
Non-Coding RNAs in Lung Cancer: Contribution of Bioinformatics Analysis to the Development of Non-Invasive Diagnostic Tools

 , ,
, ,
Abstract
:1. Introduction
2. miRNAs as Diagnostic Biomarkers
3. lncRNAs as Diagnostic Biomarkers
4. Bioinformatics Databases, Algorithms and Analysis Tools
4.1. Bioinformatics Databases
4.2. Phylogenetic Sequence-Structure Conservation
4.3. Functional Interaction Partner Analysis
4.4. miRNA Target Prediction Algorithms
4.5. lncRNA–RNA Interaction Prediction Algorithms
4.6. RNA-Protein Interaction Prediction Algorithms
4.7. Functional GO and Pathway Enrichment Analysis
4.8. Bioinformatics Tools for Integrated Functional Analysis
4.9. Promoter Analysis
4.10. Automation
5. Conclusions and Future Directions
Acknowledgments
Conflicts of Interest
References
- Wei, Y.; Zhang, X. Transcriptome analysis of distinct long non-coding RNA transcriptional fingerprints in lung adenocarcinoma and squamous cell carcinoma. Tumour Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhu, N.; Chen, X. A novel long noncoding RNA LINC01133 is upregulated in lung squamous cell cancer and predicts survival. Tumour Biol. 2015, 36, 7465–7471. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Heymach, J.V.; Lippman, S.M. Lung Cancer. N. Engl. J. Med. 2008, 359, 1367–1380. [Google Scholar] [CrossRef] [PubMed]
- GLOBOCAN project. Available online: http://globocan.iarc.fr (accessed on 13 August 2015).
- Du, L.; Schageman, J.J.; Irnov, G.L.; Hammond, S.M.; Minna, J.D.; Gazdar, A.F.; Pertsemlidis, A. MicroRNA expression distinguishes SCLC from NSCLC lung tumor cells and suggests a possible pathological relationship between SCLCs and NSCLCs. J. Exp. Clin. Cancer Res. 2010. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lin, J.; Liu, T.; Chen, T.; Pan, S.; Huang, W.; Li, S. Analysis of lncRNA expression profiles in non-small cell lung cancers (NSCLC) and their clinical subtypes. Lung Cancer 2014, 85, 110–115. [Google Scholar] [CrossRef] [PubMed]
- White, N.M.; Cabanski, C.R.; Silva-Fisher, J.M.; Dang, H.X.; Govindan, R.; Maher, C.A. Transcriptome sequencing reveals altered long intergenic non-coding RNAs in lung cancer. Genome Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Taniwaki, M.; Daigo, Y.; Ishikawa, N.; Takano, A.; Tsunoda, T.; Yasui, W.; Inai, K.; Kohno, N.; Nakamura, Y. Gene expression profiles of small-cell lung cancers: molecular signatures of lung cancer. Int. J. Oncol. 2006, 29, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Taneja, T.K.; Sharma, S.K. Markers of small cell lung cancer. World J. Surg. Oncol. 2004. [Google Scholar] [CrossRef] [PubMed]
- Bangur, C.S.; Switzer, A.; Fan, L.; Marton, M.J.; Meyer, M.R.; Wang, T. Identification of genes over-expressed in small cell lung carcinoma using suppression subtractive hybridization and cDNA microarray expression analysis. Oncogene 2002, 21, 3814–3825. [Google Scholar] [CrossRef] [PubMed]
- Coate, L.E.; John, T.; Tsao, M.S.; Shepherd, F.A. Molecular predictive and prognostic markers in non-small-cell lung cancer. Lancet Oncol. 2009, 10, 1001–1010. [Google Scholar] [CrossRef]
- Yao, Y.; Fan, Y.; Wu, J.; Wan, H.; Wang, J.; Lam, S.; Lam, W.L.; Girard, L.; Gazdar, A.F.; Wu, Z.; et al. Potential application of non-small cell lung cancer-associated autoantibodies to early cancer diagnosis. Biochem. Biophys. Res. Commun. 2012, 423, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Sozzi, G.; Boeri, M.; Rossi, M.; Verri, C.; Suatoni, P.; Bravi, F.; Roz, L.; Conte, D.; Grassi, M.; Sverzellati, N.; et al. Clinical utility of a plasma-based miRNA signature classifier within computed tomography lung cancer screening: A correlative MILD trial study. J. Clin. Oncol. 2014, 32, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Montani, F.; Marzi, M.J.; Dezi, F.; Dama, E.; Carletti, R.M.; Bonizzi, G.; Bertolotti, R.; Bellomi, M.; Rampinelli, C.; Maisonneuve, P.; et al. miR-Test: A blood test for lung cancer early detection. J. Nat. Cancer Inst. 2015. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, R.; Zhang, K.; Chen, L.-B. Long non-coding RNAs in non-small cell lung cancer as biomarkers and therapeutic targets. J. Cell. Mol. Med. 2014, 18, 2425–2436. [Google Scholar] [CrossRef] [PubMed]
- Foa, P.; Fornier, M.; Miceli, R.; Seregni, E.; Santambrogio, L.; Nosotti, M.; Cataldo, I.; Sala, M.; Caldiera, S.; Bombardieri, E. Tumour markers CEA, NSE, SCC, TPA and CYFRA 21.1 in resectable non-small cell lung cancer. Anticancer Res. 1999, 19, 3613–3618. [Google Scholar] [PubMed]
