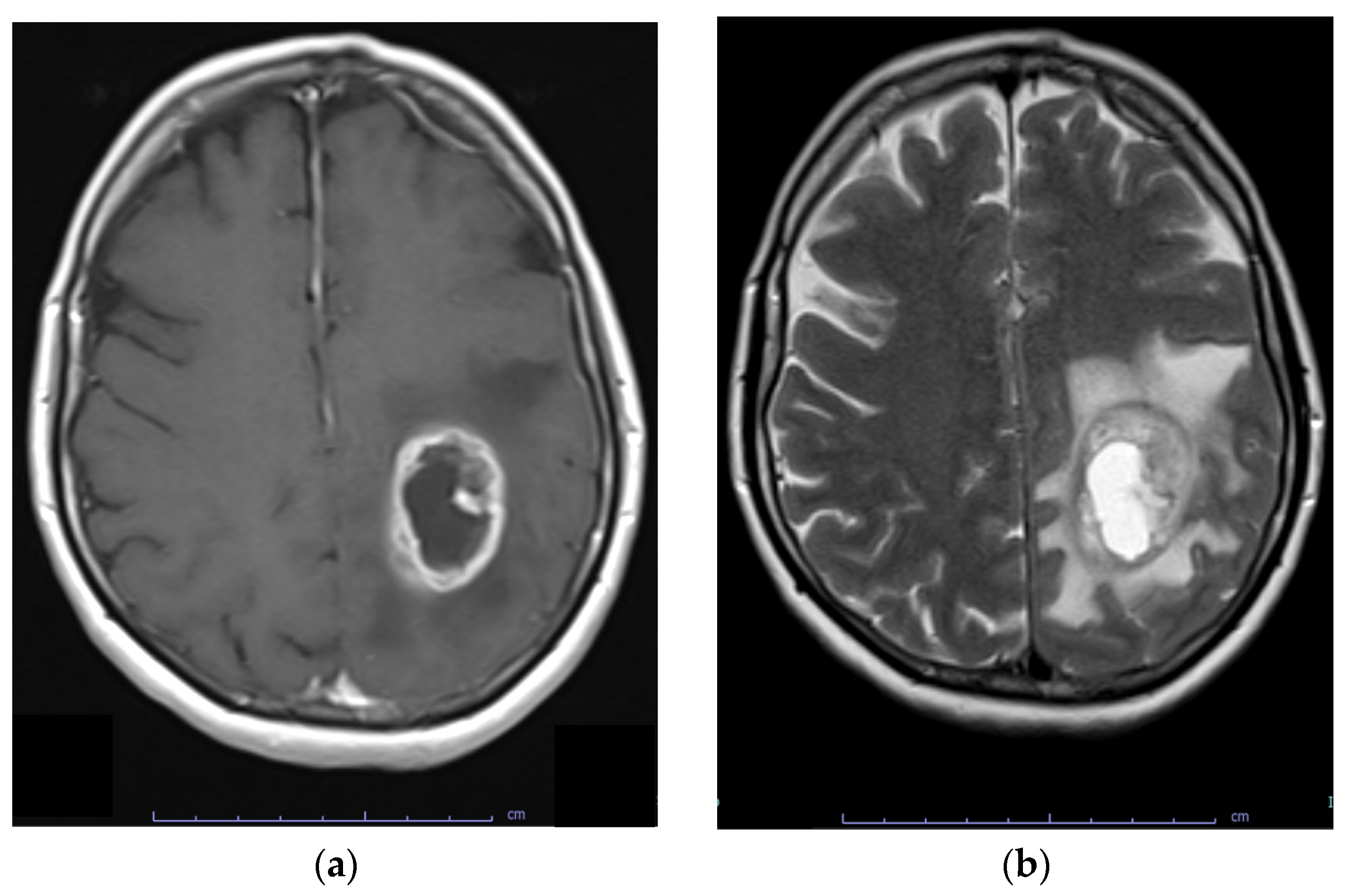
Most glioblastoma are diagnosed after the onset of symptoms because they rapidly expand or infiltrate brain structures. Indicative symptoms may include new-onset seizures, progressive headache, focal neurological symptoms, mental status changes, and signs of increased intracranial pressure [112,113]. Contrast-enhanced MRI is the diagnostic tool of choice for glioblastoma. These lesions are infiltrative and diverse, originating and spreading from the white matter. Involvement of the corpus callosum is frequently observed. Glioblastomas are poorly circumscribed and display contrast enhancement at their margin as a sign of blood–brain barrier disruption. The core of the abnormal tissue shows decreased signal intensity on T1-weighted imaging as a result of necrosis. Enhancement (gadolinium) is variable but is almost always present, typically peripheral and irregular with nodular components, and it usually surrounds the necrosis. Commonly, the surrounding area exhibits increased signal intensity on T2-weighted and fluid-attenuated inversion recovery (FLAIR) images, indicating cerebral oedema. (Figure 4). Multicentric enhancement, haemorrhage, and cystic changes are also frequent [114].
7. Current Treatment Options
7.1. Management of Newly Diagnosed Glioblastoma
Radical microsurgical resection of a glioblastoma is limited by the highly invasive nature of the tumour, with infiltrating tumour cells typically extending significant distances from the main tumour mass [115]. Nonetheless, the goal of glioblastoma surgery should be gross total resection of the enhancing solid tumour mass whenever feasible. Although some studies reported progressive improvements in outcomes as the extent of resection increased beyond 78%, in both newly diagnosed [116,117] and recurrent glioblastoma, only gross total resection was associated with improved outcomes [118,119].
The extent of resection (EOR) is a highly studied subject in relation to the surgical management of glioblastoma. Firstly, it has been demonstrated that EOR has an impact on the overall survival (OS) of patients with glioblastoma. However, there has been considerable discussion on the ideal threshold for EOR. The paper by Lacroix et al. reported that an EOR > 98% or higher resulted in a significant improvement in median survival (8.8 months vs. 13 months, p < 0.0001) [120].
Shah et al. investigated the impact of supramaximal resection or anatomic lobectomy on the survival rates of patients diagnosed with non-eloquent gliomas. A propensity-matched analysis demonstrated that supramaximal resection led to enhanced overall survival (30.7 vs. 14.1 months) and progression-free survival (17.2 vs. 8.1 months) in comparison to the gross total resection group (p < 0.001) [121].
With the intent to achieve more control of the quality of the resection, 5-aminolevulinic acid (5-ALA) was introduced in the surgery setting. 5-ALA is a natural precursor of hemoglobin and is a fluorescent dye that is preferably picked up by tumour cells after being orally administered 2–3 h prior to surgery. 5-ALA-guided surgery is highly effective in accurately detecting malignant tumour tissues during surgery. It is an intraoperative tool that may be used independently of neuronavigation to achieve the maximum extent of tumour removal without causing neurological impairment [122,123].
Following maximal safe resection, adjuvant involved-field radiotherapy (RT) with concurrent (75 mg/m2/day × 6 wk.) and adjuvant (150–200 mg/m2/day × 5 days for six 28 day-day cycles wk.) temozolomide (TMZ) is the standard treatment recommended for patients with newly diagnosed glioblastoma based on the results of the phase III, randomized EORTC-NCIC study [124].
In this study, 573 patients, with newly diagnosed glioblastoma who were aged ≤70 years and had a WHO PS ≤2 were assigned to treatment. Overall survival was 27.2% (95% CI 22.2–32.5) at 2 years, 16.0% (12.0–20.6) at 3 years, 12.1% (8.5–16.4) at 4 years, and 9.8% (6.4–14.0) at 5 years with temozolomide, versus 10.9% (7.6–14.8), 4.4% (2.4–7.2), 3.0% (1.4–5.7), and 1.9% (0.6–4.4) with radiotherapy alone (hazard ratio 0.6, 95% CI 0.5–0.7; p < 0.0001) [22,124].
The new guidelines recommend this approach with or without alternating electric field therapy (TTFields) [22]. Even though the spectrum of effects elicited remains incompletely understood, emerging data suggest that TTFields exert biophysical forces on a variety of charged and polarisable molecules to elicit a multitude of biological processes with anticancer effects, including DNA repair, autophagy, cell migration, permeability, and immunological responses. Substantial geographic variation in TTFields availability exists in the clinical practice, following cost-effectiveness continuous evaluations [125].
Even in the elderly, radiation therapy has confirmed a modest improvement in survival without a detriment in quality of life or neurocognition [126].
Patients who are elderly (≥70 years), have reduced performance status, significant medical comorbidities, or significant neurologic deficits can be treated with a diversity of established hypofractionated (higher dose per fraction over fewer total treatments) schedules ranging from 5 to 15 fractions [127,128].
7.2. Radiotherapy Considerations
For resected tumours, GTV delineation should be based on the resection cavity (if present) and any residual enhancing tumour on contrast-enhanced T1-weighted MRI, regardless of peritumoral oedema [129].
Although the European Organization for Research and Treatment of Cancer (EORTC) and the Radiotherapy and Oncology Group (RTOG) have adopted different methods for delineating target volumes in glioblastoma, both groups have formerly recommended a volumetric GTV expansion of 2 cm to generate the CTV. This margin was applied to embrace areas of potential microscopic tumour infiltration and was adjusted to respect anatomical borders, as described in other preceding glioblastoma target delineation guidelines [129,130] (Figure 5).
Recent, retrospective, and prospective studies, applying reduced GTV-to-CTV margins of 0.5–1.5 cm with either conventionally fractionated or hypofractionated radiation schedules, have shown that overall survival, progression-free survival times, and recurrence patterns are similar to those found in guidelines using current target delineation recommendations [128,131].
Proton beam therapy (PBT) enables high-dose irradiation of tumours without increasing the dose to the surrounding normal tissue by applying a sharp energy peak called the Bragg peak, which has an acute distal dose fall-off in its depth-dose distribution protecting normal surrounding tissue [132]. Recent authors demonstrated that high-dose PBT (96.6 Gy in 56 fractions by hyperfractionated concomitant boost) conferred a statistically significant survival advantage in patients with glioblastoma compared to conventional radiation therapy using propensity-matched cohorts. Although acute radiation-related toxicities were equivalent between the PBT and CRT groups, radiation necrosis as a late radiation-related toxicity was more prevalent in the PBT group [133]. The possibility of access to this radiotherapy technique is still scarce in many places around the world.
