
Exploring the Role of E6 and E7 Oncoproteins in Cervical Oncogenesis through MBD2/3-NuRD Complex Chromatin Remodeling
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Culture Conditions
2.2. Patient Sample Collection
2.3. HPV DNA Detection and Genotyping
2.4. Design for E6/E7 HPV16 shRNAs, Plasmid Construction and Cloning
2.5. Plasmid DNA Isolation and Sanger Sequencing
2.6. Transfection of E6 and E7 shRNAs in CaSki Cells (Experimental Model)
2.7. Evaluation of Cell Cycle, Proliferation, Apoptosis, and Senescence in Transfected Cells
2.8. Chromatin Immunoprecipitation (ChIP) and Sequencing
2.9. Quantitative Real-Time PCR
2.10. Western Blot Analysis
2.11. Bioinformatic Analysis and Data Visualization
2.12. Pathway, Network and Functional Analysis
2.13. Statistical Analysis
3. Results
3.1. Selection of shRNA Sequences for E6/E7 Viral Genes Silencing
3.2. E6 and E7 Viral Gene Silencing Using shRNA Sequences
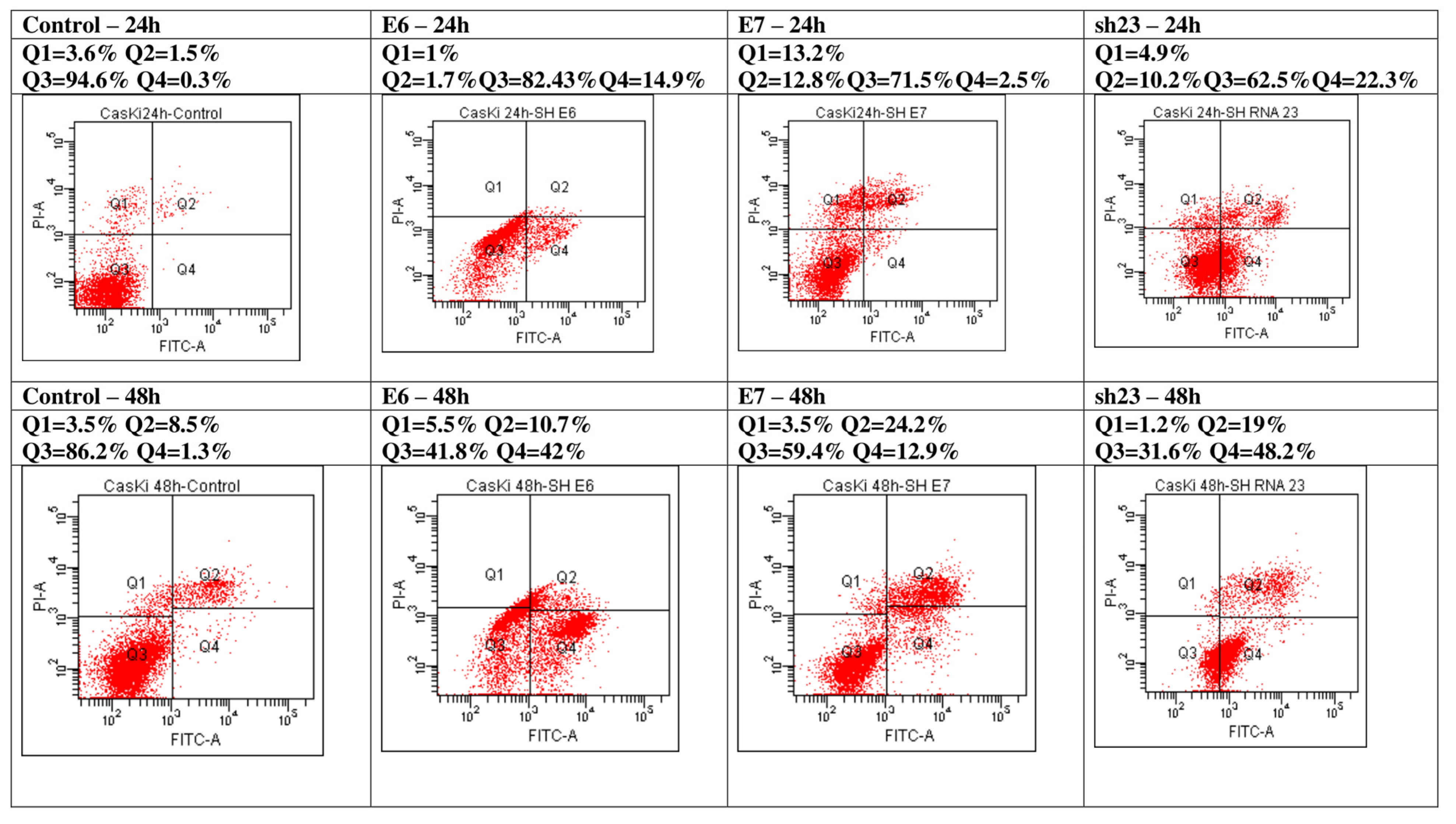
3.3. Evaluation of Apoptosis and Cell Cycle Phases in CasKi Transfected Cells
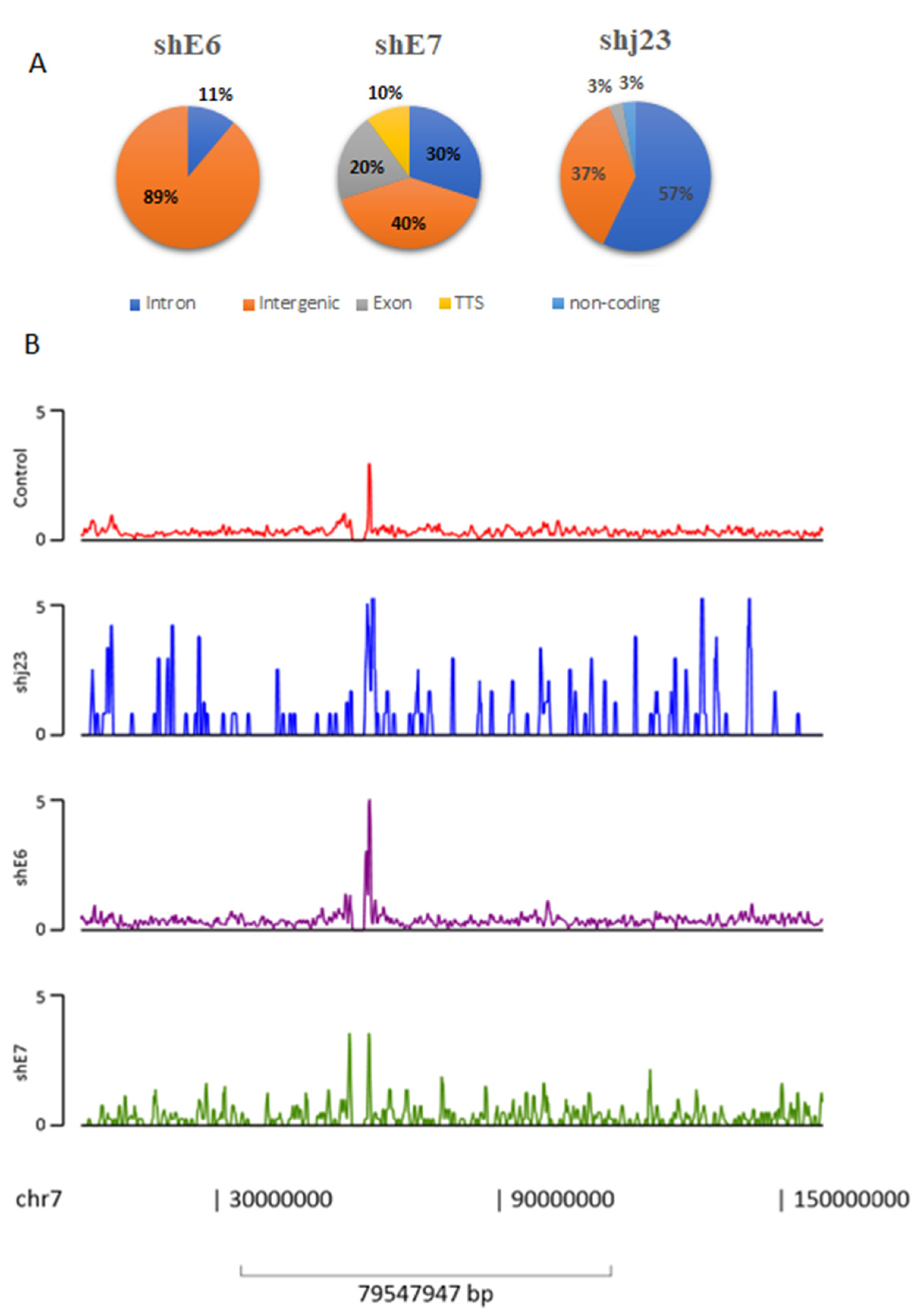
3.4. ChIP-Seq Analysis
3.5. Genome-Wide Characterization of MBD2, MBB3-NuRD Interactions
