
Alu Elements as Novel Regulators of Gene Expression in Type 1 Diabetes Susceptibility Genes?



Abstract
:1. Introduction
2. Materials and Methods
Functional Annotation and Enrichment Analysis
3. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
| Characteristics | T1D genes | CDS | Introns | 5'UTRs | 3'UTRs |
|---|---|---|---|---|---|
| Sequences | 941 | 2419 | 2403 | 2048 | 1758 |
| Total length | 20,074,601 nt | 2,568,639 nt | 64,787,126 nt | 491,916 nt | 1,287,596 nt |
| GC level | 44.54% | 55.81% | 43.65% | 59.87% | 48.10% |
| Average length | 10,531 | 1061 | 7435 | 240 | 732 |
| Max length | 32,759 | 15,018 | 32,753 | 3474 | 11,007 |
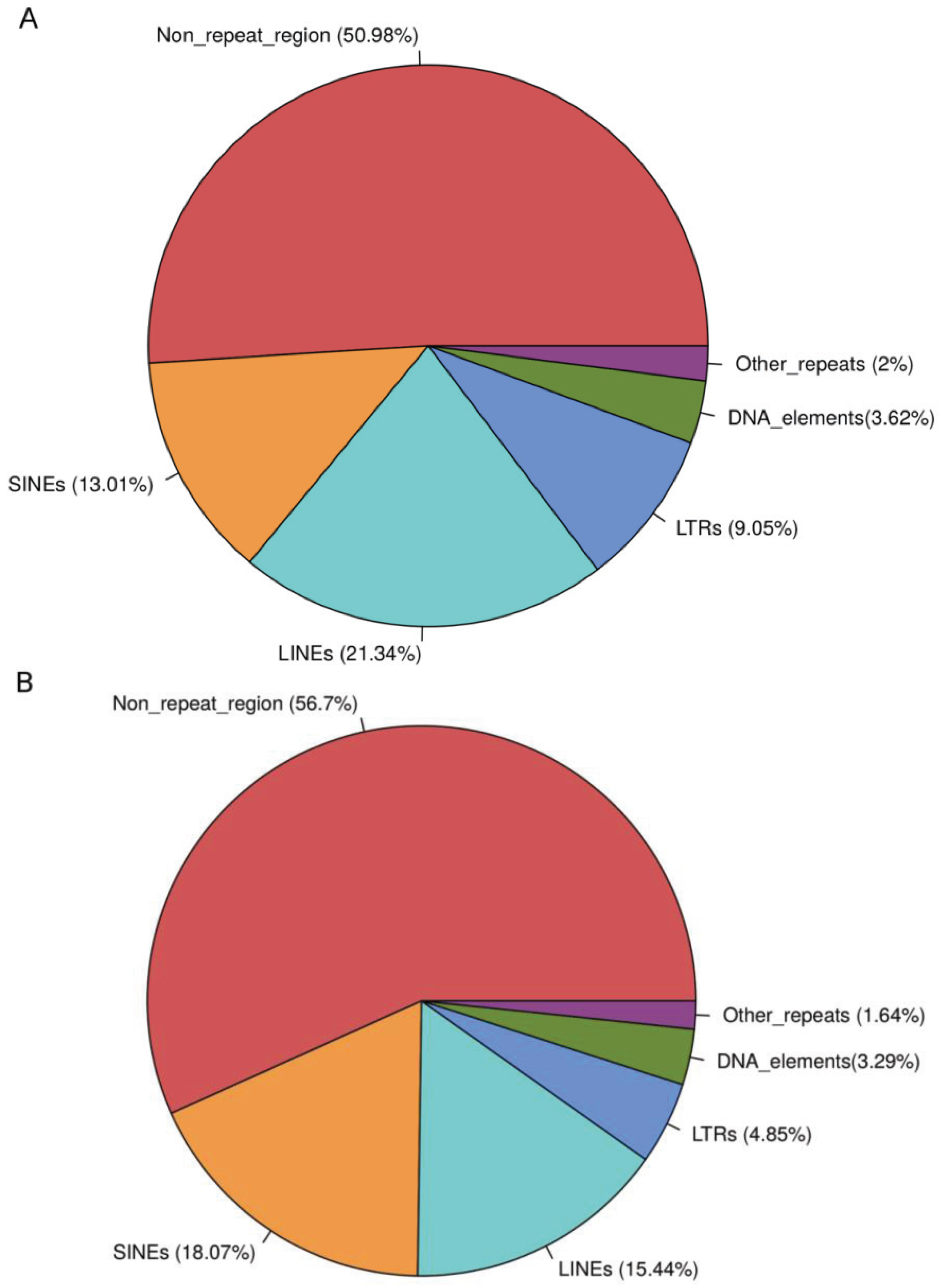
3.1. T1D Genes Are Enriched for Alu Elements

| Category | T1D genes | CDS | Introns | 5'UTRs | 3'UTRs |
|---|---|---|---|---|---|
| Total number of repeats | 37,101 | 660 | 125,952 | 582 | 1025 |
| Total number of Alus | 11,335 | 56 | 40,990 | 68 | 217 |
| Sequences harboring repeats | 81.08% | 20.95% | 92.09% | 20.41% | 30.48% |
| Sequences harboring Alus | 59.29% | 1.77% | 80.44% | 3% | 8.02% |
| One Alu element occurrence per | 1771 nt (1.7 kb) | 45,868 nt (45 kb) | 1580 nt (1.5 kb) | 7234 nt (7.2 kb) | 5933 nt (5.9 kb) |
| Percentage sequence covered by Alus | 15.03% | 0.17% | 16.53% | 1.83% | 3.88% |
3.2. Embedded Alu Elements within T1D UTRs
| T1D Gene | Total Repeats | SINEs | Alu Elements |
|---|---|---|---|
| RAD51B | 1619 | 528 | 278 |
| AFF3 | 925 | 327 | 212 |
| RPH3A | 861 | 319 | 127 |
| GLIS3 | 826 | 221 | 102 |
| CUX2 | 805 | 387 | 221 |
| DOK6 | 664 | 153 | 100 |
| BACH2 | 589 | 178 | 98 |
| SKAP2 | 515 | 195 | 137 |
| HECTD4 | 488 | 260 | 230 |
| CLEC16A | 468 | 201 | 121 |
| CTD-3088G3.8 | 427 | 212 | 151 |
| FBXL20 | 414 | 274 | 267 |
| ATXN2 | 358 | 210 | 196 |
| CFDP1 | 351 | 174 | 158 |
| PTPN2 | 290 | 144 | 125 |
| MTMR3 | 273 | 135 | 118 |
| RP11-57A19.4 | 255 | 155 | 142 |
| PTPN11 | 248 | 140 | 128 |
| CDK12 | 233 | 165 | 155 |
| MORF4L1 | 219 | 135 | 116 |
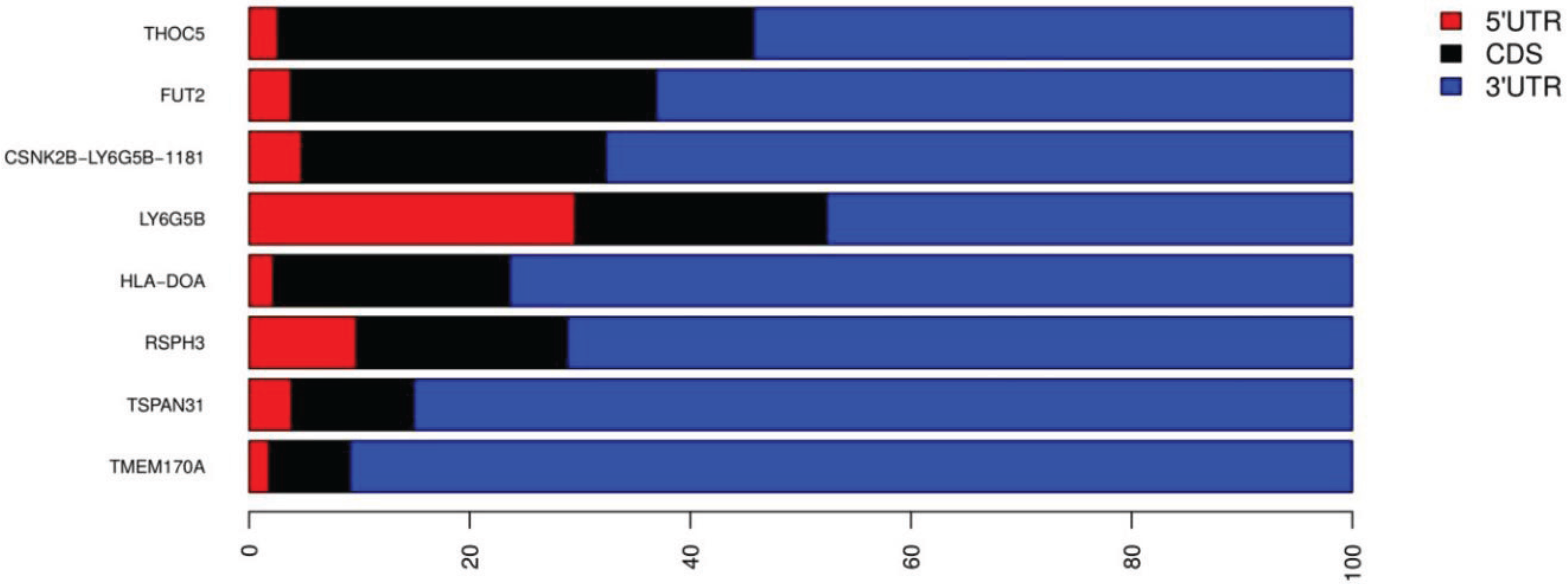
3.3. Inverted Alu Repeats (IRAlus) in T1D 3'UTRs
| Gene Name | Transcript ID | Total Alus | Alu subfamily | Alu Length | Alu Direction | Transcript Biotype |
|---|---|---|---|---|---|---|
| CEP76 | ENST00000593250 | 6 | AluSx, AluSx, AluSp, AluSp, Alu, AluSz6 | 149; 292; 293; 303; 49; 115 | a;s;s;s;s;s | Nonsense-mediated decay |