- Niklinski, J.; Furman, M. Clinical tumour markers in lung cancer. Eur. J. Cancer Prev. 1995, 4, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Kasimir-Bauer, S.; Schleucher, N.; Weber, R.; Neumann, R.; Seeber, S. Evaluation of different markers in non-small cell lung cancer: Prognostic value of clinical staging, tumour cell detection and tumour marker analysis for tumour progression and overall survival. Oncol. Rep. 2003, 10, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Tarro, G.; Perna, A.; Esposito, C. Early diagnosis of lung cancer by detection of tumor liberated protein. J. Cell. Physiol. 2005, 203, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Jacot, W.; Lhermitte, L.; Dossat, N.; Pujol, J.L.; Molinari, N.; Daurès, J.P.; Maudelonde, T.; Mangé, A.; Solassol, J. Serum proteomic profiling of lung cancer in high-risk groups and determination of clinical outcomes. J. Thoracic Oncol. 2008, 3, 840–850. [Google Scholar] [CrossRef] [PubMed]
- Solassol, J.; Maudelonde, T.; Mange, A.; Pujol, J.L. Clinical relevance of autoantibody detection in lung cancer. J. Thoracic Oncol. 2011, 6, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.G.; Johnen, G.; Casjens, S.; Bryk, O.; Pesch, B.; Jockel, K.H.; Kollmeier, J.; Brüning, T. Evaluation of long noncoding RNA MALAT1 as a candidate blood-based biomarker for the diagnosis of non-small cell lung cancer. BMC Res. Notes 2013. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef] [PubMed]
- Stahlhut, C.; Slack, F.J. Combinatorial Action of MicroRNAs let-7 and miR-34 Effectively Synergizes with Erlotinib to Suppress Non-small Cell Lung Cancer Cell Proliferation. Cell Cycle 2015, 14, 2171–2180. [Google Scholar] [CrossRef] [PubMed]
- Kasinski, A.L.; Kelnar, K.; Stahlhut, C.; Orellana, E.; Zhao, J.; Shimer, E.; Dysart, S.; Chen, X.; Bader, A.G.; Slack, F.J. A combinatorial microRNA therapeutics approach to suppressing non-small cell lung cancer. Oncogene 2015, 34, 3547–3555. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Liu, J.; Ma, S.; Zhang, S. Long Noncoding RNA MALAT-1 Can Predict Metastasis and a Poor Prognosis: A Meta-Analysis. Pathol. Oncol. Res. 2015, 21, 1259–1264. [Google Scholar] [CrossRef] [PubMed]
- Landi, M.T.; Zhao, Y.; Rotunno, M.; Koshiol, J.; Liu, H.; Bergen, A.W.; Rubagotti, M.; Goldstein, A.M.; Linnoila, I.; Marincola, F.M.; et al. MicroRNA expression differentiates histology and predicts survival of lung cancer. Clin. Cancer Res. 2010, 16, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, M.; Romano, G.; Di Leva, G.; Nuovo, G.; Jeon, YJ.; Ngankeu, A.; Sun, J.; Lovat, F.; Alder, H.; Condorelli, G.; et al. EGFR and MET receptor tyrosine kinase-altered microRNA expression induces tumorigenesis and gefitinib resistance in lung cancers. Nat. Med. 2012, 18, 74–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, J.; Meng, F.; Chan, L.W.; Cho, W.C.; Wong, S.C. Circulating Plasma MicroRNAs As Diagnostic Markers for NSCLC. Front Genet. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kunz, M.; Xiao, K.; Liang, C.; Viereck, J.; Pachel, C.; Frantz, S.; Thum, T.; Dandekar, T. Bioinformatics of cardiovascular miRNA biology. J. Mol. Cell. Cardiol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Erson, A.E.; Petty, E.M. miRNAs and cancer: New research developments and potential clinical applications. Cancer Biol. Therapy 2009, 8, 2317–2322. [Google Scholar] [CrossRef]
- Wang, Z. MicroRNA: A matter of life or death. World J. Biol. Chem. 2010, 1, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Jansson, M.D.; Lund, A.H. MicroRNA and cancer. Mol. Oncol. 2012, 6, 590–610. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Shen, Y.; Wang, M.; Yang, R.; Wang, Z.; Sui, A.; Jiao, W.; Wang, Y. Identification of plasma microRNAs as novel noninvasive biomarkers for early detection of lung cancer. Eur. J. Cancer Prev. 2013, 22, 540–548. [Google Scholar] [CrossRef] [PubMed]
- McKiernan, P.J.; Greene, C.M. High-throughput profiling for discovery of non-coding RNA biomarkers of lung disease. Expert Rev. Mol. Diagn. 2016, 16, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Geng, Q.; Fan, T.; Zhang, B.; Wang, W.; Xu, Y.; Hu, H. Five microRNAs in plasma as novel biomarkers for screening of early-stage non-small cell lung cancer. Respir. Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Zhou, K.; Zha, Y.; Chen, D.; He, J.; Ma, H.; Liu, X.; Le, H.; Zhang, Y. Diagnostic Value of Serum miR-182, miR-183, miR-210, and miR-126 Levels in Patients with Early-Stage Non-Small Cell Lung Cancer. PLoS ONE 2016, 11, e0153046. [Google Scholar] [CrossRef] [PubMed]
- Bjaanaes, M.M.; Halvorsen, A.R.; Solberg, S.; Jorgensen, L.; Dragani, T.A.; Galvan, A.; Galvan, A.; Colombo, F.; Anderlini, M.; Pastorino, U.; Kure, E.; et al. Unique microRNA-profiles in EGFR-mutated lung adenocarcinomas. Int. J. Cancer 2014, 135, 1812–1821. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.L.; Chen, H.Y.; Chang, G.C.; Chen, C.Y.; Chen, H.W.; Singh, S.; Cheng, C.L.; Yu, C.J.; Lee, Y.C.; Chen, H.S.; et al. MicroRNA signature predicts survival and relapse in lung cancer. Cancer Cell 2008, 13, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Yanaihara, N.; Caplen, N.; Bowman, E.; Seike, M.; Kumamoto, K.; Yi, M.; Stephens, R.M.; Okamoto, A.; Yokota, J.; Tanaka, T.; et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 2006, 9, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Schetter, A.J.; Mollerup, S.; Kohno, T.; Skaug, V.; Bowman, E.D.; Mathé, E.A.; Takenoshita, S.; Yokota, J.; Haugen, A.; et al. The association of microRNA expression with prognosis and progression in early-stage, non-small cell lung adenocarcinoma: A retrospective analysis of three cohorts. Clin. Cancer Res. 2011, 17, 1875–1882. [Google Scholar] [CrossRef] [PubMed]