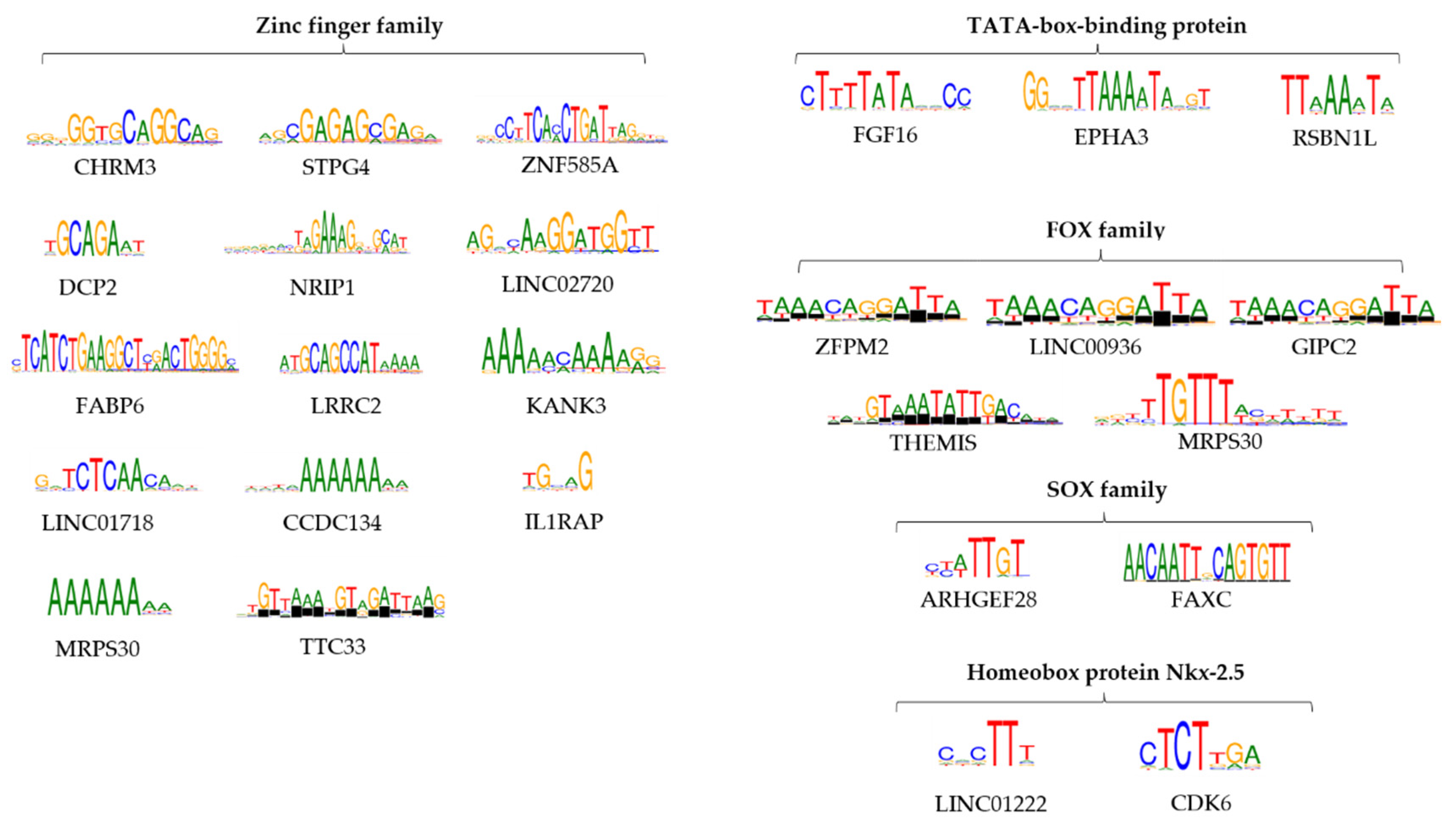
3.6. Peak Annotation
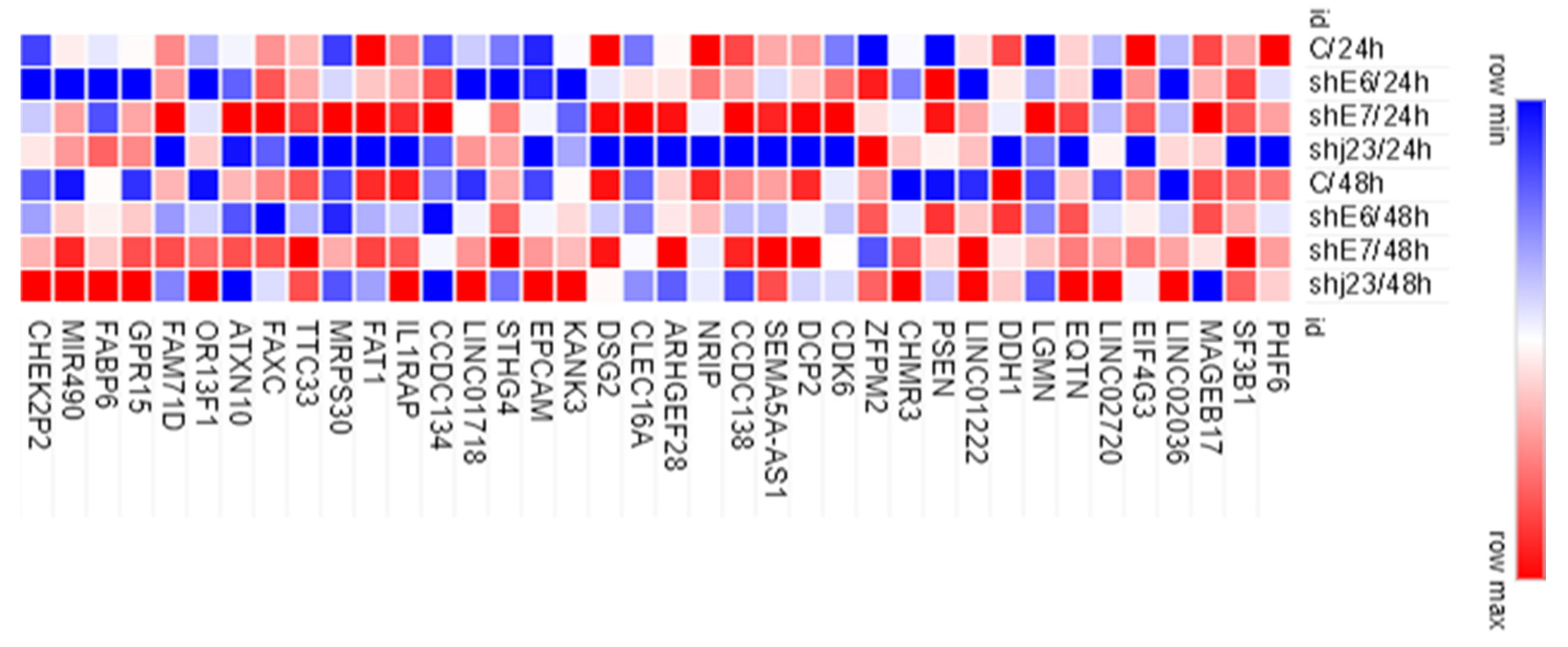
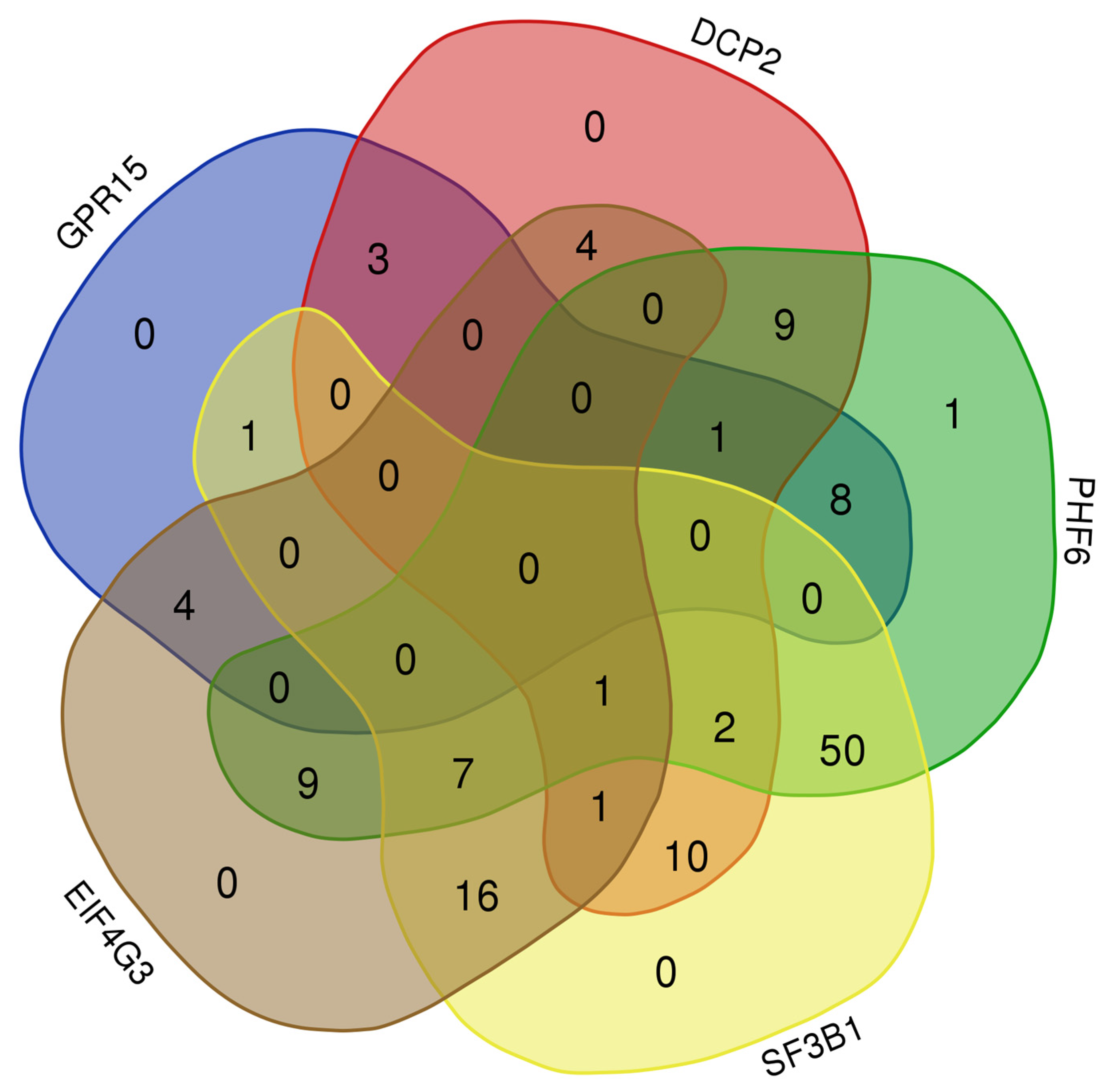
3.7. Evaluation of Gene Expression for ChIPseq-Selected Genes in Experimental Model
3.8. Distribution of HPV Genotypes in Studied Groups
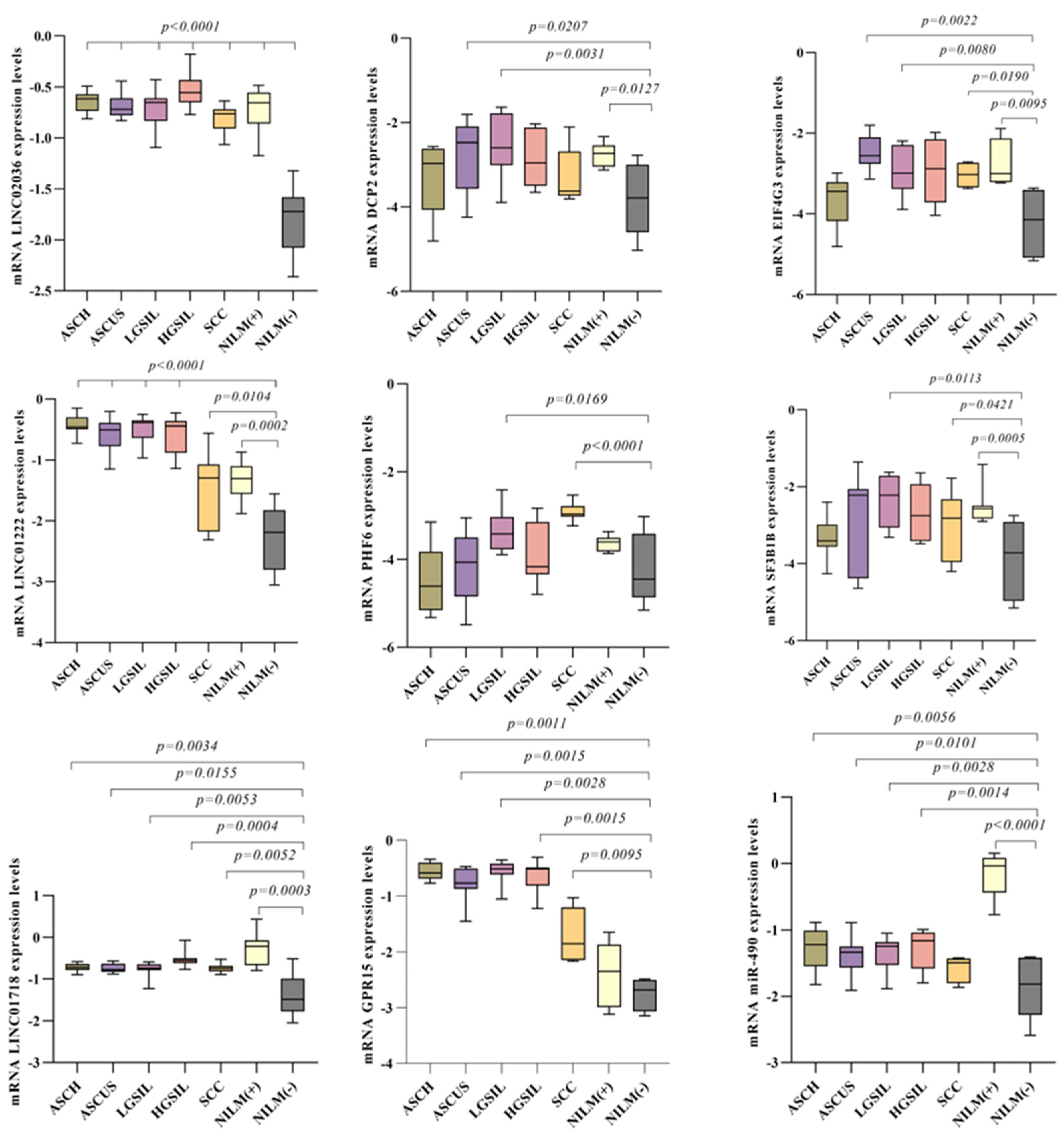
3.9. Evaluation of Selected Gene by Real-Time PCR in Patient’s Samples
3.10. Canonical Pathways, Disease and Functions, Networks
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Ervik, M.; Lam, F.; Laversanne, M.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today; International Agency for Research on Cancer: Lyon, France, 2024; Available online: https://gco.iarc.fr/today/data/factsheets/populations/900-world-fact-sheets.pdf (accessed on 19 March 2024).
