
Chromatin Dynamics in Vivo: A Game of Musical Chairs

Abstract
:1. Introduction

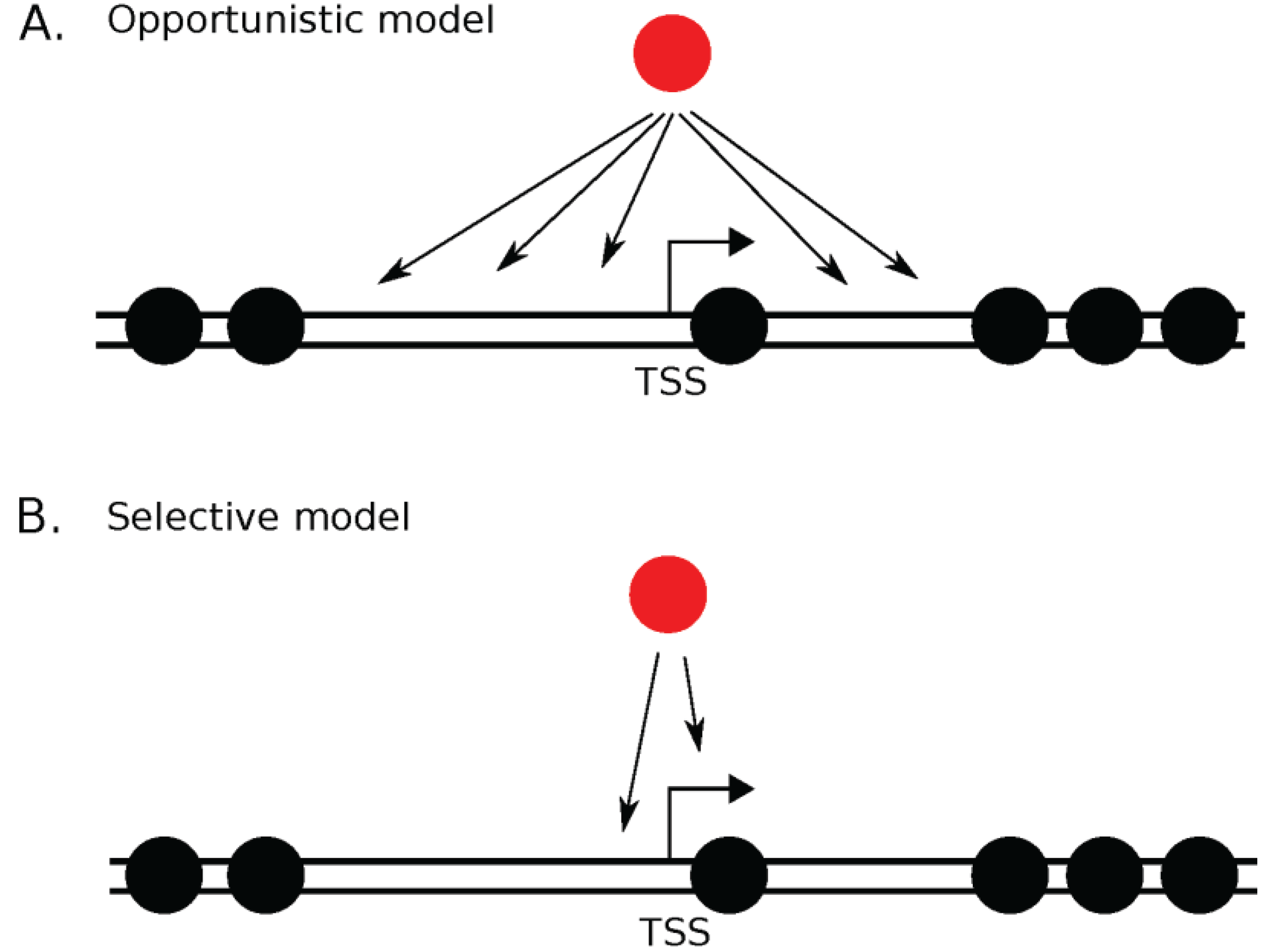{kind=link}
{kind=link}
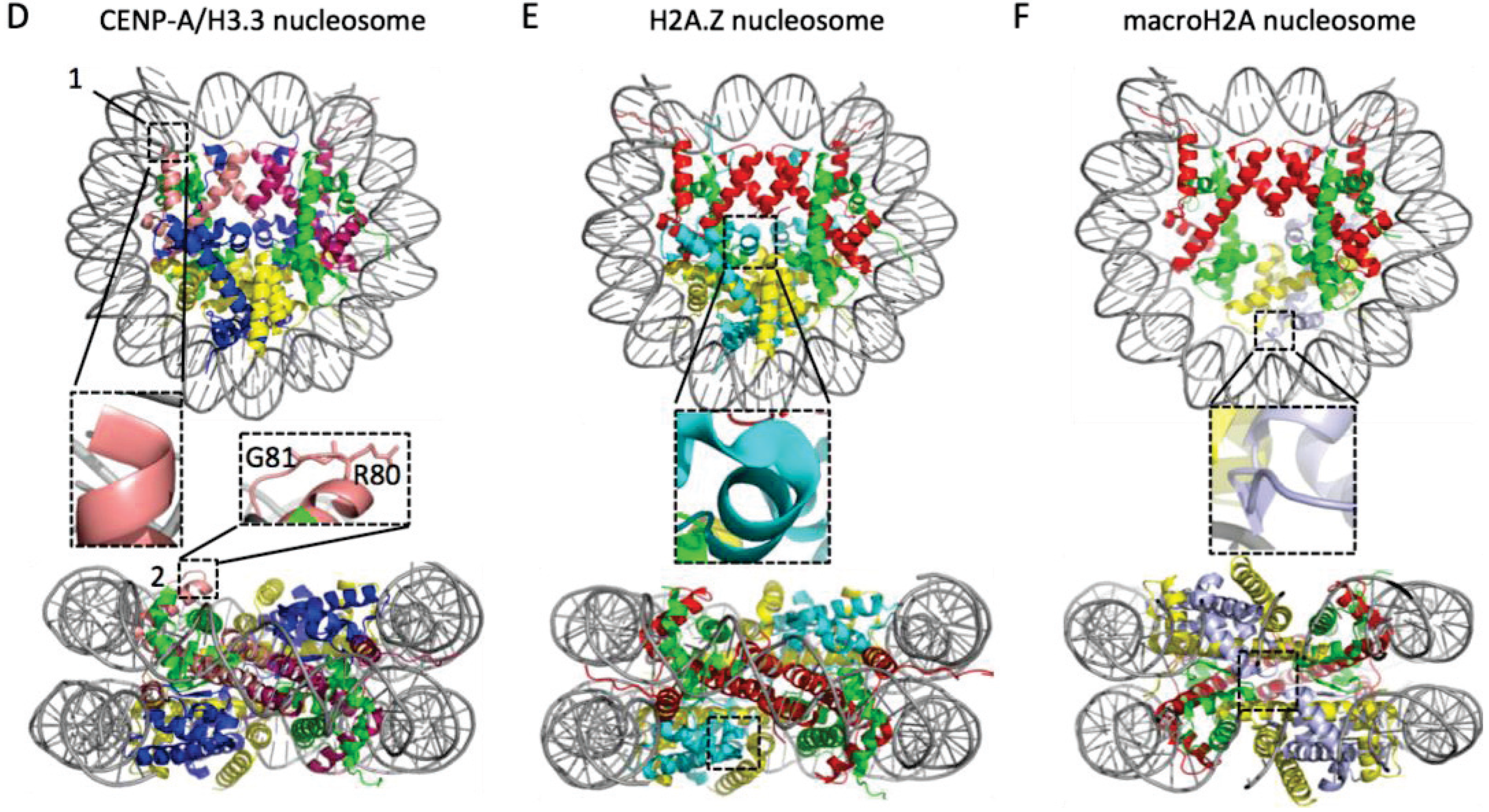{kind=link}
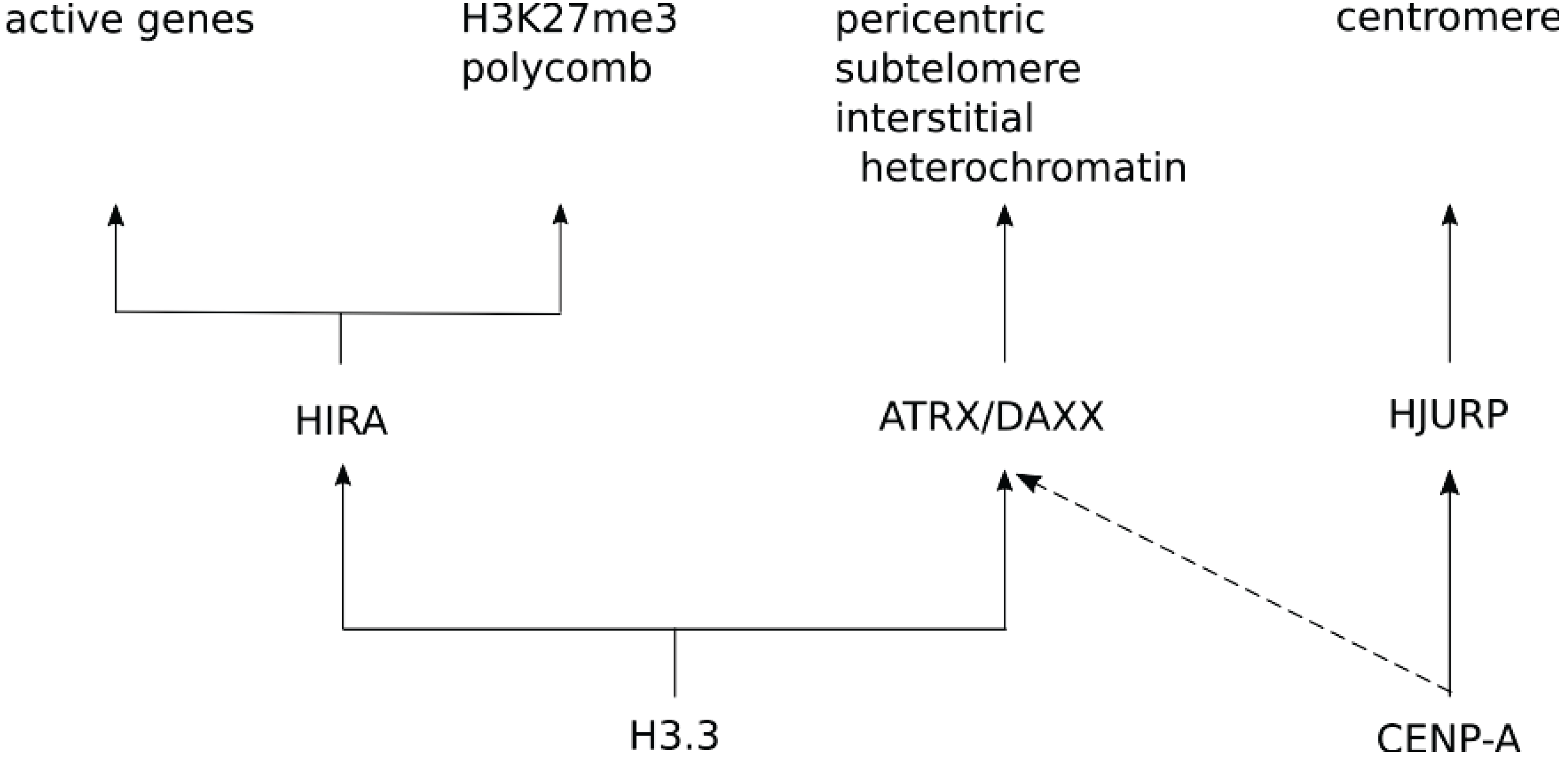{kind=link}
| Histone | Genes | Replication | Chaperone | Function | Knockout/Knockdown Phenotype | Refs. |
|---|---|---|---|---|---|---|
| H2A | HIST2H2A (cluster) | independent, dependent | FACT, NAP-1 | Canonical | N.D. | [1,15] |
| H2A.X | H2AFX | independent | FACT | Phosphorylated form marks ssDNA breaks | Genomic instability, growth retardation, immune deficiency, male infertility | [15,16,17] |
| macroH2A | H2AFY *, H2AFY2 | independent | APLF | Contains macro domain, enriched on inactivated X chromosome | Impairs pre- and postnatal growth, interferes with reproductive efficiency | [15,18,19] |
| H2A.Z | H2AFZ * | independent | Tip60, SWR1 | Contains acidic-patch, accumulation at +1 nucleosome of highly expressed genes | Embryonically lethal (E4.5–E7.5), impairs cellular proliferation, arrest in G1/S | [15,20,21,22,23,24,25,26,27] |