- Capodanno, A.; Boldrini, L.; Proietti, A.; Ali, G.; Pelliccioni, S.; Niccoli, C.; D′Incecco, A.; Cappuzzo, F.; Chella, A.; Lucchi, M.; et al. Let-7g and miR-21 expression in non-small cell lung cancer: Correlation with clinicopathological and molecular features. Int. J. Oncol. 2013, 43, 765–774. [Google Scholar] [PubMed]
- Lee, J.H.; Voortman, J.; Dingemans, A.M.; Voeller, D.M.; Pham, T.; Wang, Y.; Giaccone, G. MicroRNA expression and clinical outcome of small cell lung cancer. PLoS ONE 2011, 6, e21300. [Google Scholar] [CrossRef] [PubMed]
- Lebanony, D.; Benjamin, H.; Gilad, S.; Ezagouri, M.; Dov, A.; Ashkenazi, K.; Gefen, N.; Izraeli, S.; Rechavi, G.; Pass, H.; et al. Diagnostic assay based on hsa-miR-205 expression distinguishes squamous from nonsquamous non-small-cell lung carcinoma. J. Clin. Oncol. 2009, 27, 2030–2037. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.A.; Benjamin, H.; Cholakh, H.; Chajut, A.; Clark, D.P.; Westra, W.H. Accurate classification of non-small cell lung carcinoma using a novel microRNA-based approach. Clin. Cancer Res. 2010, 16, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Hennessey, P.T.; Sanford, T.; Choudhary, A.; Mydlarz, W.W.; Brown, D.; Adai, A.T.; Ochs, M.F.; Ahrendt, S.A.; Mambo, E.; Califano, J.A. Serum microRNA biomarkers for detection of non-small cell lung cancer. PLoS ONE 2012, 7, e32307. [Google Scholar] [CrossRef]
- Markou, A.; Sourvinou, I.; Vorkas, P.A.; Yousef, G.M.; Lianidou, E. Clinical evaluation of microRNA expression profiling in non small cell lung cancer. Lung Cancer 2013, 81, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.X.; Bian, H.B.; Wang, J.R.; Cheng, Z.X.; Wang, K.M.; De, W. Prognostic significance of serum miRNA-21 expression in human non-small cell lung cancer. J. Surg. Oncol. 2011, 104, 847–851. [Google Scholar] [CrossRef] [PubMed]
- Vosa, U.; Vooder, T.; Kolde, R.; Vilo, J.; Metspalu, A.; Annilo, T. Meta-analysis of microRNA expression in lung cancer. Int. J. Cancer 2013, 132, 2884–2893. [Google Scholar] [CrossRef] [PubMed]
- Guan, P.; Yin, Z.; Li, X.; Wu, W.; Zhou, B. Meta-analysis of human lung cancer microRNA expression profiling studies comparing cancer tissues with normal tissues. J. Exp. Clin. Cancer Res. 2012, 31. [Google Scholar] [CrossRef] [PubMed]
- Nagano, T.; Fraser, P. No-Nonsense Functions for Long Noncoding RNAs. Cell 2011, 145, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and Functions of Long Noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T. Epigenetic regulation by long noncoding RNAs. Science 2012, 338, 1435–1439. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I.; Bartel, D.P. lincRNAs: Genomics, evolution, and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [PubMed]
- Prasanth, K.V.; Spector, D.L. Eukaryotic regulatory RNAs: An answer to the “genome complexity” conundrum. Genes Dev. 2007, 21, 11–42. [Google Scholar] [CrossRef] [PubMed]
- Volders, P.J.; Helsens, K.; Wang, X.; Menten, B.; Martens, L.; Gevaert, K.; Vandesompele, J.; Mestdagh, P. LNCipedia: A database for annotated human lncRNA transcript sequences and structures. Nucleic Acids Res. 2013, 41, D246–D251. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Deb, A.; Maji, R.K.; Saha, S.; Ghosh, Z. LncRBase: An enriched resource for lncRNA Information. PLoS ONE 2014, 9, e108010. [Google Scholar] [CrossRef] [PubMed]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [PubMed]
- Signal, B.; Gloss, B.S.; Dinger, M.E. Computational approaches for functional prediction and characterisation of long noncoding RNAs. Trends Genet. 2016, 32, 620–637. [Google Scholar] [CrossRef] [PubMed]
- Kumarswamy, R.; Bauters, C.; Volkmann, I.; Maury, F.; Fetisch, J.; Holzmann, A.; Lemesle, G.; de Groote, P.; Pinet, F.; Thum, T. Circulating long noncoding RNA, LIPCAR, predicts survival in patients with heart failure. Circ. Res. 2014, 114, 1569–1575. [Google Scholar] [CrossRef] [PubMed]
- Panzitt, K.; Tschernatsch, M.M.; Guelly, C.; Moustafa, T.; Stradner, M.; Strohmaier, H.M.; Buck, C.R.; Denk, H.; Schroeder, R.; Trauner, M.; et al. Characterization of HULC, a novel gene with striking up-regulation in hepatocellular carcinoma, as noncoding RNA. Gastroenterology 2007, 132, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Durand, X.; Moutereau, S.; Xylinas, E.; de la Taille, A. Progensa PCA3 test for prostate cancer. Expert Rev. Mol. Diagn. 2011, 11, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Reis, E.M.; Verjovski-Almeida, S. Perspectives of long non-coding RNAs in cancer diagnostics. Front. Genetic. 2012. [Google Scholar] [CrossRef] [PubMed]