8. Novel Treatment Options
8.1. Targeted Therapies: From Cell Signalling Pathways to Cell Metabolism
Targeted therapies aim to interfere with the function of a specific molecule involved in the genesis of glioblastoma, affecting the cascade of reactions downstream of the pathway where this molecule acts. Despite advances in understanding the molecular mechanisms and molecular profile of glioblastoma, there has not yet been significant progress with applicability in clinical practice [27,118].
Several clinical trials (Table 2) targeted different cell signalling pathways related to the development and progression of glioblastoma.
Some of these trials use drugs that target receptor tyrosine kinases (RTKs) or their respective cell pathways, specifically directed at EGFR, MET, FGFR, BRAF mutations, neurotrophic tyrosine receptor kinases (NTRK), or the PI3K/AKT/mTOR pathway. Others target elements of cell cycle regulation and apoptosis pathways, such as RB, the p53 pathway, and TERT. Drugs acting on elements of the tumour microenvironment, such as VEGF and transforming growth factor-β (TGF-β), are also being investigated [27,134]. Targeted therapies may include inhibitors of the target molecule, vaccines, CAR T-cells, or antibodies [134].
Regarding clinical trials targeting proteins in the PI3K/AKT/mTOR pathway, specifically NCT01339052 and NCT01019434, buparlisib demonstrated minimal single-agent efficacy in patients with PI3K-activated recurrent glioblastoma, and temsirolimus was not superior to temozolomide in patients with an unmethylated MGMT promoter, respectively [135,136]. However, Wen et al., concluded that buparlisib achieved significant brain penetration, but the inhibition of the signalling pathway in the tumour tissue was incomplete, which can explain the low efficacy [135].
In the cases of certain drugs specifically targeting receptor tyrosine kinases (RTKs) in clinical trials (NCT01268566; NCT01632228; NCT01975701), the results were not promising. Despite good tolerance, their use as monotherapy or in combination with others demonstrated limited efficacy, showing a lack of clinical benefit [137,138,139].
More recently, a clinical trial (NCT04121455) showed good tolerance and safety regarding the therapeutic approach. This trial included several patients who underwent biopsy or incomplete resection of the tumour and lacked MGMT promoter hypermethylation. The combined use of radiotherapy with olaptesed pegol (NOX-A12), a CXCL12 inhibitor, demonstrated a median overall survival (OS) of 19.9 months, indicating a survival benefit for the patients [140].
Another clinical trial (NCT06102525), currently in development based on gene therapy, used an RNA-replacement enzyme that targets hTERT mRNA and replaces it with therapeutic gene RNA, contributing to the modulation of the tumour microenvironment by reducing VEGF expression, thus making it more responsive to immunotherapy.
Wick et al. randomly assigned patients with progression after chemoradiation in a 2:1 ratio to receive lomustine plus bevacizumab or lomustine alone. The combination therapy did not provide a survival advantage; the median overall survival was 9.1 months in the combination group and 8.6 months in the monotherapy group (hazard ratio for death, 0.95; 95% CI, 0.74 to 1.21; p = 0.65). PFS survival was 2.7 months longer in the combination group than in the monotherapy group: 4.2 months versus 1.5 months (hazard ratio for disease progression or death, 0.49; 95% CI, 0.39 to 0.61; p < 0.001 [141].
The BELOB trial was an open-label, three-group, phase 2 study. Adult patients with a first recurrence of a glioblastoma after temozolomide chemoradiotherapy were randomly allocated to treatment with oral lomustine 110 mg/m2 once every 6 weeks, intravenous bevacizumab 10 mg/kg once every 2 weeks, or combination treatment with lomustine 110 mg/m2 every 6 weeks and bevacizumab 10 mg/kg every 2 weeks. The 9-month overall survival was 43% in the lomustine group, 38% in the bevacizumab group, and 63% (49–75) for the combined bevacizumab and lomustine groups. These results permit the use of this combination in off-label use in some centres [142].
Lee et al. performed a systematic review on the risks and benefits of the use of anti-EGFR therapies in glioblastomas. It was observed there was no evidence of a benefit in OS with the use of anti-EGFR therapy in the first-line or recurrent setting (hazard ratio (HR) 0.89, 95% confidence interval (CI) 0.76 to 1.04). The addition of anti-EGFR therapy showed no evidence of an increase in progression-free survival (PFS) in the first-line setting. In the recurrent setting, there was an increase in PFS with the use of anti-EGFR therapy [143].
Tumour metabolism can also serve as a therapeutic target, as it may be involved in the development and progression of glioblastoma. Tumour cells show increased glycolysis, known as the Warburg effect, providing a proliferative advantage in hypoxic conditions. Alterations in other metabolic processes such as oxidative phosphorylation, the pentose phosphate pathway (PPP), lipids, amino acids, and nucleotides metabolism have also been described [144,145].
Some ongoing clinical trials related to tumour metabolism (Table 1) are still in early phases and, therefore, do not yet have available results. Clinical trials NCT03032484 and NCT02029573, despite both treatments being well-tolerated and safe, lack evidence of their effectiveness in patient survival [146,147].
Despite advances in understanding the molecular pathogenesis of glioblastoma and its impact on the development of targeted therapies, further studies should be carried out to expand the knowledge of the relationship between cell metabolism, cell signalling pathways, the tumour microenvironment, and genomic alterations. This will allow the development of new targeted therapies with a significant impact on the management of the disease.
8.2. Immunotherapies
Immunotherapy aims to use the patient’s immune system to target cancer cells. This approach has shown promising results in various types of cancer, particularly in hematological cancers. Immunotherapy may include checkpoint inhibitors, chimeric antigen receptor (CAR) T-cells, monoclonal antibodies, vaccines, and immune system modulators [148]. Regarding glioblastoma, few advances have been made and it remains in the clinical trial phase.
The brain is considered an immune-privileged organ due to the presence of the BBB, which limits the access of immune system cells and poses a challenge to the development of targeted therapies. Glioblastoma also develops an immunosuppressive microenvironment, which stimulates disease progression. Glioblastoma cells release chemokines, growth factors, and cytokines, which attract and stimulate cells with immunosuppressive function [148,149]. Glioblastoma cells also exhibit overexpression of programmed death receptor ligand 1 (PD-L1), which interacts with the receptor PD-1 on T cells, inhibiting their function and leading to a decreased immune response [149]. Hypoxia within the tumour microenvironment has been associated with increased expression of hypoxia-inducible factor 1-α (HIF1-α). Overexpression of HIF1-α can compromise T cell activity and upregulate immunomodulators such as transforming growth factor β (TGF-β) and vascular endothelial growth factor (VEGF), promoting immunosuppression [148,150].
The intricate and distinctive tumour microenvironment of glioblastoma may contribute to the reduced efficacy of immunotherapy. In this context, there are several clinical trials to determine the efficacy of immunotherapy, some of which are described in Table 3.
In the clinical trial NCT03726515, using CART-EGFRvIII cells, any clinical benefit could not be established. However, the authors concluded that these intravenously infused cells did reach the tumour and exert antigen-directed activity, although the tumour microenvironment became even more immunosuppressive after treatment with the CAR T cells [151]. In the clinical trial NCT01109095, treating progressive glioblastoma with HER2-CAR VSTs was found to be feasible and safe, resulting in clinical benefit for 8 out of 17 patients. In this cohort, the median overall survival was 24.5 months from diagnosis [152]. The combination of CAR T cells with checkpoint inhibition (NCT04003649) is also under study in the context of glioblastoma.
Vaccines have also been explored in glioblastoma treatment. The use of rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma did not demonstrate any survival benefit in a phase III trial [153]. On the other hand, a phase II trial (NCT01498328) reported favourable results in a cohort of 73 patients with recurrent EGFRvIII-positive glioblastoma [78].
The use of monoclonal antibodies such as avelumab (NCT03047473) and nivolumab (NCT02017717), belonging to the immune-checkpoint inhibitor class, failed to demonstrate efficacy for checkpoint blockade, bringing no improvement in overall survival [78,154]. On the contrary, the use of atezolizumab combined with TMZ and radiotherapy (NCT03174197) showed modest efficacy [155].
Weiss et al., 2021, combined the use of TMZ with L19TNF in newly diagnosed glioblastoma patients (NCT04443010). So far, this clinical trial is still in the recruitment phase and has not yet shown results. L19TNF represents a fully human antibody–cytokine fusion protein, composed of TNF (tumour necrosis factor) fused to the L19 antibody, which binds to a tumour-specific epitope of the extracellular matrix protein fibronectin (EDB-FN). This allows the precise delivery of therapeutic TNF doses exclusively to the tumour, without affecting other regions [156].
Although immunotherapy does not yet have clinical applicability in the case of glioblastoma, the various clinical trials may provide important information for the development of more robust clinical trials with larger sample sizes. These trials should consider addressing issues such as antigen escape, tumour heterogeneity, the tumour microenvironment, drug delivery strategies, patient selection based on tumour genetics, and multimodal approaches [148]. Furthermore, clinical trials with unmet endpoints contribute to a better understanding of the role of the tumour microenvironment in immune responses, leading to the development of new strategies and drugs with different molecular targets or the combination of different therapeutic approaches, improving outcomes and disease management.
8.3. Obstacles in Using Targeted Therapies
Despite numerous studies with several promising in vitro results regarding glioblastoma, there is still difficulty in implementing targeted therapies that are effective. Several challenges can affect the efficacy of these treatments, namely tumour heterogeneity, BBB, tumour microenvironment, redundant cell signalling pathways and the presence of glioblastoma stem cells (GSCs) [27,118].