| H2A.B | H2AFB1, H2AFB2, H2AFB3 | independent | NAP-1 | Assoc. with active genes; strongly expressed in testis | Reduced efficiency in mRNA splicing, | [15,28,29,30,31] |
| H2B | H2BFM, H2BFS, H2BFWT, HIST2H2 (cluster) | independent, dependent | NAP-1 | Canonical, monoubiquitinated form regulate transcription | N.D. | [1,15,32] |
| H3.1 | HIST3H3 (cluster) | dependent | CAF-1, ASF1a, ASF1b | Canonical | N.D. | [1,15,32] |
| H3.2 | HIST2H3C (cluster) | dependent | CAF-1, ASF1b | Canonical | N.D. | [15,32] |
| H3.3 | H3F3A, H3F3B | independent | HIRA, ASF1a, ASF1b, DEK, ARTX/DAXX | Imprinted paternal genes; active genes, accumulation in senescent cells | infertility, genome instability, defective cell division and chromosome segregation | [15,32,33,34,35,36,37,38,39,40,41,42,43] |
| CENP-A | CENPA * | independent | HJURP, DAXX, RbAP46/48 | Centromere-specific, incorporated in early G1 | Chromosome missegregation; embryonically lethal | [15,44,45,46,47,48,49,50,51] |
| H4 | HIST4H4 (cluster) | dependent | CAF-1 | Canonical | N.D. | [1,15,32] |

2. H3.3