- Vencken, S.F.; Greene, C.M.; McKiernan, P.J. Non-coding RNA as lung disease biomarkers. Thorax 2015, 70, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Diederichs, S.; Wang, W.; Boing, S.; Metzger, R.; Schneider, P.M.; Tidow, N.; Brandt, B.; Buerger, H.; Bulk, E.; et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003, 22, 8031–8041. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.; Xu, Y.; Yang, X.; Wang, J.; Hu, J.; Xu, L.; Yin, R. CCAT2 is a lung adenocarcinoma-specific long non-coding RNA and promotes invasion of non-small cell lung cancer. Tumour Biol. 2014, 35, 5375–5380. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wu, H.; Lv, N.; Wang, H.; Wang, Y.; Tang, Q.; Shao, H.; Sun, C. LncRNA CCAT2 predicts poor prognosis and regulates growth and metastasis in small cell lung cancer. Biomed. Pharmacother. 2016, 82, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.H.; Liu, Z.L.; Sun, M.; Liu, J.; Wang, Z.X.; De, W. The long non-coding RNA HOTAIR indicates a poor prognosis and promotes metastasis in non-small cell lung cancer. BMC Cancer 2013. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, H.; Hou, S.; Hu, B.; Liu, J.; Wang, J. The noncoding RNA expression profile and the effect of lncRNA AK126698 on cisplatin resistance in non-small-cell lung cancer cell. PLoS ONE 2013, 8, e65309. [Google Scholar] [CrossRef] [PubMed]
- Sui, J.; Li, Y.H.; Zhang, Y.Q.; Li, C.Y.; Shen, X.; Yao, W.Z.; Peng, H.; Hong, W.W.; Yin, L.H.; Pu, Y.P.; et al. Integrated analysis of long non-coding RNAassociated ceRNA network reveals potential lncRNA biomarkers in human lung adenocarcinoma. Int. J. Oncol. 2016, 49, 2023–2036. [Google Scholar] [PubMed]
- Tantai, J.; Hu, D.; Yang, Y.; Geng, J. Combined identification of long non-coding RNA XIST and HIF1A-AS1 in serum as an effective screening for non-small cell lung cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 7887–7895. [Google Scholar] [PubMed]
- Gong, W.J.; Yin, J.Y.; Li, X.P.; Fang, C.; Xiao, D.; Zhang, W.; Zhou, H.H.; Li, X.; Liu, Z.Q. Association of well-characterized lung cancer lncRNA polymorphisms with lung cancer susceptibility and platinum-based chemotherapy response. Tumour Biol. 2016, 37, 8349–8358. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Liu, H.; Liu, Z.; Owzar, K.; Han, Y.; Su, L.; Wei, Y.; Hung, R.J.; McLaughlin, J.; Brhane, Y.; et al. A novel genetic variant in long non-coding RNA gene NEXN-AS1 is associated with risk of lung cancer. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Loewen, G.; Jayawickramarajah, J.; Zhuo, Y.; Shan, B. Functions of lncRNA HOTAIR in lung cancer. J. Hematol. Oncol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, B.; Wang, T.; Wang, H. LncRNA MALAT1 overexpression is an unfavorable prognostic factor in human cancer: evidence from a meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 5499–5505. [Google Scholar] [PubMed]
- Hong, H.H.; Hou, L.K.; Pan, X.; Wu, C.Y.; Huang, H.; Li, B.; Nie, W. Long non-coding RNA UCA1 is a predictive biomarker of cancer. Oncotarget 2016, 7, 44442–44447. [Google Scholar] [CrossRef] [PubMed]
- He, A.; Hu, R.; Chen, Z.; Liao, X.; Li, J.; Wang, D.; Lv, Z.; Liu, Y.; Wang, F.; Mei, H. Role of long noncoding RNA UCA1 as a common molecular marker for lymph node metastasis and prognosis in various cancers: A meta-analysis. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Long, X.; Lin, M.; Bergmann, J.H.; Nanda, V.; Cowan, S.L.; Zhou, Q.; Han, Y.; Spector, D.L.; Zheng, D.; et al. Identification and initial functional characterization of a human vascular cell enriched long non-coding RNA. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Muppirala, U.K.; Honavar, V.G.; Dobbs, D. Predicting RNA-protein interactions using only sequence information. BMC Bioinform. 2011, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.B.; Hughes, T.R.; Morris, Q.D. High-throughput characterization of protein-RNA interactions. Brief. Funct. Genom. 2015, 14, 74–89. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Burge, S.W.; Bateman, A.; Daub, J.; Eberhardt, R.Y.; Eddy, S.R.; Floden, E.W.; Gardner, P.P.; Jones, T.A.; Tate, J.; et al. Rfam 12.0: Updates to the RNA families database. Nucleic Acids Res. 2015, 43, D130–D137. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Hofacker, I.L. Vienna RNA secondary structure server. Nucleic Acids Res. 2003, 31, 3429–3431. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Bernhart, S.; Hofacker, I.; Will, S.; Gruber, A.; Stadler, P. RNAalifold: Improved consensus structure prediction for RNA alignments. BMC Bioinform. 2008. [Google Scholar] [CrossRef] [PubMed]
- Havgaard, J.H.; Lyngsø, R.B.; Gorodkin, J. The foldalign web server for pairwise structural RNA alignment and mutual motif search. Nucleic Acids Res. 2005, 33, W650–W653. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Heyne, S.; Richter, A.S.; Will, S.; Backofen, R. Freiburg RNA Tools: A web server integrating INTARNA, EXPARNA and LOCARNA. Nucleic Acids Res. 2010, 38, W373–W377. [Google Scholar] [CrossRef] [PubMed]