- Pal, A.; Kundu, R. Human Papillomavirus E6 and E7: The Cervical Cancer Hallmarks and Targets for Therapy. Front. Microbiol. 2020, 10, 3116. [Google Scholar] [CrossRef]
- Chan, C.K.; Aimagambetova, G.; Ukybassova, T.; Kongrtay, K.; Azizan, A. Human Papillomavirus Infection and Cervical Cancer: Epidemiology, Screening, and Vaccination-Review of Current Perspectives. J. Oncol. 2019, 2019, 3257939. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.M.; Pillai, M.R.; Kumar, R. Prognostic Significance of Dysregulated Epigenomic and Chromatin Modifiers in Cervical Cancer. Cells 2021, 10, 2665. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.Z.; Flam, E.L.; Izumchenko, E.; Danilova, L.V.; Wulf, H.A.; Guo, T.; Singman, D.A.; Afsari, B.; Skaist, A.M.; Considine, M.; et al. Integrated Analysis of Whole-Genome ChIP-Seq and RNA-Seq Data of Primary Head and Neck Tumor Samples Associates HPV Integration Sites with Open Chromatin Marks. Cancer Res. 2017, 77, 6538–6550. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Han, J.W. Targeting epigenetics for cancer therapy. Arch. Pharm. Res. 2019, 42, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Clapier, C.R.; Iwasa, J.; Cairns, B.R.; Peterson, C.L. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 2017, 18, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Torchy, M.P.; Hamiche, A.; Klaholz, B.P. Structure and function insights into the NuRD chromatin remodeling complex. Cell Mol. Life Sci. 2015, 72, 2491–2507. [Google Scholar] [CrossRef]
- Tollefsbol, T.; Balasinor, N.H.; Parte, P.; Singh, D. Epigenetics and Reproductive Health, 1st ed.; Academic Press: Cambridge, MA, USA, 2020; ISBN 9780128197530. eISBN 9780128197547. [Google Scholar]
- Wood, K.H.; Zhou, Z. Emerging Molecular and Biological Functions of MBD2, a Reader of DNA Methylation. Front. Genet. 2016, 7, 93. [Google Scholar] [CrossRef] [PubMed]
- Bostan, M.; Petrica-Matei, G.G.; Radu, N.; Hainarosie, R.; Stefanescu, C.D.; Diaconu, C.C.; Roman, V. The Effect of Resveratrol or Curcumin on Head and Neck Cancer Cells Sensitivity to the Cytotoxic Effects of Cisplatin. Nutrients 2020, 12, 2596. [Google Scholar] [CrossRef]
- Mihaila, M.; Bostan, M.; Hotnog, D.; Ferdes, M.; Brasoveanu, L.I. Real-time analysis of quercetin, resveratrol and/or doxorubicin effects in MCF-7 cells. Rom. Biotechnol. Lett. 2013, 18, 8106–8114. [Google Scholar]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://github.com/taoliu/MACS/ (accessed on 14 January 2024).
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://homer.ucsd.edu/homer/ngs/peakMotifs.html (accessed on 16 January 2024).
- Available online: http://homer.ucsd.edu/homer/ngs/annotation.html (accessed on 16 January 2024).