| TSPAN31 | ENST00000547992 | 5 | AluSx, AluSx, FLAM_C, AluSc, FRAM | 296; 310; 111; 289; 198 | a;a;s;s;s | Protein coding |
| THOC5 | ENST00000490103 | 4 | AluSz, AluJr, AluJo, AluSx | 297; 303; 296; 275 | s;s;a;s | Protein coding |
| RSPH3 | ENST00000367069 | 4 | AluSp, AluJb, AluSc, AluSc | 306; 315; 291; 284 | s;a;s;s | Protein coding |
| TMEM170A | ENST00000357613 | 4 | AluSg4, AluJb, AluY, AluSx1 | 303; 272; 288; 300 | a;s;a;a | Protein coding |
| TMEM170A | ENST00000568559 | 4 | AluSg4, AluJb, AluY, AluSx1 | 216; 241; 288; 308 | a;s;a;a | Nonsense-mediated decay |
| FUT2 | ENST00000425340 | 3 | AluSz, AluSg, FAM | 311; 307; 156 | s;a;s | Protein coding |
| CTD-3088G3.8 | ENST00000595170 | 3 | AluSz6, AluSg, AluSx1 | 100; 75; 311 | s;a;s | Nonsense-mediated decay |
| HLA-DOA | ENST00000229829 | 2 | AluJr, AluSc | 313; 283 | s;a | Protein coding |
| CSNK2B- LY6G5B-1181 | ENST00000409691 | 2 | AluSx3, AluSz | 292; 290 | a;s | Protein coding |
| CSNK2B- LY6G5B-1181 | ENST00000375880 | 2 | AluSx3, AluSz | 292; 290 | a;s | Protein coding (Major isoform) |
| LY6G5B | ENST00000375864 | 2 | AluSx3, AluSz | 292; 290 | a;s | Protein coding |
| SUOX | ENST00000550065 | 2 | AluSq, AluSx | 216; 200 | s;a | Nonsense-mediated decay |


3.4. T1D Genes Harboring Alu Repeats Are Enriched in Immune-Mediated Processes
| Term | Count | % | p-Value | Bonferroni | |
|---|---|---|---|---|---|
| Biological Process (BP_FAT) | |||||
| 1 | antigen processing and presentation | 19 | 5.22 | 2.12e−14 | 3.98e−11 |
| 2 | immune response | 46 | 12.64 | 5.77e−13 | 1.08e−09 |
| 3 | defense response | 38 | 10.44 | 8.29e−10 | 1.56e−06 |
| 4 | positive regulation of immune system process | 22 | 6.04 | 7.71e−09 | 1.45e−05 |
| 5 | positive regulation of immune response | 16 | 4.40 | 1.43e−07 | 2.69e−04 |
| 6 | positive regulation of response to stimulus | 19 | 5.22 | 8.25e−07 | 1.55e−03 |
| 7 | response to unfolded protein | 10 | 2.75 | 9.00e−06 | 0.02 |
| 8 | regulation of T cell activation | 12 | 3.30 | 1.71e−05 | 0.03 |
| 9 | inflammatory response | 20 | 5.49 | 2.00e−05 | 0.04 |
| 10 | regulation of leukocyte activation | 14 | 3.85 | 2.16e−05 | 0.04 |
| Cellular Component (CC_FAT) | |||||
| 1 | MHC protein complex | 14 | 3.85 | 3.07e−11 | 9.33e−09 |
| 2 | plasma membrane part | 75 | 20.60 | 1.34e−07 | 4.06e−05 |
| 3 | integral to plasma membrane | 42 | 11.54 | 9.18e−05 | 0.03 |
| 4 | intrinsic to plasma membrane | 42 | 11.54 | 1.50e−04 | 0.04 |