The presence of diverse cell clones with different molecular profiles makes it challenging to employ targeted therapies for specific alterations or signalling pathways. Thus, tumour heterogeneity may account for the failures observed in in vivo clinical trials based on monotherapy [27].
The BBB is an obstacle to drug delivery to the brain, preventing the passage of 98% of all small molecules. While this barrier may be compromised in certain regions due to tumour development, other locations, typically the infiltrative tumour edge left behind after resection, may maintain its integrity preventing the passage of the therapeutic agent [27,118].
Redundant pathways share the same downstream signalling targets and, therefore, can contribute to increased therapy resistance [118]. The tumour microenvironment, characterized by hypoxia, plays a role in angiogenesis, promoting migration, invasion, and chemoresistance.
Glioblastoma stem cells (GSCs) arise from the malignant transformation of neural stem cells or through dedifferentiation of tumour cells following radiation or chemotherapy. These cells exhibit high proliferative capacity, metastatic potential, and the ability to suppress the anti-inflammatory response, contributing to a greater resistance to treatment [27].
All of these factors can contribute to the acquisition of resistance to therapies, affecting their effectiveness.
9. Conclusions
Glioblastoma is a highly malignant, and aggressive, primary brain tumour that causes substantial neurological impairment. The current standard of care comprises the optimal surgical removal of the tumour, simultaneous administration of chemotherapy and radiation therapy, and subsequent use of adjuvant chemotherapy.
Despite major advancements in comprehending the molecular pathophysiology and biology of glioblastoma, there has been limited translation of this knowledge into substantial improvements in patient outcomes. Molecular and genetic biomarker testing will become a regular part of the eligibility requirements for clinical studies. These studies are necessary to evaluate the ability of substances to cross the blood–brain barrier and their effects on the body’s response to drugs. Additionally, incorporating imaging and a wide range of biomarkers will help identify patients who are more likely to benefit from treatment based on the unique biology of their tumours.
The challenges in glioblastoma clinical trial design stem from inherent factors related to these tumours, including the limited pool of eligible patients and the subsequent challenges in conducting large randomized trials. Another challenge lies in comprehensively understanding the extensive genetic and molecular heterogeneity observed among different glioblastomas.
The assessment of response or ‘biological activity’ will involve the measurement of biomarkers at the molecular level, in addition to the use of established clinical and imaging criteria.
By incorporating a broader range of medical disciplines, spanning from laboratory research to patient care, it is expected that this heightened awareness will result in the discovery of more efficient and refined treatments.
Author Contributions
Conceptualization, D.R., P.V., J.B.M., I.M.C. and I.P.R.; formal analysis, D.R. and P.V.; writing—original draft preparation, D.R. and P.V.; writing—review and editing, J.B.M., I.M.C. and I.P.R.; supervision, I.P.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Perry, A.; Wesseling, P. Histologic Classification of Gliomas. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2016; pp. 71–95. [Google Scholar]
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. In Glioblastoma; Codon Publications: Singapore, 2017; pp. 143–153. [Google Scholar]
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of Glioblastoma: State of the Art and Future Directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; de Carvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef]
- Sidaway, P. Glioblastoma Subtypes Revisited. Nat. Rev. Clin. Oncol. 2017, 14, 587. [Google Scholar] [CrossRef]
- Grochans, S.; Cybulska, A.M.; Simińska, D.; Korbecki, J.; Kojder, K.; Chlubek, D.; Baranowska-Bosiacka, I. Epidemiology of Glioblastoma Multiforme–Literature Review. Cancers 2022, 14, 2412. [Google Scholar] [CrossRef]
- Patel, N.P.; Lyon, K.A.; Huang, J.H. The Effect of Race on the Prognosis of the Glioblastoma Patient: A Brief Review. Neurol. Res. 2019, 41, 967–971. [Google Scholar] [CrossRef]
- Kyritsis, A.P.; Bondy, M.L.; Rao, J.S.; Sioka, C. Inherited Predisposition to Glioma. Neuro-Oncology 2010, 12, 104–113. [Google Scholar] [CrossRef]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma Multiforme: A Review of Its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [CrossRef]
- Vienne-Jumeau, A.; Tafani, C.; Ricard, D. Environmental Risk Factors of Primary Brain Tumors: A Review. Rev. Neurol. 2019, 175, 664–678. [Google Scholar] [CrossRef]
- Rasheed, S.; Rehman, K.; Akash, M.S.H. An Insight into the Risk Factors of Brain Tumors and Their Therapeutic Interventions. Biomed. Pharmacother. 2021, 143, 112119. [Google Scholar] [CrossRef]
- Gatto, N.M.; Ogata, P.; Lytle, B. Farming, Pesticides, and Brain Cancer: A 20-Year Updated Systematic Literature Review and Meta-Analysis. Cancers 2021, 13, 4477. [Google Scholar] [CrossRef]
- Bielecka, J.; Markiewicz-żukowska, R. The Influence of Nutritional and Lifestyle Factors on Glioma Incidence. Nutrients 2020, 12, 1812. [Google Scholar] [CrossRef]
- Schüz, J.; Pirie, K.; Reeves, G.K.; Floud, S.; Beral, V. Cellular Telephone Use and the Risk of Brain Tumors: Update of the UK Million Women Study. J. Natl. Cancer Inst. 2022, 114, 704–711. [Google Scholar] [CrossRef]
- Inskip, P.D.; Tarone, R.E.; Hatch, E.E.; Wilcosky, T.C.; Shapiro, W.R.; Selker, R.G.; Fine, H.A.; Black, P.M.; Loeffler, J.S.; Linet, M.S. Cellular-Telephone Use and Brain Tumors. N. Engl. J. Med. 2001, 344, 79–86. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Ohgaki, H.; Kleihues, P. The Definition of Primary and Secondary Glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef]
- Sejda, A.; Grajkowska, W.; Trubicka, J.; Szutowicz, E.; Wojdacz, T.; Kloc, W.; Iżycka-Świeszewska, E. WHO CNS5 2021 Classification of Gliomas: A Practical Review and Road Signs for Diagnosing Pathologists and Proper Patho-Clinical and Neuro-Oncological Cooperation. Folia Neuropathol. 2022, 60, 137–152. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Burger, P.; Ellison, D.W.; Reifenberger, G.; von Deimling, A.; Aldape, K.; Brat, D.; Collins, V.P.; Eberhart, C.; et al. International Society of Neuropathology—Haarlem consensus guidelines for nervous system tumor classification and grading. Brain Pathol. 2014, 24, 429–435. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Torp, S.H.; Solheim, O.; Skjulsvik, A.J. The WHO 2021 Classification of Central Nervous System Tumours: A Practical Update on What Neurosurgeons Need to Know—A Minireview. Acta Neurochir. 2022, 164, 2453–2464. [Google Scholar] [CrossRef]
- Nabors, B.; Portnow, J.; Hattangadi-Gluth, J.; Horbinski, C. NCCN CNS Tumor Guidelines Update for 2023. Neuro-Oncology 2023, 25, 2114–2116. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef]
- Esemen, Y.; Awan, M.; Parwez, R.; Baig, A.; Rahman, S.; Masala, I.; Franchini, S.; Giakoumettis, D. Molecular Pathogenesis of Glioblastoma in Adults and Future Perspectives: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 2607. [Google Scholar] [CrossRef]
- Mao, H.; LeBrun, D.G.; Yang, J.; Zhu, V.F.; Li, M. Deregulated Signaling Pathways in Glioblastoma Multiforme: Molecular Mechanisms and Therapeutic Targets. Cancer Investig. 2012, 30, 48–56. [Google Scholar] [CrossRef]
- Dillon, M.; Lopez, A.; Lin, E.; Sales, D.; Perets, R.; Jain, P. Progress on Ras/MAPK Signaling Research and Targeting in Blood and Solid Cancers. Cancers 2021, 13, 5059. [Google Scholar] [CrossRef]
- El Atat, O.; Naser, R.; Abdelkhalek, M.; Habib, R.; El Sibai, M. Molecular Targeted Therapy: A New Avenue in Glioblastoma Treatment (Review). Oncol. Lett. 2022, 25, 46. [Google Scholar] [CrossRef]