3. CENP-A
4. Do CENP-A and H3.3 Compete for Chaperones?

5. H2A.Z
6. macroH2A
7. Evolution of Histone Variants
8. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [PubMed]
- Efroni, S.; Carmel, L.; Schaefer, C.G.; Buetow, K.H. Superposition of transcriptional behaviors determines gene state. PLoS ONE 2008, 3, e2901. [Google Scholar] [CrossRef] [PubMed]
- Fussner, E.; Strauss, M.; Djuric, U.; Li, R.; Ahmed, K.; Hart, M.; Ellis, J.; Bazett-Jones, D.P. Open and closed domains in the mouse genome are configured as 10-nm chromatin fibres. EMBO Rep. 2012, 13, 992–996. [Google Scholar] [CrossRef] [PubMed]
- Joti, Y.; Hikima, T.; Nishino, Y.; Kamada, F.; Hihara, S.; Takata, H.; Ishikawa, T.; Maeshima, K. Chromosomes without a 30-nm chromatin fiber. Nucleus 2012, 3, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Quénet, D.; McNally, J.G.; Dalal, Y. Through thick and thin: The conundrum of chromatin fibre folding in vivo. EMBO Rep. 2012, 13, 943–944. [Google Scholar] [CrossRef] [PubMed]
- Malik, H.S.; Henikoff, S. Phylogenomics of the nucleosome. Nat. Struct. Biol. 2003, 10, 882–891. [Google Scholar] [CrossRef] [PubMed]
- Rieder, D.; Trajanoski, Z.; McNally, J.G. Transcription factories. Front. Genet. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Belmont, A.S. Visualizing chromosome dynamics with GFP. Trends Cell Biol. 2001, 11, 250–257. [Google Scholar] [CrossRef]
- Dion, V.; Gasser, S.M. Chromatin movement in the maintenance of genome stability. Cell 2013, 152, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Hirara, S.; Pack, C.G.; Kaizu, K.; Tani, T.; Hanafusa, T.; Nozaki, T.; Takemoto, S.; Yoshimi, T.; Yokota, H.; Imamoto, N.; et al. Local nucleosome dynamics facilitate chromatin accessibility in living mammalian cells. Cell Rep. 2012, 2, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
- Van Bortle, K.; Corces, V.G. The role of chromatin insulators in nuclear architecture and genome function. Curr. Opin. Genet. Dev. 2013, 23, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Tai, P.W.; Zaidi, S.K.; Wu, H.; Grandy, R.A.; Montecino, M.; van Wijnen, A.J.; Lian, J.B.; Stein, G.S.; Stein, J.L. The dynamic architectural and epigenetic nuclear landscape: Developing the genomic almanac of biology and disease. J. Cell. Physiol. 2014, 229, 711–727. [Google Scholar] [CrossRef] [PubMed]
- Ricci, M.A.; Manzo, C.; García-Parajo, M.F.; Lakadamyali, M.; Cosma, M.P. Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo. Cell 2015, 160, 1145–1158. [Google Scholar] [CrossRef] [PubMed]
- Cremer, T.; Cremer, M.; Hübner, B.; Strickfaden, H.; Smeets, D.; Popken, J.; Sterr, M.; Markaki, Y.; Rippe, K.; Cremer, C. The 4D nucleome: Evidence for a dynamic nuclear landscape based on co-aligned active and inactive nuclear compartments. FEBS Lett. 2015. [Google Scholar] [CrossRef] [PubMed]
- Maze, I.; Noh, K.M.; Soshnev, A.A.; Allis, C.D. Every amino acid matters: Essential contributions of histone variants to mammalian development and disease. Nat. Rev. Genet. 2014, 15, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Celeste, A.; Petersen, S.; Romanienko, P.J.; Fernandez-Capetillo, O.; Chen, H.T.; Sedelnikova, O.A.; Reina-San-Martin, B.; Coppola, V.; Meffre, E.; Difilippantonio, M.J. Genomic instability in mice lacking histone H2AX. Science 2002, 296, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Heo, K.; Kim, H.; Choi, S.H.; Choi, J.; Kim, K.; Gu, J.; Lieber, M.R.; Yang, A.S.; An, W. FACT-mediated exchange of histone variant H2AX regulated by phosphorylation of H2AX and ADP-ribosylation of Spt16. Mol. Cell 2008, 30, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Pehrson, J.R.; Changolkar, L.N.; Costanzi, C.; and Leu, N.A. Mice without MacroH2A Histone Variants. Mol. Cell. Biol. 2014, 34, 4523–4533. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, P.V.; Ahel, D.; Ryan, D.P.; Weston, R.; Wiechens, N.; Kraehenbuehl, R.; Owen-Hughes, T.; Ahel, I. DNA repair factor APLF is a histone chaperone. Mol. Cell 2011, 41, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Zlatanova, J.; Thakar, A. H2A.Z: View from the top. Structure 2008, 16, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Suto, R.K.; Clarkson, M.J.; Tremethick, D.J.; Luger, K. Crystal structure of a nucleosome core particle containing the variant histone H2A.Z. Nat. Struct. Biol. 2000, 7, 1121–1124. [Google Scholar] [PubMed]
- Kalashnikova, A.A.; Porter-Goff, M.E.; Muthurajan, U.M.; Luger, K.; Hansen, J.C. The role of the nucleosome acidic patch in modulating higher order chromatin structure. J. R. Soc. Interface 2013, 10. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.A.; Garlick, J.D.; Kingston, R.E. Chromatin remodeling by imitation switch (ISWI) class ATP-dependent remodelers is stimulated by histone variant H2A.Z. J. Biol. Chem. 2010, 285, 4645–4651. [Google Scholar] [CrossRef] [PubMed]
- Meneghini, M.D.; Wu, M.; Madhani, H.D. Conserved histone variant H2A.Z protects euchromatin from the ectopic spread of silent heterochromatin. Cell 2003, 112, 725–736. [Google Scholar] [CrossRef]
- Greaves, I.K.; Rangasamy, D.; Ridgway, P.; Tremethick, D.J. H2A.Z contributes to the unique 3D structure of the centromere. Proc. Natl. Acad. Sci. USA 2007, 104, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Erdel, F.; Rippe, K. Chromatin remodelling in mammalian cells by ISWI-type complexes—Where, when and why? FEBS J. 2011, 278, 3608–3618. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.; Wang, Y.; Kallgren, S.P.; Thompson, J.; Yates, J.R., III; Jia, S. Histone variant H2A.Z regulates centromere silencing and chromosome segregation in fission yeast. J. Biol. Chem. 2010, 285, 1909–1918. [Google Scholar] [CrossRef] [PubMed]
- Okuwaki, M.; Kato, K.; Shimahara, H.; Tate, S.; Nagata, K. Assembly and disassembly of nucleosome core particles containing histone variants by human nucleosome assembly protein I. Mol. Cell Biol. 2005, 25, 10639–10651. [Google Scholar] [CrossRef] [PubMed]
- González-Romero, R.; Méndez, J.; Ausió, J.; Eirín-López, J.M. Quickly evolving histones, nucleosome stability and chromatin folding: All about histone H2A.Bbd. Gene 2008, 413, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Arimura, Y.; Kimura, H.; Oda, T.; Sato, K.; Osakabe, A.; Tachiwana, H.; Sato, Y.; Kinugasa, Y.; Ikura, T.; Sugiyama, M.; et al. Structural basis of a nucleosome containing histone H2A.B/H2A.Bbd that transiently associates with reorganized chromatin. Sci. Rep. 2013, 16. [Google Scholar] [CrossRef] [PubMed]
- Sansoni, V.; Casas-Delucchi, C.S.; Rajan, M.; Schmidt, A.; Bönisch, C.; Thomae, A.W.; Staege, M.S.; Hake, S.B.; Cardoso, M.C.; Imhof, A. The histone variant H2A.Bbd is enriched at sites of DNA synthesis. Nucleic Acids Res. 2014, 42, 6405–6420. [Google Scholar] [CrossRef] [PubMed]
- Tachiwana, H.; Osakabe, A.; Shiga, T.; Miya, Y.; Kimura, H.; Kagawa, W.; Kurumizaka, H. Structures of human nucleosomes containing major histone H3 variants. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Szenker, E.; Ray-Gallet, D.; Almouzni, G. The double face of the histone variant H3.3. Cell Res. 2011, 21, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Waterborg, J.H. Evolution of histone H3: Emergence of variants and conservation of post-translational modification sites. Biochem. Cell Biol. 2012, 90, 79–95. [Google Scholar] [PubMed]