- Langmead, B.; Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Jönsson, J.; Wang, L.; Kajitani, N.; Schwartz, S. A novel HPV16 splicing enhancer critical for viral oncogene expression and cell immortalization. Nucleic Acids Res. 2024, 11, 316–336. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards suite: From gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinform. 2016, 54, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.L.; Fan, S.; Zhang, S.Y.; Chen, W.X.; Li, Q.X.; Pan, G.K.; Zhang, H.Q.; Wang, W.W.; Weng, B.; Zhang, Z.; et al. SOX8 regulates cancer stem-like properties and cisplatin-induced EMT in tongue squamous cell carcinoma by acting on the Wnt/β-catenin pathway. Int. J. Cancer 2018, 142, 1252–1265. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Zhang, S.; Wei, H.; Pang, X.; Zhang, H. Roles of Foxp3 in the occurrence and development of cervical cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 8717–8730. [Google Scholar] [PubMed]
- Chang, X.; Zhang, J.; Huang, C.; Pang, X.; Luo, Q.; Zhang, H.; Zhang, S. Sex-determining region Y-related high mobility group box (SOX)-2 is overexpressed in cervical squamous cell carcinoma and contributes cervical cancer cell migration and invasion in vitro. Tumor Biol. 2015, 36, 7725–7733. [Google Scholar] [CrossRef] [PubMed]
- Plesa, A.; Iancu, I.V.; Botezatu, A.; Huica, I.; Stoian, M.; Anton, G. The Involvement of Epigenetic Mechanisms in HPV-Induced Cervical Cancer, in Human Papillomavirus—Research in a Global Perspective; Rajkumar, R., Ed.; InTech: London, UK, 2016. [Google Scholar]
- Bornelöv, S.; Reynolds, N.; Xenophontos, M.; Gharbi, S.; Johnstone, E.; Floyd, R.; Ralser, M.; Signolet, J.; Loos, R.; Dietmann, S.; et al. The Nucleosome Remodeling and Deacetylation Complex Modulates Chromatin Structure at Sites of Active Transcription to Fine-Tune Gene Expression. Mol. Cell 2018, 71, 56–72.e4. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, N.; Salmon-Divon, M.; Dvinge, H.; Hynes-Allen, A.; Balasooriya, G.; Leaford, D.; Behrens, A.; Bertone, P.; Hendrich, B. NuRD-mediated deacetylation of H3K27 facilitates recruitment of Polycomb Repressive Complex 2 to direct gene repression. EMBO J. 2012, 31, 593–605. [Google Scholar] [CrossRef]
- Deep, U.; Yadav, R. Motif prediction of abemaciclib in a breast cancer cell line using ChIP-Seq data analysis. Biomed. Res. Ther. 2022, 9, 4971–4985. [Google Scholar] [CrossRef]
- Günther, T.; Theiss, J.M.; Fischer, N.; Grundhoff, A. Investigation of Viral and Host Chromatin by ChIP-PCR or ChIP-Seq Analysis. Curr. Protoc. Microbiol. 2016, 40, 1E.10.1–1E.10.21. [Google Scholar] [CrossRef] [PubMed]
- Ntziachristos, P. PHF6: It is written in the stem cells. Blood 2019, 133, 2461–2462. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, F.; Ruan, K.; Zhang, J.; Mei, Y.; Wu, J.; Shi, Y. Structural and functional insights into the human Börjeson-Forssman-Lehmann syndrome-associated protein PHF6. J. Biol. Chem. 2014, 289, 10069–10083. [Google Scholar] [CrossRef] [PubMed]
- Hajjari, M.; Salavaty, A.; Crea, F.; Kee Shin, Y. The potential role of PHF6 as an oncogene: A genotranscriptomic/proteomic meta-analysis. Tumour Biol. 2016, 37, 5317–5325. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Vacas, J.M.; Herrero-Aguayo, V.; Gómez-Gómez, E.; León-González, A.J.; Sáez-Martínez, P.; Alors-Pérez, E.; Fuentes-Fayos, A.C.; Martínez-López, A.; Sánchez-Sánchez, R.; González-Serrano, T.; et al. Spliceosome component SF3B1 as novel prognostic biomarker and therapeutic target for prostate cancer. Transl. Res. 2019, 212, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Dolatshad, H.; Pellagatti, A.; Fernandez-Mercado, M.; Yip, B.H.; Malcovati, L.; Attwood, M.; Przychodzen, B.; Sahgal, N.; Kanapin, A.A.; Lockstone, H.; et al. Disruption of SF3B1 results in deregulated expression and splicing of key genes and pathways in myelodysplastic syndrome hematopoietic stem and progenitor cells. Leukemia 2015, 29, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, X.; Zhang, H.; Liu, F.; Bi, Y.; Zhang, Y.; Cheng, C.; Liu, J. Knockdown of SF3B1 inhibits cell proliferation, invasion and migration triggering apoptosis in breast cancer via aberrant splicing. Breast Cancer 2020, 27, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Popli, P.; Richters, M.M.; Chadchan, S.B.; Kim, T.H.; Tycksen, E.; Griffith, O.; Thaker, P.H.; Griffith, M.; Kommagani, R. Splicing factor SF3B1 promotes endometrial cancer progression via regulating KSR2 RNA maturation. Cell Death Dis. 2020, 11, 842. [Google Scholar] [CrossRef] [PubMed]