- Di Nunno, V.; Gatto, L.; Tosoni, A.; Bartolini, S.; Franceschi, E. Implications of BRAF V600E Mutation in Gliomas: Molecular Considerations, Prognostic Value and Treatment Evolution. Front. Oncol. 2023, 12, 1067252. [Google Scholar] [CrossRef]
- Pearson, J.R.D.; Regad, T. Targeting Cellular Pathways in Glioblastoma Multiforme. Signal Transduct. Target. Ther. 2017, 2, 17040. [Google Scholar] [CrossRef]
- Uddin, M.S.; Al Mamun, A.; Alghamdi, B.S.; Tewari, D.; Jeandet, P.; Sarwar, M.S.; Ashraf, G.M. Epigenetics of Glioblastoma Multiforme: From Molecular Mechanisms to Therapeutic Approaches. Semin. Cancer Biol. 2022, 83, 100–120. [Google Scholar] [CrossRef]
- Romani, M.; Pistillo, M.P.; Banelli, B. Epigenetic Targeting of Glioblastoma. Front. Oncol. 2018, 8, 448. [Google Scholar] [CrossRef]
- Śledzińska, P.; Bebyn, M.G.; Furtak, J.; Kowalewski, J.; Lewandowska, M.A. Prognostic and Predictive Biomarkers in Gliomas. Int. J. Mol. Sci. 2021, 22, 10373. [Google Scholar] [CrossRef]
- Chen, H.-M.; Nikolic, A.; Singhal, D.; Gallo, M. Roles of Chromatin Remodelling and Molecular Heterogeneity in Therapy Resistance in Glioblastoma. Cancers 2022, 14, 4942. [Google Scholar] [CrossRef]
- Ganguly, D.; Sims, M.; Cai, C.; Fan, M.; Pfeffer, L.M. Chromatin Remodeling Factor BRG1 Regulates Stemness and Chemosensitivity of Glioma Initiating Cells. Stem Cells 2018, 36, 1804–1815. [Google Scholar] [CrossRef] [PubMed]
- Goenka, A.; Tiek, D.M.; Song, X.; Iglesia, R.P.; Lu, M.; Hu, B.; Cheng, S.-Y. The Role of Non-Coding RNAs in Glioma. Biomedicines 2022, 10, 2031. [Google Scholar] [CrossRef]
- McClellan, B.L.; Haase, S.; Nunez, F.J.; Alghamri, M.S.; Dabaja, A.A.; Lowenstein, P.R.; Castro, M.G. Impact of Epigenetic Reprogramming on Antitumor Immune Responses in Glioma. J. Clin. Investig. 2023, 133, e163450. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, R.P.; Costello, J.F. Epigenetic Mechanisms in Glioblastoma Multiforme. Semin. Cancer Biol. 2009, 19, 188–197. [Google Scholar] [CrossRef]
- Wu, Q.; Berglund, A.E.; Etame, A.B. The Impact of Epigenetic Modifications on Adaptive Resistance Evolution in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 8324. [Google Scholar] [CrossRef]
- Do, H.; Kim, W. Roles of Oncogenic Long Non-Coding RNAs in Cancer Development. Genom. Inform. 2018, 16, e18. [Google Scholar] [CrossRef] [PubMed]
- Yadav, B.; Pal, S.; Rubstov, Y.; Goel, A.; Garg, M.; Pavlyukov, M.; Pandey, A.K. LncRNAs Associated with Glioblastoma: From Transcriptional Noise to Novel Regulators with a Promising Role in Therapeutics. Mol. Ther. Nucleic Acids 2021, 24, 728–742. [Google Scholar] [CrossRef]
- Behrooz, A.B.; Latifi-Navid, H.; da Silva Rosa, S.C.; Swiat, M.; Wiechec, E.; Vitorino, C.; Vitorino, R.; Jamalpoor, Z.; Ghavami, S. Integrating Multi-Omics Analysis for Enhanced Diagnosis and Treatment of Glioblastoma: A Comprehensive Data-Driven Approach. Cancers 2023, 15, 3158. [Google Scholar] [CrossRef]
- Rana, R.; Kumari, B.; Kumari, J.; Ganguly, N.K. Glioblastoma Diagnostics and Prognostic Biomarkers: Current Status in Medicine and Exosome Derivation. Curr. Med. Res. Pract. 2019, 9, 65–73. [Google Scholar] [CrossRef]
- Sharma, N.; Mallela, A.N.; Shi, D.D.; Tang, L.W.; Abou-Al-Shaar, H.; Gersey, Z.C.; Zhang, X.; McBrayer, S.K.; Abdullah, K.G. Isocitrate Dehydrogenase Mutations in Gliomas: A Review of Current Understanding and Trials. Neurooncol. Adv. 2023, 5, vdad053. [Google Scholar] [CrossRef] [PubMed]
- Kayabolen, A.; Yilmaz, E.; Bagci-Onder, T. IDH Mutations in Glioma: Double-Edged Sword in Clinical Applications? Biomedicines 2021, 9, 799. [Google Scholar] [CrossRef] [PubMed]
- Hasanau, T.; Pisarev, E.; Kisil, O.; Nonoguchi, N.; Le Calvez-Kelm, F.; Zvereva, M. Detection of TERT Promoter Mutations as a Prognostic Biomarker in Gliomas: Methodology, Prospects, and Advances. Biomedicines 2022, 10, 728. [Google Scholar] [CrossRef] [PubMed]
- Arita, H.; Narita, Y.; Fukushima, S.; Tateishi, K.; Matsushita, Y.; Yoshida, A.; Miyakita, Y.; Ohno, M.; Collins, V.P.; Kawahara, N.; et al. Upregulating Mutations in the TERT Promoter Commonly Occur in Adult Malignant Gliomas and Are Strongly Associated with Total 1p19q Loss. Acta Neuropathol. 2013, 126, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Guo, Y.; Zhao, D.; Zou, Q.; Yu, F.; Zhang, L.; Xu, L. Comprehensive Analysis Revealed That CDKN2A Is a Biomarker for Immune Infiltrates in Multiple Cancers. Front. Cell Dev. Biol. 2021, 9, 808208. [Google Scholar] [CrossRef]
- Clark, K.H.; Villano, J.L.; Nikiforova, M.N.; Hamilton, R.L.; Horbinski, C. 1p/19q Testing Has No Significance in the Workup of Glioblastomas. Neuropathol. Appl. Neurobiol. 2013, 39, 706–717. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, M.; Yoshimoto, K.; Ma, X.; Guan, Y.; Hata, N.; Amano, T.; Nakamizo, A.; Suzuki, S.O.; Iwaki, T.; Sasaki, T. Molecular Characteristics of Glioblastoma with 1p/19q Co-Deletion. Brain Tumor Pathol. 2012, 29, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Stichel, D.; Ebrahimi, A.; Reuss, D.; Schrimpf, D.; Ono, T.; Shirahata, M.; Reifenberger, G.; Weller, M.; Hänggi, D.; Wick, W.; et al. Distribution of EGFR Amplification, Combined Chromosome 7 Gain and Chromosome 10 Loss, and TERT Promoter Mutation in Brain Tumors and Their Potential for the Reclassification of IDHwt Astrocytoma to Glioblastoma. Acta Neuropathol. 2018, 136, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Benitez, J.A.; Ma, J.; D’Antonio, M.; Boyer, A.; Camargo, M.F.; Zanca, C.; Kelly, S.; Khodadadi-Jamayran, A.; Jameson, N.M.; Andersen, M.; et al. PTEN Regulates Glioblastoma Oncogenesis through Chromatin-Associated Complexes of DAXX and Histone H3.3. Nat. Commun. 2017, 8, 15223. [Google Scholar] [CrossRef]
- Senhaji, N.; Houssaini, A.S.; Lamrabet, S.; Louati, S.; Bennis, S. Molecular and Circulating Biomarkers in Patients with Glioblastoma. Int. J. Mol. Sci. 2022, 23, 7474. [Google Scholar] [CrossRef]
- Ludwig, K.; Kornblum, H.I. Molecular Markers in Glioma. J. Neurooncol. 2017, 134, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Kaye, A.H.; Luwor, R.B. The EGFRvIII Variant in Glioblastoma Multiforme. J. Clin. Neurosci. 2009, 16, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Shinojima, N.; Tada, K.; Shiraishi, S.; Kamiryo, T.; Kochi, M.; Nakamura, H.; Makino, K.; Saya, H.; Hirano, H.; Kuratsu, J.-I.; et al. Prognostic Value of Epidermal Growth Factor Receptor in Patients with Glioblastoma Multiforme. Cancer Res. 2003, 63, 6962–6970. [Google Scholar] [PubMed]
- Li, J.; Liang, R.; Song, C.; Xiang, Y.; Liu, Y. Prognostic Significance of Epidermal Growth Factor Receptor Expression in Glioma Patients. OncoTargets Ther. 2018, 11, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, K.; Das, B.; Singh, A.K.; Misra, A.; Misra, S.; Misra, S.S. Prognostic Significance of Epidermal Growth Factor Receptor in Patients of Glioblastoma Multiforme. J. Clin. Diagn. Res. 2017, 11, EC05. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Dessen, P.; Jourde, B.; Horstmann, S.; Nishikawa, T.; Di Patre, P.-L.; Burkhard, C.; Schüler, D.; Probst-Hensch, N.M.; Maiorka, P.C.; et al. Genetic Pathways to Glioblastoma. Cancer Res. 2004, 64, 6892–6899. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, C.; Palmer, A.; Williams, H.; Wragg, C.; Haynes, H.R.; White, P.; DeSouza, R.-M.; Williams, M.; Hopkins, K.; Kurian, K.M. EGFR and EGFRvIII Analysis in Glioblastoma as Therapeutic Biomarkers. Br. J. Neurosurg. 2015, 29, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.; Pongor, L.; Su, Y.-T.; Xi, L.; Raffeld, M.; Quezado, M.; Trepel, J.; Aldape, K.; Pommier, Y.; Wu, J. MGMT Status as a Clinical Biomarker in Glioblastoma. Trends Cancer 2020, 6, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Zhang, W.; Wang, Y.; Peng, X.; Chen, B.; Qiu, X.; Li, G.; Li, S.; Wu, C.; Yao, K.; et al. IDH Mutation and MGMT Promoter Methylation in Glioblastoma: Results of a Prospective Registry. Oncotarget 2015, 6, 40896–40906. [Google Scholar] [CrossRef]
- Radke, J.; Koch, A.; Pritsch, F.; Schumann, E.; Misch, M.; Hempt, C.; Lenz, K.; Löbel, F.; Paschereit, F.; Heppner, F.L.; et al. Predictive MGMT Status in a Homogeneous Cohort of IDH Wildtype Glioblastoma Patients. Acta Neuropathol. Commun. 2019, 7, 89. [Google Scholar] [CrossRef]