- Postberg, J.; Forcob, S.; Chang, W.J.; Lipps, H.J. The evolutionary history of histone H3 suggests a deep eukaryotic root of chromatin modifying mechanisms. BMC Evol. Biol. 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.; Henikoff, S. Epigenetic consequences of nucleosome dynamics. Cell 2002, 111, 281–284. [Google Scholar] [CrossRef]
- Tang, M.C.; Jacobs, S.A.; Mattiske, D.M.; Soh, Y.M.; Graham, A.N.; Tran, A.; Lim, S.L.; Hudson, D.F.; Kalitsis, P.; O’Bryan, M.K.; et al. Contribution of the two genes encoding histone variant h3.3 to viability and fertility in mice. PLoS Genet. 2015, 11, e1004964. [Google Scholar] [CrossRef] [PubMed]
- Couldrey, C.; Carlton, M.B.; Nolan, P.M.; Colledge, W.H.; Evans, M.J. A retroviral gene trap insertion into the histone 3.3A gene causes partial neonatal lethality, stunted growth, neuromuscular deficits and male sub-fertility in transgenic mice. Hum. Mol. Genet. 1999, 8, 2489–2495. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.M.; Yuen, B.T.; Barrilleaux, B.L.; Riggs, J.W.; O’Geen, H.; Cotterman, R.F.; Knoepfler, P.S. Endogenous mammalian histone H3.3 exhibits chromatin-related functions during development. Epigenetics Chromatin 2013, 6. [Google Scholar] [CrossRef] [PubMed]
- Szenker, E.; Lacoste, N.; Almouzni, G. A developmental requirement for HIRA-dependent H3.3 deposition revealed at gastrulation in Xenopus. Cell Rep. 2012, 1, 730–740. [Google Scholar] [CrossRef] [PubMed]
- Wen, D.; Banaszynski, L.A.; Liu, Y.; Geng, F.; Noh, K.M.; Xiang, J.; Elemento, O.; Rosenwaks, Z.; Allis, C.D.; Rafii, S. Histone variant H3.3 is an essential maternal factor for oocyte reprogramming. Proc. Natl. Acad. Sci. USA 2014, 111, 7325–7330. [Google Scholar] [CrossRef] [PubMed]
- Elsässer, S.J.; Noh, K.M.; Diaz, N.; Allis, C.D.; Banaszynski, L.A. Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature 2015, 522, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Duarte, L.F.; Young, A.R.; Wang, Z.; Wu, H.A.; Panda, T.; Kou, Y.; Kapoor, A.; Hasson, D.; Mills, N.R.; Ma’ayan, A.; et al. Histone H3.3 and its proteolytically processed form drive a cellular senescence programme. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Tachiwana, H.; Kagawa, W.; Shiga, T.; Osakabe, A.; Miya, Y.; Saito, K.; Hayashi-Takanaka, Y.; Oda, T.; Sato, M.; Park, S.Y.; et al. Crystal structure of the human centromeric nucleosome containing CENP-A. Nature 2011, 476, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Foltz, D.R.; Jansen, L.E.; Bailey, A.O.; Yates, J.R.; Bassett, E.A.; Wood, S.; Black, B.E.; Cleveland, D.W. Centromere-specific assembly of CENP-a nucleosomes is mediated by HJURP. Cell 2009, 137, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Zhou, X.; Wang, W.; Deng, W.; Fang, J.; Hu, H.; Wang, Z.; Li, S.; Cui, L.; Shen, J.; et al. Dynamic phosphorylation of CENP-A at Ser68 orchestrates its cell-cycle-dependent deposition at centromeres. Dev. Cell 2015, 32, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Niikura, Y.; Kitagawa, R.; Ogi, H.; Abdulle, R.; Pagala, V.; Kitagawa, K. CENP-A K124 Ubiquitylation is required for CENP-A deposition at the centromere. Dev. Cell 2015, 32, 589–603. [Google Scholar] [CrossRef] [PubMed]
- Lacoste, N.; Woolfe, A.; Tachiwana, H.; Garea, A.V.; Barth, T.; Cantaloube, S.; Kurumizaka, H.; Imhof, A.; Almouzni, G. Mislocalization of the centromeric histone variant CenH3/CENP-A in human cells depends on the chaperone DAXX. Mol. Cell 2014, 53, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Fachinetti, D.; Han, J.S.; McMahon, M.A.; Ly, P.; Abdullah, A.; Wong, A.J.; Cleveland, D.W. DNA sequence-specific binding of CENP-B enhances the fidelity of human centromere function. Dev. Cell 2015, 33, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Regnier, V.; Vagnarelli, P.; Fukagawa, T.; Zerjal, T.; Burns, E.; Trouche, D.; Earnshaw, W.; Brown, W. CENP-A is required for accurate chromosome segregation and sustained kinetochore association of BubR1. Mol. Cell Biol. 2005, 25, 3967–3981. [Google Scholar] [CrossRef] [PubMed]
- Furuyama, T.; Dalal, Y.; Henikoff, S. Chaperone-mediated assembly of centromeric chromatin in vitro. Proc. Natl. Acad. Sci. USA 2006, 103, 6172–6177. [Google Scholar] [CrossRef] [PubMed]
- Filipescu, D.; Müller, S.; Almouzni, G. Histone H3 variants and their chaperones during development and disease: Contributing to epigenetic control. Annu. Rev. Cell Dev. Biol. 2014, 30, 615–646. [Google Scholar] [CrossRef] [PubMed]
- Kallappagoudar, S.; Yadav, R.K.; Lowe, B.R.; Partridge, J.F. Histone H3 mutations—A special role for H3.3 in tumorigenesis? Chromosoma 2015, 124, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Appin, C.L.; Brat, D.J. Molecular pathways in gliomagenesis and their relevance to neuropathologic diagnosis. Adv. Anat. Pathol. 2015, 22, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Bender, S.; Tang, Y.; Lindroth, A.M.; Hovestadt, V.; Jones, D.T.; Kool, M.; Zapatka, M.; Northcott, P.A.; Sturm, D.; Wang, W.; et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 2013, 24, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.W.; Müller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Venneti, S.; Felicella, M.M.; Coyne, T.; Phillips, J.J.; Gorovets, D.; Huse, J.T.; Kofler, J.; Lu, C.; Tihan, T.; Sullivan, L.M.; et al. Histone 3 lysine 9 trimethylation is differentially associated with isocitrate dehydrogenase mutations in oligodendrogliomas and high-grade astrocytomas. J. Neuropathol. Exp. Neurol. 2013, 72, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.M.; Fang, D.; Gan, H.; Hashizume, R.; Yu, C.; Schroeder, M.; Gupta, N.; Mueller, S.; James, C.D.; Jenkins, R.; et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013, 27, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Behjati, S.; Tarpey, P.S.; Presneau, N.; Scheipl, S.; Pillay, N.; van Loo, P.; Wedge, D.C.; Cooke, S.L.; Gundem, G.; Davies, H.; et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet. 2013, 45, 1479–1482. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Athwal, R.K.; Walkiewicz, M.P.; Baek, S.; Fu, S.; Bui, M.; Camps, J.; Ried, T.; Sung, M.H.; Dalal, Y. CENP-A nucleosomes localize to transcription factor hotspots and subtelomeric sites in human cancer cells. Epigenetics Chromatin 2015, 8. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, S.; Ahmad, K.; Malik, H.S. The centromere paradox: Stable inheritance with rapidly evolving DNA. Science 2001, 293, 1098–1102. [Google Scholar] [CrossRef] [PubMed]
- Malik, H.S.; Henikoff, S. Major evolutionary transitions in centromere complexity. Cell 2009, 138, 1067–1082. [Google Scholar] [CrossRef] [PubMed]