| KEGG PATHWAY | Count | % | p-value | Bonferroni | |
|---|---|---|---|---|---|
| 1 | Allograft rejection | 12 | 3.30 | 5.40e−10 | 7.29e−08 |
| 2 | Type 1 diabetes mellitus | 12 | 3.30 | 3.39e−09 | 4.58e−07 |
| 3 | Antigen processing and presentation | 15 | 4.12 | 1.10e−08 | 1.48e−06 |
| 4 | Graft-versus-host disease | 11 | 3.02 | 2.28e−08 | 3.08e−06 |
| 5 | Autoimmune thyroid disease | 12 | 3.30 | 3.12e−08 | 4.21e−06 |
| 6 | Viral myocarditis | 13 | 3.57 | 1.28e−07 | 1.73e−05 |
| 7 | Intestinal immune network for IgA production | 11 | 3.02 | 2.38e−07 | 3.22e−05 |
| 8 | Cell adhesion molecules (CAMs) | 15 | 4.12 | 4.03e−06 | 5.44e−04 |
| 9 | Systemic lupus erythematosus | 13 | 3.57 | 5.11e−06 | 6.89e−04 |
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cordaux, R.; Batzer, M.A. The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 2009, 10, 691–703. [Google Scholar] [PubMed]
- Batzer, M.A.; Deininger, P.L. Alu repeats and human genomic diversity. Nat. Rev. Genet. 2002, 3, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Zhang, F.; Lupski, J.R. Mechanisms for human genomic rearrangements. Pathogenetics 2008. [Google Scholar] [CrossRef] [PubMed]
- Kolomietz, E.; Meyn, M.S.; Pandita, A.; Squire, J.A. The role of Alu repeat clusters as mediators of recurrent chromosomal aberrations in tumors. Genes Chromosomes Cancer 2002, 35, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-L.; DeCerbo, J.N.; Carmichael, G.G. Alu element-mediated gene silencing. EMBO J. 2008, 27, 1694–1705. [Google Scholar] [CrossRef] [PubMed]
- Deininger, P. Alu elements: Know the SINEs. Genome Biol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.K.; Pandey, R.; Jha, V.; Mukerji, M. Transcriptome-wide expansion of non-coding regulatory switches: Evidence from co-occurrence of Alu exonization, antisense and editing. Nucleic Acids Res. 2013, 41, 2121–2137. [Google Scholar] [CrossRef] [PubMed]
- Daskalova, E.; Baev, V.; Rusinov, V.; Minkov, I. 3'UTR-located ALU elements: Donors of potential miRNA target sites and mediators of network miRNA-based regulatory interactions. Evol. Bioinform. Online 2006, 2, 103–120. [Google Scholar]
- Lehnert, S.; van Loo, P.; Thilakarathne, P.J.; Marynen, P.; Verbeke, G.; Schuit, F.C. Evidence for co-evolution between human microRNAs and Alu-repeats. PLoS ONE 2009, 4, e4456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borchert, G.M.; Gilmore, B.L.; Spengler, R.M.; Xing, Y.; Lanier, W.; Bhattacharya, D.; Davidson, B.L. Adenosine deamination in human transcripts generates novel microRNA binding sites. Hum. Mol. Genet. 2009, 18, 4801–4807. [Google Scholar] [CrossRef] [PubMed]