- Annavarapu, S.; Gogate, A.; Pham, T.; Davies, K.; Singh, P.; Robert, N. Treatment Patterns and Outcomes for Patients with Newly Diagnosed Glioblastoma Multiforme: A Retrospective Cohort Study. CNS Oncol. 2021, 10, CNS76. [Google Scholar] [CrossRef] [PubMed]
- Chai, R.; Li, G.; Liu, Y.; Zhang, K.; Zhao, Z.; Wu, F.; Chang, Y.; Pang, B.; Li, J.; Li, Y.; et al. Predictive Value of MGMT Promoter Methylation on the Survival of TMZ Treated IDH-Mutant Glioblastoma. Cancer Biol. Med. 2021, 18, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Szylberg, M.; Sokal, P.; Śledzińska, P.; Bebyn, M.; Krajewski, S.; Szylberg, Ł.; Szylberg, A.; Szylberg, T.; Krystkiewicz, K.; Birski, M.; et al. MGMT Promoter Methylation as a Prognostic Factor in Primary Glioblastoma: A Single-Institution Observational Study. Biomedicines 2022, 10, 2030. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Zhang, L.; Wei, Q.; Shao, A. O6-Methylguanine-DNA Methyltransferase (MGMT): Challenges and New Opportunities in Glioma Chemotherapy. Front. Oncol. 2019, 9, 1547. [Google Scholar] [CrossRef] [PubMed]
- De Mattos-Arruda, L.; Mayor, R.; Ng, C.K.Y.; Weigelt, B.; Martínez-Ricarte, F.; Torrejon, D.; Oliveira, M.; Arias, A.; Raventos, C.; Tang, J.; et al. Cerebrospinal Fluid-Derived Circulating Tumour DNA Better Represents the Genomic Alterations of Brain Tumours than Plasma. Nat. Commun. 2015, 6, 8839. [Google Scholar] [CrossRef] [PubMed]
- Bagley, S.J.; Till, J.; Abdalla, A.; Sangha, H.K.; Yee, S.S.; Freedman, J.; Black, T.A.; Hussain, J.; Binder, Z.A.; Brem, S.; et al. Association of Plasma Cell-Free DNA with Survival in Patients with IDH Wild-Type Glioblastoma. Neurooncol. Adv. 2021, 3, vdab011. [Google Scholar] [CrossRef]
- Mouliere, F.; Smith, C.G.; Heider, K.; Su, J.; van der Pol, Y.; Thompson, M.; Morris, J.; Wan, J.C.M.; Chandrananda, D.; Hadfield, J.; et al. Fragmentation Patterns and Personalized Sequencing of Cell-free DNA in Urine and Plasma of Glioma Patients. EMBO Mol. Med. 2021, 13, e12881. [Google Scholar] [CrossRef] [PubMed]
- Eisenbarth, D.; Wang, Y.A. Glioblastoma Heterogeneity at Single Cell Resolution. Oncogene 2023, 42, 2155–2165. [Google Scholar] [CrossRef] [PubMed]
- Eibl, R.H.; Schneemann, M. Liquid Biopsy and Glioblastoma. Explor. Target. Antitumor Ther. 2023, 4, 28–41. [Google Scholar] [CrossRef]
- Pentsova, E.I.; Shah, R.H.; Tang, J.; Boire, A.; You, D.; Briggs, S.; Omuro, A.; Lin, X.; Fleisher, M.; Grommes, C.; et al. Evaluating Cancer of the Central Nervous System through Next-Generation Sequencing of Cerebrospinal Fluid. J. Clin. Oncol. 2016, 34, 2404–2415. [Google Scholar] [CrossRef]
- Martínez-Ricarte, F.; Mayor, R.; Martínez-Sáez, E.; Rubio-Pérez, C.; Pineda, E.; Cordero, E.; Cicuéndez, M.; Poca, M.A.; López-Bigas, N.; Ramon y Cajal, S.; et al. Molecular Diagnosis of Diffuse Gliomas through Sequencing of Cell-Free Circulating Tumor DNA from Cerebrospinal Fluid. Clin. Cancer Res. 2018, 24, 2812–2819. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Diplas, B.H.; Chen, X.; Wu, Y.; Xiao, X.; Jiang, L.; Geng, Y.; Xu, C.; Sun, Y.; Zhang, P.; et al. Molecular Profiling of Tumors of the Brainstem by Sequencing of CSF-Derived Circulating Tumor DNA. Acta Neuropathol. 2019, 137, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Hickman, R.A.; Miller, A.M.; Arcila, M.E. Cerebrospinal Fluid: A Unique Source of Circulating Tumor DNA with Broad Clinical Applications. Transl. Oncol. 2023, 33, 101688. [Google Scholar] [CrossRef] [PubMed]
- Bark, J.M.; Kulasinghe, A.; Chua, B.; Day, B.W.; Punyadeera, C. Circulating Biomarkers in Patients with Glioblastoma. Br. J. Cancer 2020, 122, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Mo, F.; Pellerino, A.; Soffietti, R.; Rudà, R. Blood–Brain Barrier in Brain Tumors: Biology and Clinical Relevance. Int. J. Mol. Sci. 2021, 22, 12654. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Turner, S.; Peters, K.B.; Desjardins, A.; Gururangan, S.; Sampson, J.H.; McLendon, R.E.; Herndon, J.E.; Jones, L.W.; Kirkpatrick, J.P.; et al. A Review of VEGF/VEGFR-Targeted Therapeutics for Recurrent Glioblastoma. J. Natl. Compr. Cancer Netw. 2011, 9, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Schwaederle, M.; Husain, H.; Fanta, P.T.; Piccioni, D.E.; Kesari, S.; Schwab, R.B.; Banks, K.C.; Lanman, R.B.; Talasaz, A.; Parker, B.A.; et al. Detection Rate of Actionable Mutations in Diverse Cancers Using a Biopsy-Free (Blood) Circulating Tumor Cell DNA Assay. Oncotarget 2016, 7, 9707–9717. [Google Scholar] [CrossRef]
- Piccioni, D.E.; Achrol, A.S.; Kiedrowski, L.A.; Banks, K.C.; Boucher, N.; Barkhoudarian, G.; Kelly, D.F.; Juarez, T.; Lanman, R.B.; Raymond, V.M.; et al. Analysis of Cell-Free Circulating Tumor DNA in 419 Patients with Glioblastoma and Other Primary Brain Tumors. CNS Oncol. 2019, 8, CNS34. [Google Scholar] [CrossRef]
- Bagley, S.J.; Nabavizadeh, S.A.; Mays, J.J.; Till, J.E.; Ware, J.B.; Levy, S.; Sarchiapone, W.; Hussain, J.; Prior, T.; Guiry, S.; et al. Clinical Utility of Plasma Cell-Free DNA in Adult Patients with Newly Diagnosed Glioblastoma: A Pilot Prospective Study. Clin. Cancer Res. 2020, 26, 397–407. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef]
- Oshi, M.; Murthy, V.; Takahashi, H.; Huyser, M.; Okano, M.; Tokumaru, Y.; Rashid, O.M.; Matsuyama, R.; Endo, I.; Takabe, K. Urine as a Source of Liquid Biopsy for Cancer. Cancers 2021, 13, 2652. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, T.R. A Case of Cancer in Which Cells Similar to Those in the Tumours Were Seen in the Blood after Death. Aus. Med. J. 1869, 14, 146–149. [Google Scholar]
- Alba-Bernal, A.; Lavado-Valenzuela, R.; Domínguez-Recio, M.E.; Jiménez-Rodriguez, B.; Queipo-Ortuño, M.I.; Alba, E.; Comino-Méndez, I. Challenges and Achievements of Liquid Biopsy Technologies Employed in Early Breast Cancer. EBioMedicine 2020, 62, 103100. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Shen, L.; Luo, M.; Zhang, K.; Li, J.; Yang, Q.; Zhu, F.; Zhou, D.; Zheng, S.; Chen, Y.; et al. Circulating Tumor Cells: Biology and Clinical Significance. Signal Transduct. Target. Ther. 2021, 6, 404. [Google Scholar] [CrossRef] [PubMed]
- Lessi, F.; Morelli, M.; Franceschi, S.; Aretini, P.; Menicagli, M.; Marranci, A.; Pasqualetti, F.; Gambacciani, C.; Pieri, F.; Grimod, G.; et al. Innovative Approach to Isolate and Characterize Glioblastoma Circulating Tumor Cells and Correlation with Tumor Mutational Status. Int. J. Mol. Sci. 2023, 24, 10147. [Google Scholar] [CrossRef] [PubMed]
- Krol, I.; Castro-Giner, F.; Maurer, M.; Gkountela, S.; Szczerba, B.M.; Scherrer, R.; Coleman, N.; Carreira, S.; Bachmann, F.; Anderson, S.; et al. Detection of Circulating Tumour Cell Clusters in Human Glioblastoma. Br. J. Cancer 2018, 119, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.; Powter, B.; Po, J.W.; Cooper, A.; Garrett, C.; Koh, E.-S.; Sheridan, M.; van Gelder, J.; Darwish, B.; Mckechnie, S.; et al. Isolation of Circulating Tumor Cells from Glioblastoma Patients by Direct Immunomagnetic Targeting. Appl. Sci. 2020, 10, 3338. [Google Scholar] [CrossRef]
- Qi, Y.; Sun, Q.; Deng, G.; Zhang, H.; Xu, Y.; Li, Y.; Huang, S.; Li, Y.; Ye, Z.; Wang, Y.; et al. Identifying Circulating Glioma Cells and Their Clusters as Diagnostic Markers by a Novel Detection Platform. Clin. Transl. Med. 2021, 11, e318. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Holtschmidt, J.; Auer, M.; Heitzer, E.; Lamszus, K.; Schulte, A.; Matschke, J.; Langer-Freitag, S.; Gasch, C.; Stoupiec, M.; et al. Hematogenous Dissemination of Glioblastoma Multiforme. Sci. Transl. Med. 2014, 6, 247ra101. [Google Scholar] [CrossRef]
- Sullivan, J.P.; Nahed, B.V.; Madden, M.W.; Oliveira, S.M.; Springer, S.; Bhere, D.; Chi, A.S.; Wakimoto, H.; Rothenberg, S.M.; Sequist, L.V.; et al. Brain Tumor Cells in Circulation Are Enriched for Mesenchymal Gene Expression. Cancer Discov. 2014, 4, 1299–1309. [Google Scholar] [CrossRef]
- Gatto, L.; Franceschi, E.; Di Nunno, V.; Tosoni, A.; Lodi, R.; Brandes, A.A. Liquid Biopsy in Glioblastoma Management: From Current Research to Future Perspectives. Oncologist 2021, 26, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Saenz-Antoñanzas, A.; Auzmendi-Iriarte, J.; Carrasco-Garcia, E.; Moreno-Cugnon, L.; Ruiz, I.; Villanua, J.; Egaña, L.; Otaegui, D.; Samprón, N.; Matheu, A. Liquid Biopsy in Glioblastoma: Opportunities, Applications and Challenges. Cancers 2019, 11, 950. [Google Scholar] [CrossRef] [PubMed]