- Steffen, P.; Voss, B.; Rehmsmeier, M.; Reeder, J.; Giegerich, R. RNAshapes: An integrated RNA analysis package based on abstract shapes. Bioinformatics 2006, 22, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Seibel, P.N.; Müller, T.; Dandekar, T.; Schultz, J.; Wolf, M. 4SALE—A tool for synchronous RNA sequence and secondary structure alignment and editing. BMC Bioinform. 2006, 7, 1–7. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Carbon, S.; Ireland, A.; Mungall, C.J.; Shu, S.; Marshall, B.; Lewis, S.; Web Presence Working Group. AmiGO: Online access to ontology and annotation data. Bioinformatics 2009, 25, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Poudel, S.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. PANTHER version 10: Expanded protein families and functions, and analysis tools. Nucleic Acids Res. 2016, 44, D336–D342. [Google Scholar] [CrossRef] [PubMed]
- Croft, D.; O’Kelly, G.; Wu, G.; Haw, R.; Gillespie, M.; Matthews, L.; Caudy, M.; Garapati, P.; Gopinath, G.; Jassal, B.; et al. Reactome: A database of reactions, pathways and biological processes. Nucleic Acids Res. 2011, 39, D691–D697. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Furumichi, M.; Tanabe, M.; Hirakawa, M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010, 38, D355–D360. [Google Scholar] [CrossRef] [PubMed]
- Kutmon, M.; Riutta, A.; Nunes, N.; Hanspers, K.; Willighagen Egon, L.; Bohler, A.; Mélius, J.; Waagmeester, A.; Sinha, S.R.; Miller, R.; et al. WikiPathways: Capturing the full diversity of pathway knowledge. Nucleic Acids Res. 2016, 44, D488–D494. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Lotia, S.; Pico, A.R.; Bader, G.D.; Ideker, T. A travel guide to Cytoscape plugins. Nat. Methods 2012, 9, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-H.; Liu, S.; Zhou, H.; Qu, L.-H.; Yang, J.-H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [PubMed]
- Wingender, E.; Chen, X.; Hehl, R.; Karas, H.; Liebich, I.; Matys, V.; Meinhardt, T.; Prüß, M.; Reuter, I.; Schacherer, F. TRANSFAC: An integrated system for gene expression regulation. Nucleic Acids Res. 2000, 28, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Sandelin, A.; Alkema, W.; Engstrom, P.; Wasserman, W.W.; Lenhard, B. JASPAR: An open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004, 32, D91–D94. [Google Scholar] [CrossRef] [PubMed]
- Messeguer, X.; Escudero, R.; Farre, D.; Nunez, O.; Martinez, J.; Alba, M.M. PROMO: Detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 2002, 18, 333–334. [Google Scholar] [CrossRef] [PubMed]
- Cartharius, K.; Frech, K.; Grote, K.; Klocke, B.; Haltmeier, M.; Klingenhoff, A.; Frisch, M.; Bayerlein, M.; Werner, T. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics 2005, 21, 2933–2942. [Google Scholar] [CrossRef] [PubMed]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D.S. MicroRNA targets in Drosophila. Genome Biol. 2003. [Google Scholar] [CrossRef]
- Muckstein, U.; Tafer, H.; Hackermuller, J.; Bernhart, S.H.; Stadler, P.F.; Hofacker, I.L. Thermodynamics of RNA-RNA binding. Bioinformatics 2006, 22, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Busch, A.; Richter, A.S.; Backofen, R. IntaRNA: efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics 2008, 24, 2849–2856. [Google Scholar] [CrossRef] [PubMed]
- RNAcentral Consortium. RNAcentral: An international database of ncRNA sequences. Nucleic Acids Res. 2015, 43, D123–D129. [Google Scholar]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Wang, Y.; Hao, Y.; Juan, L.; Teng, M.; Zhang, X.; Li, M.; Wang, G.; Liu, Y. miR2Disease: A manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009, 37, D98–D104. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Shih, I.H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of mammalian microRNA targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef]
- Krek, A.; Grun, D.; Poy, M.N.; Wolf, R.; Rosenberg, L.; Epstein, E.J.; MacMenamin, P.; Da Piedade, I.; Gunsalus, K.C.; Stoffel, M.; et al. Combinatorial microRNA target predictions. Nat. Genet. 2005, 37, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Kertesz, M.; Iovino, N.; Unnerstall, U.; Gaul, U.; Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007, 39, 1278–1284. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wang, Z.; Wang, D.; Qiu, C.; Liu, M.; Chen, X.; Zhang, Q.; Yan, G.; Cui, Q. LncRNADisease: A database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 2013, 41, D983–D986. [Google Scholar] [CrossRef] [PubMed]