- Drinnenberg, I.A.; de Young, D.; Henikoff, S.; Malik, H.S. Recurrent loss of CenH3 is associated with independent transitions to holocentricity in insects. Elife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Lowell, J.E.; Cross, G.A. A variant histone H3 is enriched at telomeres in Trypanosoma brucei. J. Cell Sci. 2004, 117, 5937–5947. [Google Scholar] [CrossRef] [PubMed]
- Berriman, M.; Ghedin, E.; Hertz-Fowler, C.; Blandin, G.; Renauld, H.; Bartholomeu, D.C.; Lennard, N.J.; Caler, E.; Hamlin, N.E.; Haas, B.; et al. The genome of the African trypanosome Trypanosoma brucei. Science 2005, 309, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Akiyoshi, B.; Gull, K. Discovery of unconventional kinetochores in kinetoplastids. Cell 2014, 156, 1247–1258. [Google Scholar] [CrossRef] [PubMed]
- Talbert, P.B.; Ahmad, K.; Almouzni, G.; Ausió, J.; Berger, F.; Bhalla, P.L.; Bonner, W.M.; Cande, W.Z.; Chadwick, B.P.; Chan, S.W.; et al. A unified phylogeny-based nomenclature for histone variants. Epigenetics Chromatin 2012, 5. [Google Scholar] [CrossRef] [PubMed]
- Maheshwari, S.; Tan, E.H.; West, A.; Franklin, F.C.; Comai, L.; Chan, S.W. Naturally occurring differences in CENH3 affect chromosome segregation in zygotic mitosis of hybrids. PLoS Genet. 2015, 11, e1004970. [Google Scholar] [CrossRef] [PubMed]
- Black, B.E.; Cleveland, D.W. Epigenetic centromere propagation and the nature of CENP-A nucleosomes. Cell 2011, 144, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Bui, M.; Dimitriadis, E.K.; Hoischen, C.; An, E.; Quénet, D.; Giebe, S.; Nita-Lazar, A.; Diekmann, S.; Dalal, Y. Cell-cycle-dependent structural transitions in the human CENP-A nucleosome in vivo. Cell 2012, 150, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Hasson, D.; Panchenko, T.; Salimian, K.J.; Salman, M.U.; Sekulic, N.; Alonso, A.; Warburton, P.E.; Black, B.E. The octamer is the major form of CENP-A nucleosomes at human centromeres. Nat. Struct. Mol. Biol. 2013, 20, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Walkiewicz, M.P.; Dimitriadis, E.K.; Dalal, Y. CENP-A octamers do not confer a reduction in nucleosome height by AFM. Nat. Struct. Mol. Biol. 2014, 21, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, S.; Ramachandran, S.; Krassovsky, K.; Bryson, T.D.; Codomo, C.A.; Brogaard, K.; Widom, J.; Wang, J.P.; Henikoff, J.G. The budding yeast Centromere DNA Element II wraps a stable Cse4 hemisome in either orientation in vivo. Elife 2014, 3, e01861. [Google Scholar] [CrossRef] [PubMed]
- Kingston, I.J.; Yung, J.S.; Singleton, M.R. Biophysical characertization of the centromere-specific nulceosome from budding yeast. J. Biol. Chem. 2011, 286, 4021–4026. [Google Scholar] [CrossRef] [PubMed]
- Dechasse, M.L.; Wyns, K.; Li, M.; Hall, M.A.; Wang, M.D.; Luger, K. Structure of Scm3-mediated assembly of budding yeast centromeric nucleosomes. Nat. Commun. 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Furuyama, T.; Codomo, C.A.; Henikoff, S. Reconstitution of hemisomes on budding yeast centromeric DNA. Nucleic Acid Res. 2013, 41, 5769–5783. [Google Scholar] [CrossRef] [PubMed]
- Yoda, K.; Ando, S.; Morishita, S.; Houmura, K.; Hashimoto, K.; Takeyasu, K.; Okazaki, T. Human centromere protein A (CENP-A) can replace histone H3 in nucleosome reconstitution in vitro. Proc. Natl Acad. Sci. U.S.A. 2000, 97, 7266–7271. [Google Scholar] [CrossRef] [PubMed]
- Sekulic, N.; Bassett, E.A.; Rogers, D.J.; Black, D.E. The structure of (CENP-A-H4)(2) reveals physical features that mark centromeres. Nature 2010, 467, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Miell, M.D.; Fuller, C.J.; Guse, A.; Barysz, H.M.; Downes, A.; Owen-Hughes, T.; Rappsilber, T.; Straight, A.F.; Allshire, R.C. CENP-A confers a reduction in height on octameric nucleosomes. Nat. Struct. Mol. Biol. 2013, 20, 763–765. [Google Scholar] [CrossRef] [PubMed]
- Dunleavy, E.M.; Roche, D.; Tagami, H.; Lacoste, N.; Ray-Gallet, D.; Nakamura, Y.; Daigo, Y.; Nakatani, Y.; Amounzi-Pettinotti, G. HJURP is a cell-cycle-dependent maintenance and deposition factor of CENP-A at centromeres. Cell 2009, 137, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.; Hajj, B.; Chen, J.; Mizuguchi, G.; Xiao, H.; Wei, D.; Dahan, M.; Wu, C. Imaging the fate of histone Cse4 reveals de novo replacement in S phase and subsequent stable residence at centromeres. Elife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Schuh, M.; Lehner, C.F.; Heidmann, S. Incorporation of Drosophila CID/CENP-A and CENP-C into centromeres during early embryonic anaphase. Curr. Biol. 2007, 17, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Jansen, L.E.; Black, B.E.; Foltz, D.R.; Cleveland, D.W. Propagation of centromeric chromatin requires exit from mitosis. J. Cell Biol. 2007, 176, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.C.; Bodor, D.L.; Stellfox, M.E.; Martins, N.M.; Hochegger, H.; Foltz, D.R.; Jansen, L.E. Cdk activity couples epigenetic centromere inheritance to cell-cycle progression. Dev. Cell 2012, 22, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.O.; Panchenko, T.; Sathyan, K.M.; Petkowski, J.J.; Pai, P.J.; Bai, D.L.; Russell, D.H.; Macara, I.G.; Shabanowitz, J.; Hunt, D.F.; et al. Posttranslational modification of CENP-A influences the conformation of centromeric chromatin. Proc. Natl. Acad. Sci. USA 2013, 110, 11827–11832. [Google Scholar] [CrossRef] [PubMed]
- Zeitlin, S.G.; Shebly, R.D.; Sullivan, K.F. CENP-A is phosphorylated by Aurora B kinase and plays an unexpected role in completion of cytokinesis. J. Cell Biol. 2001, 155, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Chan, F.L.; Marshall, O.J.; Saffery, R.; Kim, B.W.; Earle, E.; Choo, K.H.; Wong, L.H. Active transcription and essential role of RNA polymerase II at the centromere during mitosis. Proc. Natl. Acad. Sci. USA 2012, 109, 1979–1984. [Google Scholar] [CrossRef] [PubMed]
- Quénet, D.; Dalal, Y. A long non-coding RNA is required for targeting centromeric protein A to the human centromere. Elife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Mendiburo, M.J.; Padeken, J.; Fülöp, S.; Schepers, A.; Heun, P. Drosophila CENH3 is sufficient for centromere formation. Science 2011, 334, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Dechassa, M.L.; Bettini, E.; Ledoux, M.B.; Belisario, C.; Heun, P.; Luger, K.; Mellone, B.G. CAL1 is the Drosophila CENP-A assembly factor. J. Cell Biol. 2014, 204, 313–329. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Browers, S.; Lipinszki, Z.; Palladino, J.; Trusiak, S.; Bettini, E.; Rosin, L.; Przewloka, M.R.; Glover, D.M.; O’Neill, R.J.; et al. Establishment of centromeric chromatin by the CENP-A assembly factor CAL1 requires FACT-mediated transcription. Dev. Cell 2015, 34, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Felsenfeld, G. Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev. 2007, 21, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Gassmann, R.; Rechtsteiner, A.; Yuen, K.W.; Muroyama, A.; Egelhofer, T.; Gaydos, L.; Barron, F.; Maddox, P.; Essex, A.; Monen, J.; et al. An inverse relationship to germline transcription defines centromeric chromatin in C. elegans. Nature 2012, 484, 534–537. [Google Scholar] [CrossRef] [PubMed]