- Grover, D.; Majumder, P.P.; Rao, C.B.; Brahmachari, S.K.; Mukerji, M. Nonrandom distribution of Alu elements in genes of various functional categories: Insight from analysis of human chromosomes 21 and 22. Mol. Biol. Evol. 2003, 20, 1420–1424. [Google Scholar] [CrossRef] [PubMed]
- Häsler, J.; Strub, K. Alu elements as regulators of gene expression. Nucleic Acids Res. 2006, 34, 5491–5497. [Google Scholar] [CrossRef] [PubMed]
- Maas, S.; Kawahara, Y.; Tamburro, K.M.; Nishikura, K. A-to-I RNA editing and human disease. RNA Biol. 2006, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Capshew, C.R.; Dusenbury, K.L.; Hundley, H.A. Inverted Alu dsRNA structures do not affect localization but can alter translation efficiency of human mRNAs independent of RNA editing. Nucleic Acids Res. 2012, 40, 8637–8645. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Maquat, L.E. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3' UTRs via Alu elements. Nature 2011, 470, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Kapusta, A.; Kronenberg, Z.; Lynch, V.J.; Zhuo, X.; Ramsay, L.; Bourque, G.; Yandell, M.; Feschotte, C. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS Genet. 2013, 9, e1003470. [Google Scholar] [CrossRef] [PubMed]
- Deininger, P.L.; Batzer, M.A. Alu repeats and human disease. Mol. Genet. Metab. 1999, 67, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Wallace, M.R.; Andersen, L.B.; Saulino, A.M.; Gregory, P.E.; Glover, T.W.; Collins, F.S. A de novo Alu insertion results in neurofibromatosis type 1. Nature 1991, 353, 864–866. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, A.; Dunbar, T.; Chen, P.; Godmilow, L.; Ganguly, T. Exon skipping caused by an intronic insertion of a young Alu Yb9 element leads to severe hemophilia A. Hum. Genet. 2003, 113, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.H.; Kaur, S.; Brorsson, C.A.; Pociot, F. Effects of GWAS-associated genetic variants on lncRNAs within IBD and T1D candidate loci. PLoS ONE 2014, 9, e105723. [Google Scholar] [CrossRef] [PubMed]