- Franks, T.M.; Lykke-Andersen, J. The control of mRNA decapping and P-body formation. Mol. Cell 2008, 32, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Arribas-Layton, M.; Wu, D.; Lykke-Andersen, J.; Song, H. Structural and functional control of the eukaryotic mRNA decapping machinery. Biochim. Biophys. Acta 2013, 1829, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Song, M.G.; Li, Y.; Kiledjian, M. Multiple mRNA decapping enzymes in mammalian cells. Mol. Cell 2010, 40, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.K.; Unutmaz, D.; KewalRamani, V.N.; Littman, D.R. Expression cloning of new receptors used by simian and human immunodeficiency viruses. Nature 1997, 388, 296–300. [Google Scholar] [CrossRef]
- Unutmaz, D.; KewalRamani, V.N.; Littman, D.R. G protein-coupled receptors in HIV and SIV entry: New perspectives on lentivirus-host interactions and on the utility of animal models. Semin. Immunol. 1998, 10, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Ocón, B.; Pan, J.; Dinh, T.T.; Chen, W.; Ballet, R.; Bscheider, M.; Habtezion, A.; Tu, H.; Zabel, B.A.; Butcher, E.C. A Mucosal and Cutaneous Chemokine Ligand for the Lymphocyte Chemoattractant Receptor GPR15. Front. Immunol. 2017, 8, 1111. [Google Scholar] [CrossRef] [PubMed]
- Reimann, E.; Lättekivi, F.; Keermann, M.; Abram, K.; Kõks, S.; Kingo, K.; Fazeli, A. Multicomponent Biomarker Approach Improves the Accuracy of Diagnostic Biomarkers for Psoriasis Vulgaris. Acta Derm. Venereol. 2019, 99, 1258–1265. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Das, B. eIF4G-an integrator of mRNA metabolism? FEMS Yeast Res. 2016, 16, fow087. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Wu, Y.L.; Wang, Y.X.; Zhu, F.F. Characterization of the microRNA expression profile of cervical squamous cell carcinoma metastases. Asian Pac. J. Cancer Prev. 2014, 15, 1675–1679. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| shRNA | Targeted Nucleotide Sequence in Full-Length E6 and E7 HPV16 Transcript | shRNA Nucleotide Sequence (5′-3′) |
|---|---|---|
| shRNA-1 | 272–292 E6 | GGGAATCCATATGCTGTATGT |
| shRNA-2 | 503–523 E6^E7 joint | GGTCGATGTATGTCTTGTTGC |
| shRNA-3 | 189–209 E6 | AATGTGTGTACTGCAAGCAAC |
| shRNA-4 | 702–722 E7 | GGACAGAGCCCATTACAATAT |
| shRNA-5 | 709–729 E7 | GCCCATTACAATATTGTAACC |
| Total Number of Cases, n = 74 (100%) | |||||||
|---|---|---|---|---|---|---|---|
| NILM HPV− | NILM HPV+ | ASCUS HPV+ | LGSIL HPV+ | ASCH HPV+ | HGSIL HPV+ | SCC HPV+ | |
| Number of cases | n = 11 | n = 9 | n = 12 | n = 12 | n = 11 | n = 10 | n = 9 |
| (% from total number of cases) | (14.86%) | (12.16%) | (16.21%) | (16.21%) | (14.86%) | (13.51%) | (12.16%) |
| Age (Mean ± SD) | 33.88 ± 4.224 | 36.22 ± 7.496 | 32.00 ± 6.663 | 31.45 ± 8.042 | 36.00 ± 8.790 | 37.13 ± 9.062 | 62.44 ± 14.910 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fudulu, A.; Diaconu, C.C.; Iancu, I.V.; Plesa, A.; Albulescu, A.; Bostan, M.; Socolov, D.G.; Stoian, I.L.; Balan, R.; Anton, G.; et al. Exploring the Role of E6 and E7 Oncoproteins in Cervical Oncogenesis through MBD2/3-NuRD Complex Chromatin Remodeling. Genes 2024, 15, 560. https://doi.org/10.3390/genes15050560
Fudulu A, Diaconu CC, Iancu IV, Plesa A, Albulescu A, Bostan M, Socolov DG, Stoian IL, Balan R, Anton G, et al. Exploring the Role of E6 and E7 Oncoproteins in Cervical Oncogenesis through MBD2/3-NuRD Complex Chromatin Remodeling. Genes. 2024; 15(5):560. https://doi.org/10.3390/genes15050560
Chicago/Turabian StyleFudulu, Alina, Carmen Cristina Diaconu, Iulia Virginia Iancu, Adriana Plesa, Adrian Albulescu, Marinela Bostan, Demetra Gabriela Socolov, Irina Liviana Stoian, Raluca Balan, Gabriela Anton, and et al. 2024. "Exploring the Role of E6 and E7 Oncoproteins in Cervical Oncogenesis through MBD2/3-NuRD Complex Chromatin Remodeling" Genes 15, no. 5: 560. https://doi.org/10.3390/genes15050560