- Terai, G.; Iwakiri, J.; Kameda, T.; Hamada, M.; Asai, K. Comprehensive prediction of lncRNA–RNA interactions in human transcriptome. BMC Genom. 2016, 17, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ma, W.; Zeng, P.; Wang, J.; Geng, B.; Yang, J.; Cui, Q. LncTar: A tool for predicting the RNA targets of long noncoding RNAs. Brief Bioinform. 2015, 16, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Wang, J.; Liu, C.; Zhang, Y.; Shi, B.; Zhu, X.; Zhang, Z.; Skogerbø, G.; Chen, L.; Lu, H.; et al. NPInter: The noncoding RNAs and protein related biomacromolecules interaction database. Nucleic Acids Res. 2006, 34, D150–D152. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, B.C.; Westbrook, J.; Ghosh, S.; Petrov, A.I.; Sweeney, B.; Zirbel, C.L.; Leontis, N.B.; Berman, H.M. The Nucleic Acid Database: New features and capabilities. Nucleic Acids Res. 2014, 42, D114–D122. [Google Scholar] [CrossRef] [PubMed]
- Stark, C.; Breitkreutz, B.J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef] [PubMed]
- Hermjakob, H.; Montecchi-Palazzi, L.; Lewington, C.; Mudali, S.; Kerrien, S.; Orchard, S.; Vingron, M.; Roechert, B.; Roepstorff, P.; Valencia, A.; et al. IntAct: An open source molecular interaction database. Nucleic Acids Res. 2004, 32, D452–D455. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, S.; Hino, K.; Saito, A.; Miyano, S.; Miyamoto-Sato, E. PRD: A protein-RNA interaction database. Bioinformation 2012, 8, 729–730. [Google Scholar] [CrossRef] [PubMed]
- Orchard, S.; Kerrien, S.; Abbani, S.; Aranda, B.; Bhate, J.; Bidwell, S.; Bridge, A.; Briganti, L.; Brinkman, F.S.; Cesareni, G.; et al. Protein interaction data curation: The International Molecular Exchange (IMEx) consortium. Nat. Methods 2012, 9, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.U. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.A.; Walia, R.R.; Terribilini, M.; Ferguson, J.; Zheng, C.; Honavar, V.; et al. PRIDB: A protein–RNA interface database. Nucleic Acids Res. 2011, 39, D277–D282. [Google Scholar] [CrossRef] [PubMed]
- Kunz, M.; Liang, C.; Nilla, S.; Cecil, A.; Dandekar, T. The drug-minded protein interaction database (DrumPID) for efficient target analysis and drug development. Database 2016. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, M.; Agostini, F.; Masin, M.; Tartaglia, G.G. Predicting protein associations with long noncoding RNAs. Nat. Methods 2011, 8, 444–445. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Gromiha, M.M.; Raghava, G.P. Prediction of RNA binding sites in a protein using SVM and PSSM profile. Proteins 2008, 71, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.T.; Yura, K.; Go, N. Amino acid residue doublet propensity in the protein-RNA interface and its application to RNA interface prediction. Nucleic Acids Res. 2006, 34, 6450–6460. [Google Scholar] [CrossRef] [PubMed]
- Towfic, F.; Caragea, C.; Gemperline, D.C.; Dobbs, D.; Honavar, V. Struct-NB: Predicting protein-RNA binding sites using structural features. Int. J. Data Min. Bioinform. 2010, 4, 21–43. [Google Scholar] [CrossRef]
- Wang, Y.; Xue, Z.; Shen, G.; Xu, J. PRINTR: Prediction of RNA binding sites in proteins using SVM and profiles. Amino Acids 2008, 35, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Yu, N.; Choi, I.; Kim, W.; Lee, S. lncRNAtor: A comprehensive resource for functional investigation of long non-coding RNAs. Bioinformatics 2014, 30, 2480–2485. [Google Scholar] [CrossRef] [PubMed]
- Dalli, D.; Wilm, A.; Mainz, I.; Steger, G. STRAL: Progressive alignment of non-coding RNA using base pairing probability vectors in quadratic time. Bioinformatics 2006, 22, 1593–1599. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.P.; Cho, J.H.; Hood, L.; Franco, O.L.; Pereira, R.W.; Wang, K. A Review of computational tools in microRNA discovery. Front. Genet. 2013. [Google Scholar] [CrossRef] [PubMed]
- Sankoff, D. Simultaneous solution of the RNA folding, alignment and protosequence problems. SIAM J. Appl. Math. 1985, 45, 810–825. [Google Scholar] [CrossRef]
- Hofacker, I.L.; Fekete, M.; Stadler, P.F. Secondary structure prediction for aligned RNA sequences. J. Mol. Biol. 2002, 319, 1059–1066. [Google Scholar] [CrossRef]
- Havgaard, J.H.; Gorodkin, J. RNA structural alignments, part I: Sankoff-based approaches for structural alignments. Methods Mol. Biol. 2014, 1097, 275–290. [Google Scholar] [PubMed]
- Reeder, J.; Giegerich, R. Consensus shapes: An alternative to the Sankoff algorithm for RNA consensus structure prediction. Bioinformatics 2005, 21, 3516–3523. [Google Scholar] [CrossRef] [PubMed]
- Sethupathy, P.; Megraw, M.; Hatzigeorgiou, A.G. A guide through present computational approaches for the identification of mammalian microRNA targets. Nat. Methods 2006, 3, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Friedersdorf, M.B.; Keene, J.D. Advancing the functional utility of PAR-CLIP by quantifying background binding to mRNAs and lncRNAs. Genome Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Muppirala, U.K.; Lewis, B.A.; Dobbs, D. Computational tools for investigating RNA-protein interaction partners. J. Comput. Sci. Syst. Biol. 2013, 6, 182–187. [Google Scholar]
- Cirillo, D.; Agostini, F.; Tartaglia, G.G. Predictions of protein–RNA interactions. Wiley Interdiscip. Rev. 2013, 3, 161–175. [Google Scholar] [CrossRef]