- Husain, A.; Mishra, S.; Hadi, R.; Sahu, A.; Kumari, S.; Rastogi, M.; Khurana, R.; Shukla, S.; Siddiqui, M.H.; Husain, N. Dynamics of Cell-Free DNA in Predicting Response in Adult Diffuse Glioma on Chemoradiotherapy. Cancer Genet. 2022, 268–269, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Fontanilles, M.; Marguet, F.; Beaussire, L.; Magne, N.; Pépin, L.-F.; Alexandru, C.; Tennevet, I.; Hanzen, C.; Langlois, O.; Jardin, F.; et al. Cell-Free DNA and Circulating TERT Promoter Mutation for Disease Monitoring in Newly-Diagnosed Glioblastoma. Acta Neuropathol. Commun. 2020, 8, 179. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan, K.; Yekula, A.; Small, J.L.; Rosh, Z.S.; Kang, K.M.; Wang, L.; Lau, S.; Zhang, H.; Lee, H.; Bettegowda, C.; et al. TERT Promoter Mutation Analysis for Blood-Based Diagnosis and Monitoring of Gliomas. Clin. Cancer Res. 2021, 27, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Liu, Z.; Zhu, Y.; Ma, L. Genome-Wide Methylation Analysis of Circulating Tumor DNA: A New Biomarker for Recurrent Glioblastom. Heliyon 2023, 9, e14339. [Google Scholar] [CrossRef] [PubMed]
- Zanganeh, S.; Abbasgholinejad, E.; Doroudian, M.; Esmaelizad, N.; Farjadian, F.; Benhabbour, S.R. The Current Landscape of Glioblastoma Biomarkers in Body Fluids. Cancers 2023, 15, 3804. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Medarova, Z.; Moore, A. Role of MicroRNAs in Glioblastoma. Oncotarget 2021, 12, 1707–1723. [Google Scholar] [CrossRef]
- Makowska, M.; Smolarz, B.; Romanowicz, H. MicroRNAs (MiRNAs) in Glioblastoma Multiforme (GBM)—Recent Literature Review. Int. J. Mol. Sci. 2023, 24, 3521. [Google Scholar] [CrossRef]
- Khristov, V.; Lin, A.; Freedman, Z.; Staub, J.; Shenoy, G.; Mrowczynski, O.; Rizk, E.; Zacharia, B.; Connor, J. Tumor-Derived Biomarkers in Liquid Biopsy of Glioblastoma. World Neurosurg. 2023, 170, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Su, J.; Long, W.; Qin, C.; Wang, X.; Xiao, K.; Li, Y.; Xiao, Q.; Wang, J.; Pan, Y.; et al. LINC00470 Promotes Tumour Proliferation and Invasion, and Attenuates Chemosensitivity through the LINC00470/MiR-134/Myc/ABCC1 Axis in Glioma. J. Cell. Mol. Med. 2020, 24, 12094–12106. [Google Scholar] [CrossRef] [PubMed]
- Chargaff, E.; West, R. The Biological Significance of the Thromboplastic Protein of Blood. J. Biol. Chem. 1946, 166, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding Light on the Cell Biology of Extracellular Vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Irmer, B.; Chandrabalan, S.; Maas, L.; Bleckmann, A.; Menck, K. Extracellular Vesicles in Liquid Biopsies as Biomarkers for Solid Tumors. Cancers 2023, 15, 1307. [Google Scholar] [CrossRef] [PubMed]
- Del Bene, M.; Osti, D.; Faletti, S.; Beznoussenko, G.V.; DiMeco, F.; Pelicci, G. Extracellular Vesicles: The Key for Precision Medicine in Glioblastoma. Neuro-Oncology 2022, 24, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Nguyen, H.P.T.; Jones, J.J.; Stylli, S.S.; Whitehead, C.A.; Paradiso, L.; Luwor, R.B.; Areeb, Z.; Hanssen, E.; Cho, E.; et al. Extracellular Vesicles Secreted by Glioma Stem Cells Are Involved in Radiation Resistance and Glioma Progression. Int. J. Mol. Sci. 2022, 23, 2770. [Google Scholar] [CrossRef] [PubMed]
- Tzaridis, T.; Weller, J.; Bachurski, D.; Shakeri, F.; Schaub, C.; Hau, P.; Buness, A.; Schlegel, U.; Steinbach, J.; Seidel, C.; et al. A Novel Serum Extracellular Vesicle Protein Signature to Monitor Glioblastoma Tumor Progression. Int. J. Cancer 2023, 152, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Garcia, L.M.C.; Peterson, T.E.; Cepeda, M.A.; Johnson, A.J.; Parney, I.F. Isolation and Analysis of Plasma-Derived Exosomes in Patients with Glioma. Front. Oncol. 2019, 9, 651. [Google Scholar] [CrossRef]
- Weller, M.; van den Bent, M.; Tonn, J.C.; Stupp, R.; Preusser, M.; Cohen-Jonathan-Moyal, E.; Henriksson, R.; Le Rhun, E.; Balana, C.; Chinot, O.; et al. European Association for Neuro-Oncology (EANO) Guideline on the Diagnosis and Treatment of Adult Astrocytic and Oligodendroglial Gliomas. Lancet Oncol. 2017, 18, e315–e329. [Google Scholar] [CrossRef]
- Wirsching, H.-G.; Galanis, E.; Weller, M. Glioblastoma. In Malignant Brain Tumors; Springer: Berlin/Heidelberg, Germany, 2016; pp. 381–397. [Google Scholar]
- Ly, K.I.; Wen, P.Y.; Huang, R.Y. Imaging of Central Nervous System Tumors Based on the 2016 World Health Organization Classification. Neurol. Clin. 2020, 38, 95–113. [Google Scholar] [CrossRef] [PubMed]
- Sahm, F.; Capper, D.; Jeibmann, A.; Habel, A.; Paulus, W.; Dirk, T.; von Deimling, A. Addressing Diffuse Glioma as a Systemic Brain Disease with Single-Cell Analysis. Arch. Neurol. 2012, 69, 523. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, M.M.; Recinos, P.F.; Nowacki, A.S.; Schroeder, J.L.; Angelov, L.; Barnett, G.H.; Vogelbaum, M.A. Residual Tumor Volume Versus Extent of Resection: Predictors of Survival after Surgery for Glioblastoma. J. Neurosurg. 2014, 121, 1115–1123. [Google Scholar] [CrossRef] [PubMed]
- Kreth, F.-W.; Thon, N.; Simon, M.; Westphal, M.; Schackert, G.; Nikkhah, G.; Hentschel, B.; Reifenberger, G.; Pietsch, T.; Weller, M.; et al. Gross Total but Not Incomplete Resection of Glioblastoma Prolongs Survival in the Era of Radiochemotherapy. Ann. Oncol. 2013, 24, 3117–3123. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in Adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) Consensus Review on Current Management and Future Directions. Neuro-Oncology 2020, 22, 1073–1113. [Google Scholar] [CrossRef]
- Suchorska, B.; Weller, M.; Tabatabai, G.; Senft, C.; Hau, P.; Sabel, M.C.; Herrlinger, U.; Ketter, R.; Schlegel, U.; Marosi, C.; et al. Complete Resection of Contrast-Enhancing Tumor Volume Is Associated with Improved Survival in Recurrent Glioblastoma—Results from the DIRECTOR Trial. Neuro-Oncology 2016, 18, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, M.; Abi-Said, D.; Fourney, D.R.; Gokaslan, Z.L.; Shi, W.; DeMonte, F.; Lang, F.F.; McCutcheon, I.E.; Hassenbusch, S.J.; Holland, E.; et al. A Multivariate Analysis of 416 Patients with Glioblastoma Multiforme: Prognosis, Extent of Resection, and Survival. J. Neurosurg. 2001, 95, 190–198. [Google Scholar] [CrossRef]
- Shah, A.H.; Mahavadi, A.; Di, L.; Sanjurjo, A.; Eichberg, D.G.; Borowy, V.; Figueroa, J.; Luther, E.; de la Fuente, M.I.; Semonche, A.; et al. Survival Benefit of Lobectomy for Glioblastoma: Moving towards Radical Supramaximal Resection. J. Neuro-Oncol. 2020, 148, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Stummer, W.; Pichlmeier, U.; Meinel, T.; Wiestler, O.D.; Zanella, F.; Reulen, H.-J. Fluorescence-Guided Surgery with 5-Aminolevulinic Acid for Resection of Malignant Glioma: A Randomised Controlled Multicentre Phase III Trial. Lancet Oncol. 2006, 7, 392–401. [Google Scholar] [CrossRef]
- Hadjipanayis, C.G.; Widhalm, G.; Stummer, W. What Is the Surgical Benefit of Utilizing 5-Aminolevulinic Acid for Fluorescence-Guided Surgery of Malignant Gliomas? Neurosurgery 2015, 77, 663–673. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of Radiotherapy with Concomitant and Adjuvant Temozolomide versus Radiotherapy Alone on Survival in Glioblastoma in a Randomised Phase III Study: 5-Year Analysis of the EORTC-NCIC Trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Guzauskas, G.F.; Pollom, E.L.; Stieber, V.W.; Wang, B.C.M.; Garrison, L.P., Jr. Tumor Treating Fields and Maintenance Temozolomide for Newly-Diagnosed Glioblastoma: A Cost-Effectiveness Study. J. Med. Econ. 2019, 22, 1006–1013. [Google Scholar] [CrossRef]
- Keime-Guibert, F.; Chinot, O.; Taillandier, L.; Cartalat-Carel, S.; Frenay, M.; Kantor, G.; Guillamo, J.-S.; Jadaud, E.; Colin, P.; Bondiau, P.-Y.; et al. Radiotherapy for Glioblastoma in the Elderly. N. Engl. J. Med. 2007, 356, 1527–1535. [Google Scholar] [CrossRef]
- Kotecha, R.; Odia, Y.; Khosla, A.A.; Ahluwalia, M.S. Key Clinical Principles in the Management of Glioblastoma. JCO Oncol. Pract. 2023, 19, 180–189. [Google Scholar] [CrossRef]