- Steiner, F.A.; Henikoff, S. Holocentromeres are dispersed point centromeres localized at transcription factor hotspots. Elife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Dunleavy, E.M.; Almouzni, G.; Karpen, G.H. H3.3 is deposited at centromeres in S phase as a placeholder for newly assembled CENP-A in G1 phase. Nucleus 2011, 2, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Monen, J.; Hattersley, N.; Muroyama, A.; Stevens, D.; Oegema, K.; Desai, A. Separase cleaves the N-Tail of the CENP-A related protein CPAR-1 at the meiosis I metaphase-anaphase transition in C. elegans. PLoS ONE 2015, 10, e0125382. [Google Scholar] [CrossRef] [PubMed]
- Neumann, P.; Navrátilová, A.; Schroeder-Reiter, E.; Koblížková, A.; Steinbauerová, V.; Chocholová, E.; Novák, P.; Wanner, G.; Macas, J. Stretching the rules: Monocentric chromosomes with multiple centromere domains. PLoS Genet. 2012, 8, e1002777. [Google Scholar] [CrossRef] [PubMed]
- Neumann, P.; Pavlíková, Z.; Koblížková, A.; Fuková, I.; Jedličková, V.; Novák, P.; Macas, J. Centromeres off the hook: Massive changes in centromere size and structure following duplication of CenH3 gene in Fabeae species. Mol. Biol. Evol. 2015. [Google Scholar] [CrossRef]
- Hu, Z.; Huang, G.; Sadanandam, A.; Gu, S.; Lenburg, M.E.; Pai, M.; Bayani, N.; Blakely, E.A.; Gray, J.W.; Mao, J.H. The expression level of HJURP has an independent prognostic impact and predicts the sensitivity to radiotherapy in breast cancer. Breast Cancer Res. 2010, 12. [Google Scholar] [CrossRef] [PubMed]
- Valente, V.; Serafim, R.B.; de Oliveira, L.C.; Adorni, F.S.; Torrieri, R.; Tirapelli, D.P.; Espreafico, E.M.; Oba-Shinjo, S.M.; Marie, S.K.; Paçó-Larson, M.L.; et al. Modulation of HJURP (Holliday Junction-Recognizing Protein) levels is correlated with glioblastoma cells survival. PLoS ONE 2013, 8, e62200. [Google Scholar] [CrossRef] [PubMed]
- De Tayrac, M.; Saikali, S.; Aubry, M.; Bellaud, P.; Boniface, R.; Quillien, V.; Mosser, J. Prognostic significance of EDN/RB, HJURP, p60/CAF-1 and PDLI4, four new markers in high-grade gliomas. PLoS ONE 2013, 8, e73332. [Google Scholar] [CrossRef] [PubMed]
- Montes de Oca, R.; Gurard-Levin, Z.A.; Berger, F.; Rehman, H.; Martel, E.; Corpet, A.; de Koning, L.; Vassias, I.; Wilson, L.O.; Meseure, D.; et al. The histone chaperone HJURP is a new independent prognostic marker for luminal a breast carcinoma. Mol. Oncol. 2015, 9, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.H.; Henry, I.M.; Ravi, M.; Bradnam, K.R.; Mandakova, T.; Marimuthu, M.P.; Korf, I.; Lysak, M.A.; Comai, L.; Chan, S.W. Catastrophic chromosomal restructuring during genome elimination in plants. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- McAinsh, A.D.; Meraldi, P. The CCAN complex: Linking centromere specification to control of kinetochore-microtubule dynamics. Semin. Cell Dev. Biol. 2011, 22, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Fujita, R.; Otake, K.; Arimura, Y.; Horikoshi, N.; Miya, Y.; Shiga, T.; Osakabe, A.; Tachiwana, H.; Ohzeki, J.; Larionov, V.; et al. Stable complex formation of CENP-B with the CENP-A nucleosome. Nucleic Acids Res. 2015, 43, 4909–4922. [Google Scholar] [CrossRef] [PubMed]
- Falk, S.J.; Guo, L.Y.; Sekulic, N.; Smoak, E.M.; Mani, T.; Logsdon, G.A.; Gupta, K.; Jansen, L.E.; van Duyne, G.D.; Vinogradov, S.A.; et al. Chromosomes. CENP-C reshapes and stabilizes CENP-A nucleosomes at the centromere. Science 2015, 348, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; de Oca Luna, R.M.; Liu, G.; Lozano, G.; Cummings, C.; Mancini, M.; Ouspenski, I.; Brinkley, B.R.; May, G.S. The cenpB gene is not essential in mice. Chromosoma 1998, 107, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Hudson, D.F.; Fowler, K.J.; Earle, E.; Saffery, R.; Kalitsis, P.; Trowell, H.; Hill, J.; Wreford, N.G.; de Kretser, D.M.; Cancilla, M.R.; et al. Centromere protein B null mice are mitotically and meiotically normal but have lower body and testis weights. J. Cell Biol. 1998, 141, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Ohzeki, J.; Nakano, M.; Yoda, K.; Brinkley, W.R.; Larionov, V.; Masumoto, H. CENP-B controls centromere formation depending on the chromatin context. Cell 2007, 131, 1287–1300. [Google Scholar] [CrossRef] [PubMed]
- Marshall, O.J.; Choo, K.H. Putative CENP-B paralogues are not present at mammalian centromeres. Chromosoma 2012, 121, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Schueler, M.G.; Swanson, W.; Thomas, P.J.; Green, E.D. Adaptive evolution of foundation kinetochore proteins in primates. Mol. Biol. Evol. 2010, 27, 1585–1597. [Google Scholar] [CrossRef] [PubMed]
- Au, W.C.; Dawson, A.R.; Rawson, D.W.; Taylor, S.B.; Baker, R.E.; Basrai, M.A. A novel role of the N terminus of budding yeast histone H3 variant Cse4 in ubiquitin-mediated proteolysis. Genetics 2013, 194, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Boeckmann, L.; Takahashi, Y.; Au, W.C.; Mishra, P.K.; Choy, J.S.; Dawson, A.R.; Szeto, M.Y.; Waybright, T.J.; Heger, C.; McAndrew, C.; et al. Phosphorylation of centromeric histone H3 variant regulates chromosome segregation in Saccharomyces cerevisiae. Mol. Biol. Cell 2013, 24, 2034–2044. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.K.; Guo, J.; Dittman, L.E.; Haase, J.; Yeh, E.; Bloom, K.; Basrai, M.A. Pat1 protects centromere-specific histone H3 variant Cse4 from Psh1-mediated ubiquitination. Mol. Biol. Cell 2015, 26, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- Arimura, Y.; Shirayama, K.; Horikoshi, N.; Fujita, R.; Taguchi, H.; Kagawa, W.; Fukagawa, T.; Almouzni, G.; Kurumizaka, H. Crystal structure and stable property of the cancer-associated heterotypic nucleosome containing CENP-A and H3.3. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.D.; Banaszynski, L.A.; Noh, K.M.; Lewis, P.W.; Elsaesser, S.J.; Stadler, S.; Dewell, S.; Law, M.; Guo, X.; Li, X.; et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 2010, 140, 678–691. [Google Scholar] [CrossRef] [PubMed]
- Voon, H.P.; Hughes, J.R.; Rode, C.; de La Rosa-Velázquez, I.A.; Jenuwein, T.; Feil, R.; Higgs, D.R.; Gibbons, R.J. ATRX plays a key role in maintaining silencing at interstitial heterochromatic loci and imprinted genes. Cell Rep. 2015, 11, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Banaszynski, L.A.; Wen, D.; Dewell, S.; Whitcomb, S.J.; Lin, M.; Diaz, N.; Elsässer, S.J.; Chapgier, A.; Goldberg, A.D.; Canaani, E.; et al. Hira-dependent histone H3.3 deposition facilitates PRC2 recruitment at developmental loci in ES cells. Cell 2013, 155, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.M.; Georgiou, A.; Szutorisz, H.; Maia e Silva, A.; Pombo, A.; Barahona, I.; Dargelos, E.; Canzonetta, C.; Dillon, N. Variant histone H3.3 marks promoters of transcriptionally active genes during mammalian cell division. EMBO Rep. 2005, 6, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Wirbelauer, C.; Bell, O.; Schübeler, D. Variant histone H3.3 is deposited at sites of nucleosomal displacement throughout transcribed genes while active histone modifications show a promoter-proximal bias. Genes Dev. 2005, 19, 1761–1766. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, V.; Mazumder, A.; Surface, L.E.; Butty, V.L.; Fields, P.A.; Alwan, A.; Torrey, L.; Thai, K.K.; Levine, S.S.; Bathe, M.; et al. H2A.Z acidic patch couples chromatin dynamics to regulation of gene expression programs during ESC differentiation. PLoS Genet. 2013, 9, e1003725. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ayrapetov, M.K.; Xu, C.; Gursoy-Yuzugullu, O.; Hu, Y.; Price, B.D. Histone H2A.Z controls a critical chromatin remodeling step required for DNA double-strand break repair. Mol. Cell 2012, 48, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Sharma, U.; Stefanova, D.; Holmes, S.G. Histone variant H2A.Z functions in sister chromatid cohesion in Saccharomyces cerevisiae. Mol. Cell Biol. 2013, 33, 3473–3481. [Google Scholar] [CrossRef] [PubMed]