- Hammell, M. Computational methods to identify miRNA targets. Seminars Cell Dev. Biol. 2010, 21, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, J.; Cairns, M.J. Identifying miRNAs, targets and functions. Brief. Bioinform. 2014, 15, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The microRNA.org resource: Targets and expression. Nucleic Acids Res. 2008, 36, D149–D153. [Google Scholar] [CrossRef] [PubMed]
- Kiriakidou, M.; Nelson, P.T.; Kouranov, A.; Fitziev, P.; Bouyioukos, C.; Mourelatos, Z.; Hatzigeorgiou, A. A combined computational-experimental approach predicts human microRNA targets. Genes Dev. 2004, 18, 1165–1178. [Google Scholar] [CrossRef] [PubMed]
- Maziere, P.; Enright, A.J. Prediction of microRNA targets. Drug Discov. Today 2007, 12, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Yue, D.; Liu, H.; Huang, Y. Survey of computational algorithms for microRNA Target prediction. Curr. Genom. 2009, 10, 478–492. [Google Scholar] [CrossRef] [PubMed]
- Rajewsky, N. microRNA target predictions in animals. Nat. Genet. 2006, 38, S8–S13. [Google Scholar] [CrossRef] [PubMed]
- Gusev, Y.; Schmittgen, T.D.; Lerner, M.; Postier, R.; Brackett, D. Computational analysis of biological functions and pathways collectively targeted by co-expressed microRNAs in cancer. BMC Bioinform. 2007. [Google Scholar] [CrossRef] [PubMed]
- Stark, A.; Brennecke, J.; Bushati, N.; Russell, R.B.; Cohen, S.M. Animal MicroRNAs confer robustness to gene expression and have a significant impact on 3′UTR evolution. Cell 2005, 123, 1133–1146. [Google Scholar] [CrossRef] [PubMed]
- Marshall, O.J. PerlPrimer: Cross-platform, graphical primer design for standard, bisulphite and real-time PCR. Bioinformatics 2004, 20, 2471–2472. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Varani, G. Engineering RNA-binding proteins for biology. FEBS J. 2013, 280, 3734–3754. [Google Scholar] [CrossRef] [PubMed]
- Walia, R.R.; Caragea, C.; Lewis, B.A.; Towfic, F.; Terribilini, M.; El-Manzalawy, Y.; Dobbs, D.; Honavar, V. Protein-RNA interface residue prediction using machine learning: An assessment of the state of the art. BMC Bioinform. 2012. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Cai, Q.; Sun, F.; Zhong, G.; Wang, P.; Liu, H.; Luo, J.; Yu, H.; Huang, J.; Lin, T. linc-UBC1 physically associates with polycomb repressive complex 2 (PRC2) and acts as a negative prognostic factor for lymph node metastasis and survival in bladder cancer. Biochim. Biophys. Acta 2013, 1832, 1528–1537. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Ge, M.; Zhang, Y.; Peng, C.; Wang, M. Predicting long noncoding RNA and protein interactions using heterogeneous network model. Biomed. Res. Int. 2015. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Jiang, P.; Lu, Z.H. RISP: A web-based server for prediction of RNA-binding sites in proteins. Comput. Methods Programs Biomed. 2008, 90, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yang, Y.; Zhou, Y. Highly accurate and high-resolution function prediction of RNA binding proteins by fold recognition and binding affinity prediction. RNA Biol. 2011, 8, 988–996. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Sidiropoulos, K.; Garapati, P.; Gillespie, M.; Hausmann, K.; Haw, R. The Reactome pathway Knowledgebase. Nucleic Acids Res. 2016, 44, D481–D487. [Google Scholar] [CrossRef] [PubMed]
- Herwig, R.; Hardt, C.; Lienhard, M.; Kamburov, A. Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat. Protoc. 2016, 11, 1889–1907. [Google Scholar] [CrossRef] [PubMed]
- Tipney, H.; Hunter, L. An introduction to effective use of enrichment analysis software. Hum. Genom. 2010, 4, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Khatri, P.; Sirota, M.; Butte, A.J. Ten Years of pathway analysis: Current Approaches and outstanding challenges. PLoS Comput. Biol. 2012, 8, e1002375. [Google Scholar] [CrossRef] [PubMed]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef] [PubMed]
- Bader, G.D.; Hogue, C.W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; et al. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Bonnici, V.; Russo, F.; Bombieri, N.; Pulvirenti, A.; Giugno, R. Comprehensive reconstruction and visualization of non-coding regulatory networks in human. Front. Bioeng. Biotechnol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Stormo, G.D. DNA binding sites: Representation and discovery. Bioinformatics 2000, 16, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Giardine, B.; Riemer, C.; Hardison, R.C.; Burhans, R.; Elnitski, L.; Shah, P.; Zhang, Y.; Blankenberg, D.; Albert, I.; Taylor, J. Galaxy: A platform for interactive large-scale genome analysis. Genome Res. 2005, 15, 1451–1455. [Google Scholar] [CrossRef] [PubMed]
- Goodstadt, L. Ruffus: A lightweight Python library for computational pipelines. Bioinformatics 2010, 26, 2778–2779. [Google Scholar] [CrossRef] [PubMed]