- Perry, J.R.; Laperriere, N.; O’Callaghan, C.J.; Brandes, A.A.; Menten, J.; Phillips, C.; Fay, M.; Nishikawa, R.; Cairncross, J.G.; Roa, W.; et al. Short-Course Radiation plus Temozolomide in Elderly Patients with Glioblastoma. N. Engl. J. Med. 2017, 376, 1027–1037. [Google Scholar] [CrossRef]
- Niyazi, M.; Andratschke, N.; Bendszus, M.; Chalmers, A.J.; Erridge, S.C.; Galldiks, N.; Lagerwaard, F.J.; Navarria, P.; af Rosenschöld, P.M.; Ricardi, U.; et al. ESTRO-EANO Guideline on Target Delineation and Radiotherapy Details for Glioblastoma. Radiother. Oncol. 2023, 184, 109663. [Google Scholar] [CrossRef] [PubMed]
- Niyazi, M.; Brada, M.; Chalmers, A.J.; Combs, S.E.; Erridge, S.C.; Fiorentino, A.; Grosu, A.L.; Lagerwaard, F.J.; Minniti, G.; Mirimanoff, R.-O.; et al. ESTRO-ACROP Guideline “Target Delineation of Glioblastomas”. Radiother. Oncol. 2016, 118, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, B.J.; Dobelbower, M.C.; Ennis, W.H.; Bag, A.K.; Markert, J.M.; Fiveash, J.B. Patterns of Failure for Glioblastoma Multiforme Following Limited-Margin Radiation and Concurrent Temozolomide. Radiat. Oncol. 2014, 9, 130. [Google Scholar] [CrossRef] [PubMed]
- Jette, D.; Chen, W. Creating a Spread-out Bragg Peak in Proton Beams. Phys. Med. Biol. 2011, 56, N131–N138. [Google Scholar] [CrossRef]
- Matsuda, M.; Mizumoto, M.; Kohzuki, H.; Sugii, N.; Sakurai, H.; Ishikawa, E. High-Dose Proton Beam Therapy ver-sus Conventional Fractionated Radiation Therapy for Newly Diagnosed Glioblastoma: A Propensity Score Match-ing Analysis. Radiat. Oncol. 2023, 18, 38. [Google Scholar] [CrossRef]
- Yang, K.; Wu, Z.; Zhang, H.; Zhang, N.; Wu, W.; Wang, Z.; Dai, Z.; Zhang, X.; Zhang, L.; Peng, Y.; et al. Glioma Targeted Therapy: Insight into Future of Molecular Approaches. Mol. Cancer 2022, 21, 39. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Touat, M.; Alexander, B.M.; Mellinghoff, I.K.; Ramkissoon, S.; McCluskey, C.S.; Pelton, K.; Haidar, S.; Basu, S.S.; Gaffey, S.C.; et al. Buparlisib in Patients with Recurrent Glioblastoma Harboring Phosphatidylinositol 3-Kinase Pathway Activation: An Open-Label, Multicenter, Multi-Arm, Phase II Trial. J. Clin. Oncol. 2019, 37, 741–750. [Google Scholar] [CrossRef]
- Wick, W.; Gorlia, T.; Bady, P.; Platten, M.; van den Bent, M.J.; Taphoorn, M.J.B.; Steuve, J.; Brandes, A.A.; Hamou, M.-F.; Wick, A.; et al. Phase II Study of Radiotherapy and Temsirolimus Versus Radiochemotherapy with Temozolomide in Patients with Newly Diagnosed Glioblastoma without MGMT Promoter Hypermethylation (EORTC 26082). Clin. Cancer Res. 2016, 22, 4797–4806. [Google Scholar] [CrossRef] [PubMed]
- Phuphanich, S.; Raizer, J.; Chamberlain, M.; Canelos, P.; Narwal, R.; Hong, S.; Miday, R.; Nade, M.; Laubscher, K. Phase II Study of MEDI-575, an Anti-Platelet-Derived Growth Factor-α Antibody, in Patients with Recurrent Glioblastoma. J. Neuro-Oncol. 2017, 131, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.; Finocchiaro, G.; Belda-Iniesta, C.; Recht, L.; Brandes, A.A.; Pineda, E.; Mikkelsen, T.; Chinot, O.L.; Balana, C.; Macdonald, D.R.; et al. Randomized, Double-Blind, Placebo-Controlled, Multicenter Phase II Study of Onartuzumab Plus Bevacizumab Versus Placebo Plus Bevacizumab in Patients with Recurrent Glioblastoma: Efficacy, Safety, and Hepatocyte Growth Factor and O6-Methylguanine–DNA Methyltransferase Biomarker Analyses. J. Clin. Oncol. 2017, 35, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Lassman, A.B.; Sepúlveda-Sánchez, J.M.; Cloughesy, T.F.; Gil-Gil, M.J.; Puduvalli, V.K.; Raizer, J.J.; De Vos, F.Y.F.; Wen, P.Y.; Butowski, N.A.; Clement, P.M.J.; et al. Infigratinib in Patients with Recurrent Gliomas and FGFR Alterations: A Multicenter Phase II Study. Clin. Cancer Res. 2022, 28, 2270–2277. [Google Scholar] [CrossRef] [PubMed]
- Giordano, F.A.; Layer, J.P.; Leonardelli, S.; Friker, L.L.; Seidel, C.; Schaub, C.; Turiello, R.; Sperk, E.; Grau, F.; Paech, D.; et al. Radiotherapy and Olaptesed Pegol (NOX-A12) in Partially Resected or Biopsy-Only MGMT-Unmethylated Glioblastoma: Interim Data from the German Multicenter Phase 1/2 GLORIA Trial. J. Clin. Oncol. 2022, 40, 2050. [Google Scholar] [CrossRef]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef] [PubMed]
- Taal, W.; Oosterkamp, H.M.; Walenkamp, A.M.E.; Dubbink, H.J.; Beerepoot, L.V.; Hanse, M.C.J.; Buter, J.; Honkoop, A.H.; Boerman, D.; de Vos, F.Y.F.; et al. Single-Agent Bevacizumab or Lomustine Versus a Combination of Bevacizumab Plus Lomustine in Patients with Recurrent Glioblastoma (BELOB Trial): A Randomised Controlled Phase 2 Trial. Lancet Oncol. 2014, 15, 943–953. [Google Scholar] [CrossRef]
- Lee, A.; Arasaratnam, M.; Chan, D.L.H.; Khasraw, M.; Howell, V.M.; Wheeler, H. Anti-Epidermal Growth Factor Receptor Therapy for Glioblastoma in Adults. Cochrane Database Syst. Rev. 2020, 5, CD013238. [Google Scholar] [CrossRef]
- Hernández, A.; Domènech, M.; Muñoz-Mármol, A.M.; Carrato, C.; Balana, C. Glioblastoma: Relationship between Metabolism and Immunosuppressive Microenvironment. Cells 2021, 10, 3529. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, C.; Reita, D.; Martin, S.; Entz-Werle, N.; Dontenwill, M. Glioblastoma Metabolism: Insights and Therapeutic Strategies. Int. J. Mol. Sci. 2023, 24, 9137. [Google Scholar] [CrossRef] [PubMed]
- Kelly, W.; Duque, A.E.D.; Michalek, J.; Konkel, B.; Caflisch, L.; Chen, Y.; Pathuri, S.C.; Madhusudanannair-Kunnuparampil, V.; Floyd, J.; Brenner, A. Phase II Investigation of TVB-2640 (Denifanstat) with Bevacizumab in Patients with First Relapse High-Grade Astrocytoma. Clin. Cancer Res. 2023, 29, 2419–2425. [Google Scholar] [CrossRef] [PubMed]
- Altwairgi, A.K.; Alghareeb, W.A.; AlNajjar, F.H.; Alhussain, H.; Alsaeed, E.; Balbaid, A.A.O.; Aldanan, S.; Orz, Y.; Alsharm, A.A. Atorvastatin in Combination with Radiotherapy and Temozolomide for Glioblastoma: A Prospective Phase II Study. Investig. New Drugs 2021, 39, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Sener, U.; Ruff, M.W.; Campian, J.L. Immunotherapy in Glioblastoma: Current Approaches and Future Perspectives. Int. J. Mol. Sci. 2022, 23, 7046. [Google Scholar] [CrossRef] [PubMed]
- Bausart, M.; Préat, V.; Malfanti, A. Immunotherapy for Glioblastoma: The Promise of Combination Strategies. J. Exp. Clin. Cancer Res. 2022, 41, 35. [Google Scholar] [CrossRef] [PubMed]
- Himes, B.T.; Geiger, P.A.; Ayasoufi, K.; Bhargav, A.G.; Brown, D.A.; Parney, I.F. Immunosuppression in Glioblastoma: Current Understanding and Therapeutic Implications. Front. Oncol. 2021, 11, 770561. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A Single Dose of Peripherally Infused EGFRvIII-Directed CAR T Cells Mediates Antigen Loss and Induces Adaptive Resistance in Patients with Recurrent Glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor–Modified Virus-Specific T Cells for Progressive Glioblastoma. JAMA Oncol. 2017, 3, 1094. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with Temozolomide for Patients with Newly Diagnosed, EGFRvIII-Expressing Glioblastoma (ACT IV): A Randomised, Double-Blind, International Phase 3 Trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef]
- Jacques, F.H.; Nicholas, G.; Lorimer, I.A.J.; Foko, V.S.; Prevost, J.; Dumais, N.; Milne, K.; Nelson, B.H.; Woulfe, J.; Jansen, G.; et al. Avelumab in Newly Diagnosed Glioblastoma. Neurooncol. Adv. 2021, 3, vdab118. [Google Scholar] [CrossRef] [PubMed]
- Weathers, S.-P.S.; Kamiya-Matsuoka, C.; Harrison, R.A.; Liu, D.D.; Dervin, S.; Yun, C.; Loghin, M.E.; Penas-Prado, M.; Majd, N.; Yung, W.K.A.; et al. Phase I/II Study to Evaluate the Safety and Clinical Efficacy of Atezolizumab (Atezo; APDL1) in Combination with Temozolomide (TMZ) and Radiation in Patients with Newly Diagnosed Glioblastoma (GBM). J. Clin. Oncol. 2020, 38, 2511. [Google Scholar] [CrossRef]
- Weiss, T.; Hemmerle, T.; Roth, P.; Neri, D.; Weller, M. 378TiP A Phase I/II/IIb Study to Evaluate the Safety and Efficacy of the Tumor-Targeting Human Antibody-Cytokine Fusion Protein L19TNF Plus Standard Temozolomide Chemoradiotherapy in Patients with Newly Diagnosed Glioblastoma. Ann. Oncol. 2021, 32, S528. [Google Scholar] [CrossRef]
Figure 1.