- Bruce, K.; Myers, F.A.; Mantouvalou, E.; Lefevre, P.; Greaves, I.; Bonifer, C.; Tremethick, D.J.; Thorne, A.W.; Crane-Robinson, C. The replacement histone H2A.Z in a hyperacetylated form is a feature of active genes in the chicken. Nucleic Acids Res. 2005, 33, 5633–5639. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Mora, F.; Song, J.Z.; Statham, A.L.; Strbenac, D.; Robinson, M.D.; Nair, S.S.; Patterson, K.I.; Tremethick, D.J.; Stirzaker, C.; Clark, S.J. Acetylation of H2A.Z is a key epigenetic modification associated with gene deregulation and epigenetic remodeling in cancer. Genome Res. 2012, 22, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Teves, S.S.; Henikoff, S. Heat shock reduces stalled RNA polymerase II and nucleosome turnover genome-wide. Genes Dev. 2011, 25, 2387–2397. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Wang, Y.; Li, G. Dynamics of histone variant H3.3 and its coregulation with H2A.Z at enhancers and promoters. Nucleus 2014, 5, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Thatcher, T.H.; Gorovsky, M.A. Phylogenetic analysis of the core histones H2A, H2B, H3, and H4. Nucleic Acids Res. 1994, 22, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Baldi, S.; Becker, P.B. The variant histone H2A.V of Drosophila—Three roles, two guises. Chromosoma 2013, 122, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Vernì, F.; Cenci, G. The Drosophila histone variant H2A.V works in concert with HP1 to promote kinetochore-driven microtubule formation. Cell Cycle 2015, 14, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Van Daal, A.; White, E.M.; Elgin, S.C.; Gorovsky, M.A. Conservation of intron position indicates separation of major and variant H2As is an early event in the evolution of eukaryotes. J. Mol. Evol. 1990, 30, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, B.; Gorovsky, M.A. Essential and nonessential histone H2A variants in Tetrahymena thermophila. Mol. Cell Biol. 1996, 16, 4305–4311. [Google Scholar] [PubMed]
- Faast, R.; Thonglairoam, V.; Schulz, T.C.; Beall, J.; Wells, J.R.; Taylor, H.; Matthaei, K.; Rathjen, P.D.; Tremethick, D.J.; Lyons, I. Histone variant H2A.Z is required for early mammalian development. Curr. Biol. 2001, 11, 1183–1187. [Google Scholar] [CrossRef]
- Wu, R.S.; Kohn, K.W.; Bonner, W.M. Metabolism of ubiquitinated histones. J. Biol. Chem. 1981, 256, 5916–5920. [Google Scholar] [PubMed]
- Li, B.; Pattenden, S.G.; Lee, D.; Gutiérrez, J.; Chen, J.; Seidel, C.; Gerton, J.; Workman, J.L. Preferential occupancy of histone variant H2AZ at inactive promoters influences local histone modifications and chromatin remodeling. Proc. Natl. Acad. Sci. USA 2005, 102, 18385–18390. [Google Scholar] [CrossRef] [PubMed]
- Raisner, R.M.; Hartley, P.D.; Meneghini, M.D.; Bao, M.Z.; Liu, C.L.; Schreiber, S.L.; Rando, O.J.; Madhani, H.D. Histone variant H2A.Z marks the 5' ends of both active and inactive genes in euchromatin. Cell 2005, 123, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Roberts, D.N.; Cairns, B.R. Genome-wide dynamics of Htz1, a histone H2A variant that poises repressed/basal promoters for activation through histone loss. Cell 2005, 123, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, A.; Mizuguchi, G.; FitzGerald, P.C.; Wei, D.; Wang, F.; Huang, Y.; Luk, E.; Woodcock, C.L.; Wu, C. Nucleosome-free region dominates histone acetylation in targeting SWR1 to promoters for H2A.Z replacement. Cell 2013, 154, 1232–1245. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Radman-Livaja, M.; Rando, O.J.; Peterson, C.L. A histone acetylation switch regulates H2A.Z deposition by the SWR-C remodeling enzyme. Science 2013, 340, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.M.; Ramachandran, S.; Henikoff, S. Nucleosomes are context-specific, H2A.Z-modulated barriers to RNA polymerase. Mol. Cell 2014, 53, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Obri, A.; Ouararhni, K.; Papin, C.; Diebold, M.L.; Padmanabhan, K.; Marek, M.; Stoll, I.; Roy, L.; Reilly, P.T.; Mak, T.W.; et al. ANP32E is a histone chaperone that removes H2A.Z from chromatin. Nature 2014, 505, 648–653. [Google Scholar] [CrossRef] [PubMed]
- Jeronimo, C.; Watanabe, S.; Kaplan, C.D.; Peterson, C.L.; Robert, F. The histone chaperones FACT and Spt6 restrict H2A.Z from intragenic locations. Mol. Cell 2015, 58, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Nekrasov, M.; Amrichova, J.; Parker, B.J.; Soboleva, T.A.; Jack, C.; Williams, R.; Huttley, G.A.; Tremethick, D.J. Histone H2A.Z inheritance during the cell cycle and its impact on promoter organization and dynamics. Nat. Struct. Mol. Biol. 2012, 19, 1076–1083. [Google Scholar] [CrossRef] [PubMed]
- Ogiyama, Y.; Ohno, Y.; Kubota, Y.; Ishii, K. Epigenetically induced paucity of histone H2A.Z stabilizes fission-yeast ecoptic centromeres. Nat. Struct. Mol. Biol. 2013, 20, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, D.; Greaves, I.; Tremethick, D.J. RNA interference demonstrates a novel role for H2A.Z in chromosome segregation. Nat. Struct. Mol. Biol. 2004, 11, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Vanoosthuyse, V.; Fillingham, J.; Roguev, A.; Watt, S.; Kislinger, T.; Treyer, A.; Carpenter, L.R.; Bennett, C.S.; Emili, A.; et al. An acetylated form of histone H2A.Z regulates chromosome architecture in Schizosaccharomyces pombe. Nat. Struct. Mol. Biol. 2009, 16, 1286–1293. [Google Scholar] [CrossRef] [PubMed]
- Costanzi, C.; Pehrson, J.R. Histone macroH2A1 is concentrated in the inactive X chromosome of female mammals. Nature 1998, 393, 599–601. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, S.; Gundimella, S.K.; Caron, C.; Perche, P.Y.; Pehrson, J.R.; Khochbin, S.; Luger, K. Structural characterization of the histone variant macroH2A. Mol. Cell Biol. 2005, 25, 7616–7624. [Google Scholar] [CrossRef] [PubMed]
- Aygün, O.; Mehta, S.; Grewal, S.I. HDAC-mediated suppression of histone turnover promotes epigenetic stability of heterochromatin. Nat. Struct. Mol. Biol. 2013, 20, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Yelagandula, R.; Stroud, H.; Holec, S.; Zhou, K.; Feng, S.; Zhong, X.; Muthurajan, U.M.; Nie, X.; Kawashima, T.; Groth, M.; et al. The histone variant H2A.W defines heterochromatin and promotes chromatin condensation in Arabidopsis. Cell 2014, 158, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Kustatscher, G.; Hothorn, M.; Pugieux, C.; Scheffzek, K.; Ladurner, A.G. Splicing regulates NAD metabolite binding to histone macroH2A. Nat. Struct. Mol. Biol. 2005, 12, 624–625. [Google Scholar] [CrossRef] [PubMed]