- Groop, L.; Pociot, F. Genetics of diabetes—Are we missing the genes or the disease? Mol. Cell. Endocrinol. 2014, 382, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, R.J.; Kähäri, A.; Haider, S.; Zamora, J.; Proctor, G.; Spudich, G.; Almeida-King, J.; Staines, D.; Derwent, P.; Kerhornou, A.; et al. Ensembl BioMarts: A hub for data retrieval across taxonomic space. Database Oxf. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-3.0. 1996–2010. Available online: http://www.repeatmasker.org (accessed on 20 May 2015).
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Bernhart, S.H.; Höner Zu Siederdissen, C.; Flamm, C.; Hofacker, I.L.; Stadler, P.F.; Tafer, H. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Clayton, D.G.; Concannon, P.; Akolkar, B.; Cooper, J.D.; Erlich, H.A.; Julier, C.; Morahan, G.; Nerup, J.; Nierras, C.; et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 2009, 41, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.-H.; Yeh, C.-T. A gene expression restriction network mediated by sense and antisense Alu sequences located on protein-coding messenger RNAs. BMC Genomics 2013, 14, 325. [Google Scholar] [CrossRef] [PubMed]
- Athanasiadis, A.; Rich, A.; Maas, S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2004, 2, e391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levanon, E.Y.; Eisenberg, E.; Yelin, R.; Nemzer, S.; Hallegger, M.; Shemesh, R.; Fligelman, Z.Y.; Shoshan, A.; Pollock, S.R.; Sztybel, D.; et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat. Biotechnol. 2004, 22, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Scadden, A.D.J. Inosine-containing dsRNA binds a stress-granule-like complex and downregulates gene expression in trans. Mol. Cell 2007, 28, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Vitali, P.; Scadden, A.D.J. Double-stranded RNAs containing multiple IU pairs are sufficient to suppress interferon induction and apoptosis. Nat. Struct. Mol. Biol. 2010, 17, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaur, S.; Pociot, F. Alu Elements as Novel Regulators of Gene Expression in Type 1 Diabetes Susceptibility Genes? Genes 2015, 6, 577-591. https://doi.org/10.3390/genes6030577
Kaur S, Pociot F. Alu Elements as Novel Regulators of Gene Expression in Type 1 Diabetes Susceptibility Genes? Genes. 2015; 6(3):577-591. https://doi.org/10.3390/genes6030577
Chicago/Turabian StyleKaur, Simranjeet, and Flemming Pociot. 2015. "Alu Elements as Novel Regulators of Gene Expression in Type 1 Diabetes Susceptibility Genes?" Genes 6, no. 3: 577-591. https://doi.org/10.3390/genes6030577