Simplified schematic representation of the PI3K/AKT/mTOR, RAS/RAF/MAPK, RB and p53 pathways and how they interact. The activation of the PI3K/AKT/mTOR and RAS/RAF/MAPK pathways by the binding of growth factors to the RTKs plays an important role in cell proliferation. MDM2 and MDM4 are negative regulators of p53. p14 inhibits MDM2, contributing to p53 expression. These pathways interact in different ways, highlighting potential targets of therapeutic intervention. Parts of the figure were drawn using Servier Medical Art licensed under a Creative Commons Attribution 3.0 Unported License.
Figure 1.
Simplified schematic representation of the PI3K/AKT/mTOR, RAS/RAF/MAPK, RB and p53 pathways and how they interact. The activation of the PI3K/AKT/mTOR and RAS/RAF/MAPK pathways by the binding of growth factors to the RTKs plays an important role in cell proliferation. MDM2 and MDM4 are negative regulators of p53. p14 inhibits MDM2, contributing to p53 expression. These pathways interact in different ways, highlighting potential targets of therapeutic intervention. Parts of the figure were drawn using Servier Medical Art licensed under a Creative Commons Attribution 3.0 Unported License.

Figure 2.
Non-coding RNAs (ncRNAs) classification and some lncRNAs and miRNAS with oncogenic (blue) and tumor suppressor (green) potential in glioblastoma. Housekeeping ncRNAs include transfer RNA (tRNA), ribosomal RNA (rRNA), small nuclear RNA (snRNA) and small nucleolar RNA (snoRNA). Regulatory RNAs include long non-coding RNAs and small non-coding RNAs. MicroRNAs (miRNAs), PIWI-interacting RNAs (piRNAs), and tRNA-derived small RNAs (tsRNAs) are small non-coding RNAs.
Figure 2.
Non-coding RNAs (ncRNAs) classification and some lncRNAs and miRNAS with oncogenic (blue) and tumor suppressor (green) potential in glioblastoma. Housekeeping ncRNAs include transfer RNA (tRNA), ribosomal RNA (rRNA), small nuclear RNA (snRNA) and small nucleolar RNA (snoRNA). Regulatory RNAs include long non-coding RNAs and small non-coding RNAs. MicroRNAs (miRNAs), PIWI-interacting RNAs (piRNAs), and tRNA-derived small RNAs (tsRNAs) are small non-coding RNAs.

Figure 3.
Glioblastoma management overview. Glioblastoma diagnosis is based on MRI and biopsy samples. Current standard treatment consists of maximal surgical resection followed by radiation and adjuvant TMZ. Liquid biopsy emerges as a promising tool in the management of this disease, ultimately contributing to the development of targeted therapies. Parts of the figure were drawn using Servier Medical Art licensed under a Creative Commons Attribution 3.0 Unported License.
Figure 3.
Glioblastoma management overview. Glioblastoma diagnosis is based on MRI and biopsy samples. Current standard treatment consists of maximal surgical resection followed by radiation and adjuvant TMZ. Liquid biopsy emerges as a promising tool in the management of this disease, ultimately contributing to the development of targeted therapies. Parts of the figure were drawn using Servier Medical Art licensed under a Creative Commons Attribution 3.0 Unported License.

Figure 4.
Contrast-enhanced MRI of a glioblastoma. (a) Contrast-enhanced T1 image.; (b) T2 image showing oedema.
Figure 4.
Contrast-enhanced MRI of a glioblastoma. (a) Contrast-enhanced T1 image.; (b) T2 image showing oedema.

Figure 5.
(a) Isodose distribution first course to 40 Gy intensity-modulated radiation therapy (IMRT)/volumetric modulated arc therapy (VMAT) radiation technique; (b) isodose distribution boost dose to 60 Gy (IMRT)/(VMAT) radiation technique.
Figure 5.
(a) Isodose distribution first course to 40 Gy intensity-modulated radiation therapy (IMRT)/volumetric modulated arc therapy (VMAT) radiation technique; (b) isodose distribution boost dose to 60 Gy (IMRT)/(VMAT) radiation technique.

Table 2.
Examples of clinical trials using targeted therapies in glioblastoma.
| Trial | Molecular Target | Therapy | Status | |
|---|---|---|---|---|
| Cell signalling pathways | NCT01339052 | PI3K | Buparlisib | Completed (phase II) |
| NCT00943826 | VEGF | Bevacizumab + TMZ | Completed (phase III) | |
| NCT01019434 | mTOR | Temsirolimus + RT | Completed (phase II) | |
| NCT05222802 | EGFR | ERAS-801 | Recruiting (phase I) | |
| NCT02981940 | CDK4/6 | Abemaciclib | Active, not recruiting (phase II) | |
| NCT01268566 | PDGFR | MEDI-575 | Completed (phase II) | |
| NCT01632228 | MET | Onartuzumab | Completed (phase II) | |
| NCT05376800 | MDM2 | Brigimadlin | Recruiting (phase 0/Ia) | |
| NCT01975701 | FGFR | BGJ398 | Completed (phase II) | |
| NCT02340156 | p53 | SGT-53 + TMZ | Terminated (phase II) | |
| NCT02345824 | CDK4/6 | Ribociclib | Unknown status (phase I) | |
| NCT04121455 | CXCL12 | NOX-A12 + RT | Active, not recruiting (phase I/II) | |
| NCT06102525 | hTERT | RZ-001 + Valganciclovir | Not yet recruiting (phase I/IIa) | |
| NCT01582269 | TGF-β | Galunisertib | Active, not recruiting (phase II) | |
| Tumour cell metabolism | NCT04825275 | HK2—glucose metabolism | Posaconazole | Recruiting (phase 0) |
| NCT04587830 | Arginine metabolism | ADI-PEG 20 + RT + TMZ | Recruiting (phase Ib) | |
| NCT03032484 | FASN—lipid metabolism | TVB-2640 + Bevacizumab | Completed (phase II) | |
| NCT04869449 | HK2—glucose metabolism | Ketoconazole | Recruiting (early phase I) | |
| NCT02029573 | HMGCR—cholesterol Metabolism | Atorvastatin + RT + TMZ | Completed (phase II) |
Table 3.
Examples of clinical trials using immunotherapy in glioblastoma.
| Trial | Molecular Target | Type of Therapy | Status |
|---|---|---|---|
| NCT03726515 | EGFRvIII | CAR T + Pembrolizumab | Completed (phase I) |
| NCT00895180 | PDGFR | Monoclonal antibody | Completed (phase II) |
| NCT01454596 | EGFRvIII | CAR T | Completed (phase I/II) |
| NCT00128635 | DNA–histone H1 complex | Monoclonal antibody | All completed (phase I and phase II the NCT00004017) |
| NCT00004017 | |||
| NCT00509301 | |||
| NCT01498328 | EGFRvIII | Vaccine | Completed (phase II) |
| NCT01109095 | HER2 | CAR T | Completed (phase I) |
| NCT04214392 | CLTX | CAR T | Recruiting (phase I) |
| NCT03174197 | PD-1/PD-L1 | Monoclonal antibody with a checkpoint inhibitor function + RT + TMZ | Active, not recruiting (phase I/II) |
| NCT04443010 | EDB-FN | Cytokines + TMZ | Recruiting (phase I/II) |
| NCT03047473 | PD-1/PD-L1 | Monoclonal antibody with a checkpoint inhibitor function | Completed (phase II) |
| NCT02049489 | CD133 | Vaccine | Completed (phase I) |
| NCT05024175 | EGFRvIII | CAR-T | Not yet recruiting (phase I) |
| NCT04047706 | PD-1/PD-L1 | Monoclonal antibody with a checkpoint inhibitor function | Active, not recruiting (phase I) |
| NCT02017717 | PD-1/PD-L1 | Monoclonal antibody with a checkpoint inhibitor function | Active, not recruiting (phase III) |
| NCT04661384 | IL13Rα2 | CAR T | Recruiting (phase I) |
| NCT01480479 | EGFRvIII | Vaccine + TMZ | Completed (phase III) |
| NCT04003649 | IL13Rα2 | CAR T with checkpoint inhibition | Recruiting (phase I) |
TMZ—Temozolomide; CAR T—Chimeric antigen receptor T-cell.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Roda, D.; Veiga, P.; Melo, J.B.; Carreira, I.M.; Ribeiro, I.P. Principles in the Management of Glioblastoma. Genes 2024, 15, 501. https://doi.org/10.3390/genes15040501
AMA Style
Roda D, Veiga P, Melo JB, Carreira IM, Ribeiro IP. Principles in the Management of Glioblastoma. Genes. 2024; 15(4):501. https://doi.org/10.3390/genes15040501
Chicago/Turabian StyleRoda, Domingos, Pedro Veiga, Joana Barbosa Melo, Isabel Marques Carreira, and Ilda Patrícia Ribeiro. 2024. "Principles in the Management of Glioblastoma" Genes 15, no. 4: 501. https://doi.org/10.3390/genes15040501
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.