- Timinszky, G.; Till, S.; Hassa, P.O.; Hothorn, M.; Kustatscher, G.; Nijmeijer, B.; Colombelli, J.; Altmeyer, M.; Stelzer, E.H.; Scheffzek, K.; et al. A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat. Struct. Mol. Biol. 2009, 16, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Nusinow, D.A.; Sharp, J.A.; Morris, A.; Salas, S.; Plath, K.; Panning, B. The histone domain of macroH2A1 contains several dispersed elements that are each sufficient to direct enrichment on the inactive X chromosome. J. Mol. Biol. 2007, 371, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, F.; Santoro, R. The expanding role of PARPs in the establishment and maintenance of heterochromatin. FEBS J. 2013, 280, 3508–3518. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ruiz, P.D.; Novikov, L.; Casill, A.D.; Park, J.W.; Gamble, M.J. MacroH2A1.1 and PARP-1 cooperate to regulate transcription by promoting CBP-mediated H2B acetylation. Nat. Struct. Mol. Biol. 2014, 21, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, M.D.; Vatsellas, G.; Polyzos, A.; Mantouvalou, E.; Sianidis, G.; Maraziotis, I.; Agelopoulos, M.; Thanos, D. Composite macroH2A/NRF-1 nucleosomes suppress noise and generate robustness in gene expression. Cell Rep. 2015, 11, 1090–1101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, X.; Marshall, J.B.; Zhong, C.X.; Dawe, R.K. Phosphoserines on maize CENTROMERIC HISTONE H3 and histone H3 demarcate the centromere and pericentromere during chromosome segregation. Plant Cell 2005, 17, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Sporn, J.C.; Kustatscher, G.; Hothorn, T.; Collado, M.; Serrano, M.; Muley, T.; Schnabel, P.; Ladurner, A.G. Histone macroH2A isoforms predict the risk of lung cancer recurrence. Oncogene 2009, 28, 3423–3428. [Google Scholar] [CrossRef] [PubMed]
- Timinszky, G.; Ladurner, A.G. PARP1 and CBP lose their footing in cancer. Nat. Struct. Mol. Biol. 2014, 21, 947–948. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Goldberg, M.S.; Cumberland, L.K.; Ratnakumar, K.; Segura, M.F.; Emanuel, P.O.; Menendez, S.; Vardabasso, C.; LeRoy, G.; Vidal, C.I. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 2010, 468, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Kummar, S.; Chen, A.; Parchment, R.E.; Kinders, R.J.; Ji, J.; Tomaszewski, J.E.; Doroshow, J.H. Advances in using PARP inhibitors to treat cancer. BMC Med. 2012, 10. [Google Scholar] [CrossRef] [PubMed]
- Schiewer, M.J.; Goodwin, J.F.; Han, S.; Brenner, J.C.; Augello, M.A.; Dean, J.L.; Liu, F.; Planck, J.L.; Ravindranathan, P.; Chinnaiyan, A.M.; et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012, 2, 1134–1149. [Google Scholar] [CrossRef] [PubMed]
- Green, A.R.; Caracappa, D.; Benhasouna, A.A.; Alshareeda, A.; Nolan, C.C.; Macmillan, R.D.; Madhusudan, S.; Ellis, I.O.; Rakha, E.A. Biological and clinical significance of PARP1 protein expression in breast cancer. Breast Cancer Res. Treat. 2015, 149, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Skene, P.J.; Henikoff, S. Histone variants in pluripotency and disease. Development 2013, 140, 2513–2524. [Google Scholar] [CrossRef] [PubMed]
- Vardabasso, C.; Hasson, D.; Ratnakumar, K.; Chung, C.Y.; Duarte, L.F.; Bernstein, E. Histone variants: Emerging players in cancer biology. Cell. Mol. Life Sci. 2014, 71, 379–404. [Google Scholar] [CrossRef] [PubMed]
- Vassetzky, Y.S.; Hair, A.; Razin, S.V. Rearrangement of chromatin domains in cancer and development. J. Cell. Biochem. Suppl. 2000, 35, 54–60. [Google Scholar] [CrossRef]
- De la Espina, S.M.D.; Alverca, E.; Cuadrado, A.; Franca, S. Organization of the genome and gene expression in a nuclear environment lacking histones and nucleosomes: The amazing dinoflagellates. Eur. J. Cell Biol. 2005, 84, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.H.; Kwok, A.C.; Tsang, J.S.; Wong, J.T. Alveolata histone-like proteins have different evolutionary origins. J. Evol. Biol. 2006, 19, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Gornik, S.G.; Ford, K.L.; Mulhern, T.D.; Bacic, A.; McFadden, G.I.; Waller, R.F. Loss of nucleosomal DNA condensation coincides with appearance of a novel nuclear protein in dinoflagellates. Curr. Biol. 2012, 22, 2303–2312. [Google Scholar] [CrossRef] [PubMed]
- Sandman, K.; Reeve, J.N. Archaeal histones and the origin of the histone fold. Curr. Opin. Microbiol. 2006, 9, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.L.; Grayling, R.A.; Lurz, R.; Reeve, J.N. Archaeal nucleosomes. Proc. Natl. Acad. Sci. USA 1997, 94, 12633–12637. [Google Scholar] [CrossRef] [PubMed]
- Decanniere, K.; Babu, A.M.; Sandman, K.; Reeve, J.N.; Heinemann, U. Crystal structures of recombinant histones HMfA and HMfB from the hyperthermophilic archaeon Methanothermus fervidus. J. Mol. Biol. 2000, 303, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Arents, G.; Moudrianakis, E.N. The histone fold: A ubiquitous architectural motif utilized in DNA compaction and protein dimerization. Proc. Natl. Acad. Sci. USA 1995, 92, 11170–11174. [Google Scholar] [CrossRef] [PubMed]
- Koster, M.J.; Snel, B.; Timmers, H.T. Genesis of chromatin and transcription dynamics in the origin of species. Cell 2015, 161, 724–736. [Google Scholar] [CrossRef] [PubMed]
- Nabeel-Shah, S.; Ashraf, K.; Pearlman, R.E.; Fillingham, J. Molecular evolution of NASP and conserved histone H3/H4 transport pathway. BMC Evol. Biol. 2014, 14, 139. [Google Scholar] [CrossRef] [PubMed]
- Zovkic, I.B.; Paulukaitis, B.S.; Day, J.J.; Etikala, D.M.; Sweatt, J.D. Histone H2A.Z subunit exchange controls consolidation of recent and remote memory. Nature 2014, 515, 582–586. [Google Scholar] [CrossRef] [PubMed]
- Bacolla, A.; Wells, R.D. Non-B DNA conformations, genomic rearrangements, and human disease. J. Biol. Chem. 2004, 279, 47411–47414. [Google Scholar] [CrossRef] [PubMed]
- Wells, R.D. Discovery of the role of non-B DNA structures in mutagenesis and human genomic disorders. J. Biol. Chem. 2009, 284, 8997–9009. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melters, D.P.; Nye, J.; Zhao, H.; Dalal, Y. Chromatin Dynamics in Vivo: A Game of Musical Chairs. Genes 2015, 6, 751-776. https://doi.org/10.3390/genes6030751
Melters DP, Nye J, Zhao H, Dalal Y. Chromatin Dynamics in Vivo: A Game of Musical Chairs. Genes. 2015; 6(3):751-776. https://doi.org/10.3390/genes6030751
Chicago/Turabian StyleMelters, Daniël P., Jonathan Nye, Haiqing Zhao, and Yamini Dalal. 2015. "Chromatin Dynamics in Vivo: A Game of Musical Chairs" Genes 6, no. 3: 751-776. https://doi.org/10.3390/genes6030751
APA StyleMelters, D. P., Nye, J., Zhao, H., & Dalal, Y. (2015). Chromatin Dynamics in Vivo: A Game of Musical Chairs. Genes, 6(3), 751-776. https://doi.org/10.3390/genes6030751