- Koster, J.; Rahmann, S. Snakemake—A scalable bioinformatics workflow engine. Bioinformatics 2012, 28, 2520–2522. [Google Scholar] [CrossRef] [PubMed]
- Dooley, D.M.; Petkau, A.J.; Van Domselaar, G.; Hsiao, W.W. Sequence database versioning for command line and Galaxy bioinformatics servers. Bioinformatics 2016, 32, 1275–1277. [Google Scholar] [CrossRef] [PubMed]
- Boettiger, C. An introduction to Docker for reproducible research, with examples from the R environment. ACM SIGOPS Oper. Syst. Rev. 2015. [Google Scholar] [CrossRef]
- Sundaresan, T.K.; Haber, D.A. Fantastic voyage: The future of cancer diagnostics. Lancet Oncol. 2015, 16, 1596–1598. [Google Scholar] [CrossRef]
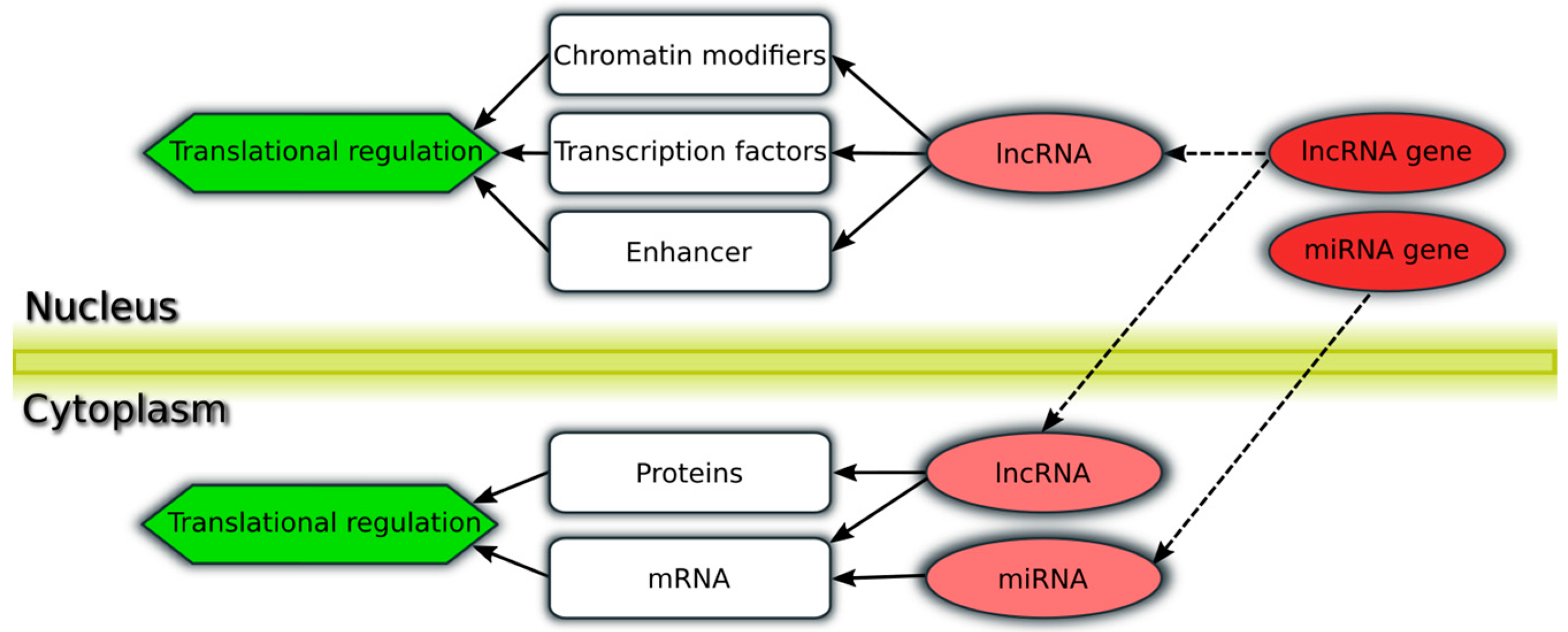
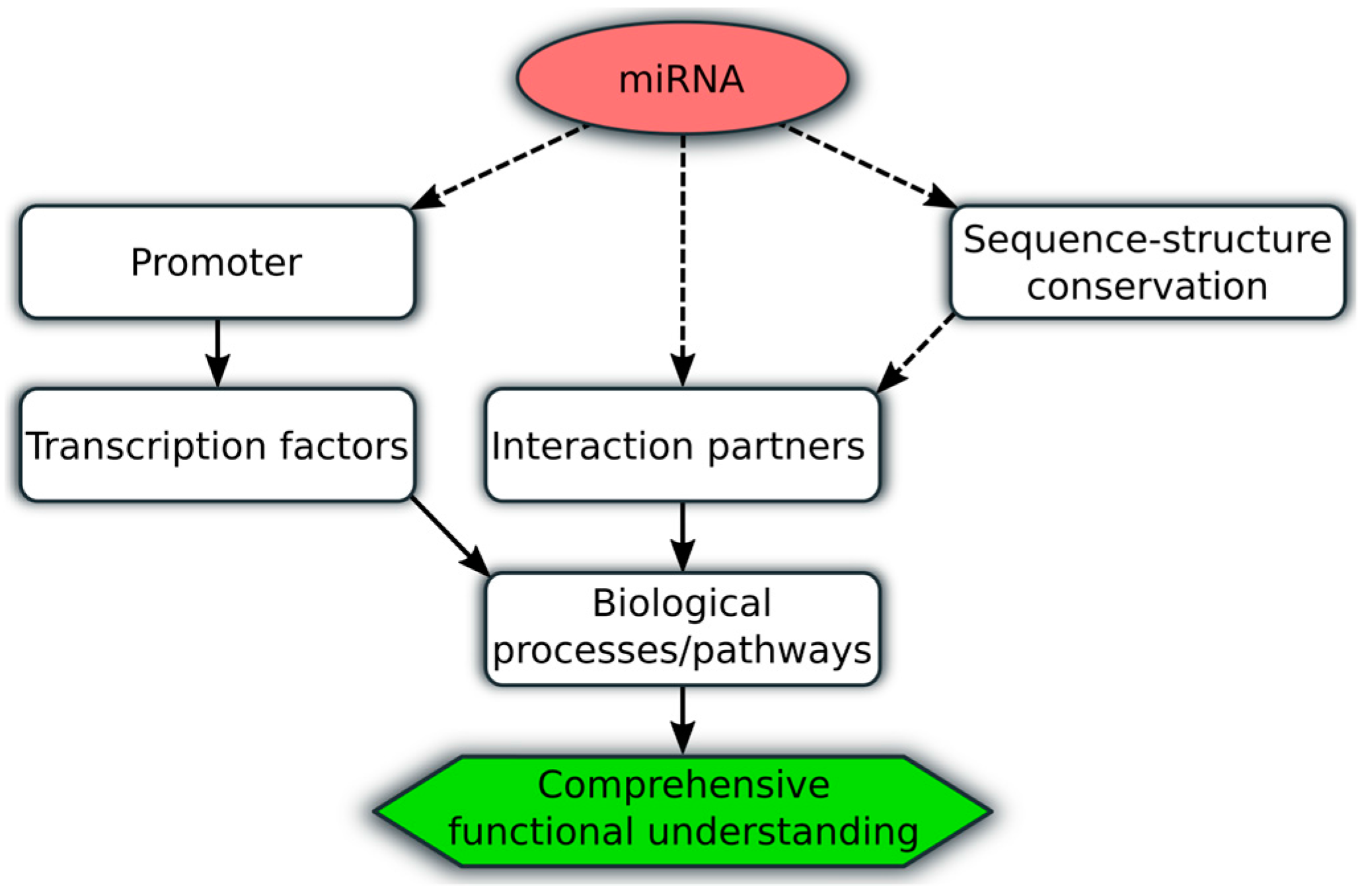


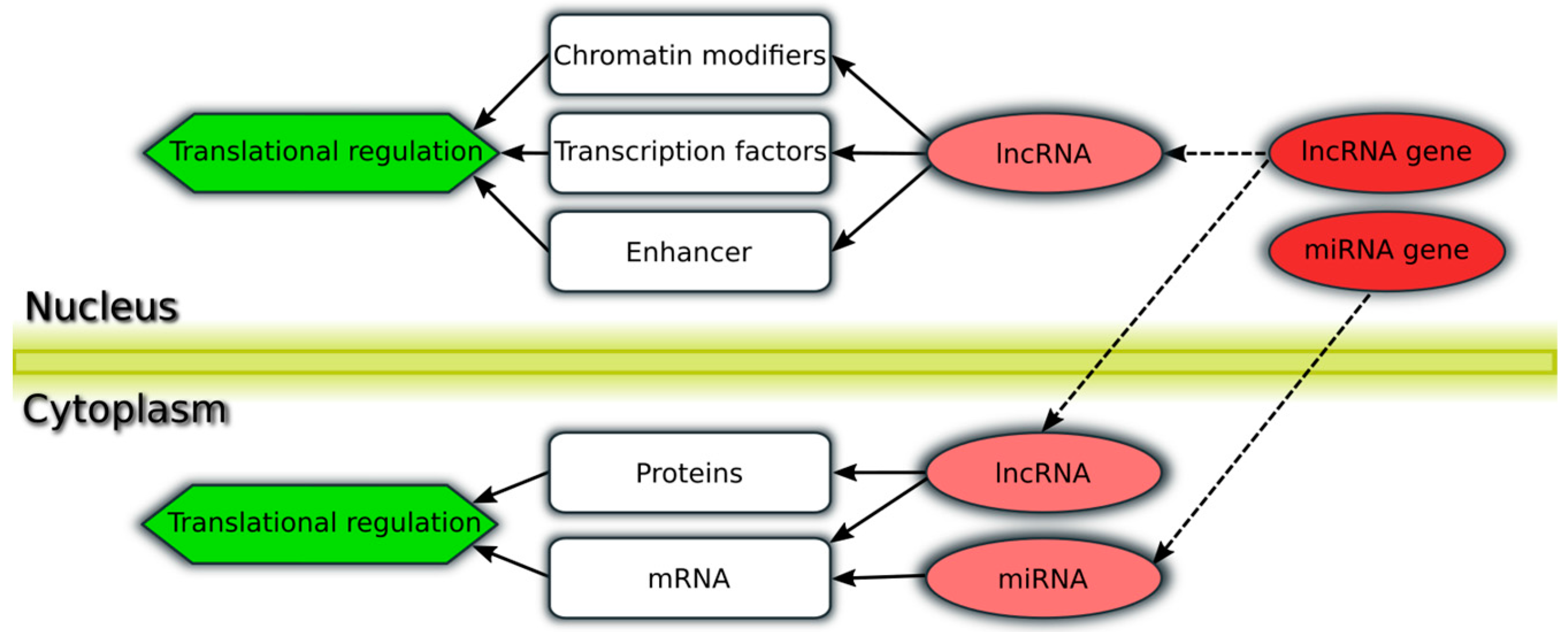{kind=link}
{kind=link}
{kind=link}
| Study (Ref.) | Patient Cohort | Important Reported Findings |
|---|---|---|
| Tang et al. (2013) [37] | Training set: 62 patients/60 healthy smokers Test set: 34 malignant tumor patients, 30 patients with benign pulmonary nodules, 32 healthy smokers | Plasma miRNA-21, miRNA-145 and miRNA-155 have strong potential as novel noninvasive biomarkers for early detection of lung cancer |
| Yu et al. (2008) [42] | 112 NSCLC patients: AC (55), SQ (50), others (7) | five-miRNA signature (miRNA-221, let-7a, miRNA-137, miRNA-372, miRNA-182 *) for NSCLC treatment prediction outcome |
| Yanaihara et al. (2006) [43] | AC (65), AC normal (65); SQ (39), SQ normal (39) Stage classification: AC: stage I (41), stage II (8), stages III and IV (16); SQ: stage I (24), stage II (9), stages III and IV (6) | miRNA-155 and let-7a-2 correlates with poor survival; miRNA-205, miRNA-99b, miRNA-203, miRNA-202, miRNA-102 and miRNA-204-prec differently expressed in AC and SQ; miRNA-21, miRNA-191, miRNA-155, miRNA-210, miRNA-126 * and miRNA-224 differentiate AC and SQ |
| Geng et al. (2014) [39] | Training set: 25 NSCLC patients: AC (8), SQ (13), others (4); stage: I (9), II (16); 25 healthy controls Test sample: 126 NSCLC patients: AC (45), SQ (64), others (17); stage: I (54), II (72); 42 NCPD (non-cancerous pulmonary disease) patients; 60 healthy controls | Plasma miRNA-20a, miRNA-145, miRNA-21, miRNA-223 and miRNA-221 as potential biomarkers in early-stage NSCLC |
| Zhu et al. (2016) [40] | 112 NSCLC patients: AC (90), SQ (22); lymph node metastasis: negative (95), positive (17); stage: 0 (0), IA, IB (82), IIA, IIB (15), IIIA, IIIB (10); 104 controls (20 current healthy smokers, 23 pneumonia patients, 21 gastric cancer patients, 40 healthy controls) | Serum miRNA-182, miRNA-183, miRNA-210 and miRNA-126 levels serve as a diagnostic biomarker for NSCLC early detection; serum levels distinguish NSCLC or early-stage NSCLC from current smokers without lung cancer and pneumonia or gastric cancer patients |
| Bjaanaes et al. (2014) [41] | 154 resected AC: stage: IA (45), IB (46), IIA (24), IIB (12), IIIA (26), IV (1); EGFR status: mutated (22), wt (130), not tested (2); KRAS status: mutated (52), wt (96), not tested (6); 20 normal tissue samples; independent cohort of 103 lung cancer patients | 129 significantly differentially expressed miRNAs in AC compared with normal lung tissue; 17 differentially expressed miRNAs between EGFR-mutated and EGFR wildtype AC; miRNA-9, miRNA-21, miRNA-126, miRNA-133a and miRNA-500a deregulated in AC compared with normal lung tissue |
| Saito et al. (2011) [44] | 317 AC patient tissues: stage: I (220), II (76), III (21) | miRNA-21 is associated with disease progression and survival in stage I AC |
| Capodanno et al. (2013) [45] | 80 NSCLC patients: EGFR status: Wt: AC (32), SQ (20), LCC and other (3); stage: I (9), II (20), III (13), IV (3); mutated: AC (22), SQ (0), LCC and other (1); stage: I (3), II (11), III (1), IV (3) KRAS status: Wt: AC (39), SQ (20), LCC and other (3); stage: I (9), II (27), III (9), IV (4); mutated: AC (15), SQ (0), LCC and other (1); stage: I (3), II (4), III (5), IV (2) | let-7g and miRNA-21 combined with KRAS mutational status are useful biomarkers for NSCLC patients |
| Du et al. (2010) [5] | 19 lung cancer cell lines: 7 NSCLC (2 AC, 3 SQ, 2 other); 9 SCLC; 3 immortalized normal | 41 out of 136 differentially expressed miRNAs distinguish NSCLC and SCLC (e.g., miRNA-17-5p, miRNA-135, miRNA-103, miRNA-107, miRNA-301 and miRNA-338 altered in SCLC relative to NSCLC); miRNA-29a/b/c, miRNA-24, miRNA-21 and miRNA-221/222 down-regulated in SCLC |
| Lee et al. (2011) [46] | 26 NSCLC cell lines, 14 SCLC cell lines, 31 SCLC tumors | miRNA-21, miRNA-29b, miRNA-34a/b/c, miRNA-155 and let-7a not related to SCLC patients |
| Landi et al. (2010) [30] | 290 tissue samples: AC (165): stage: I (65), II (43), III (46), IV (11); SQ (125): stage: I (52), II (42), III (30), IV (1) | 34 miRNAs differentiate AC from SQ in male smoker patients; five-miRNA signature predicts survival for SQ |
| Lebanony et al. (2009) [47] | Training set: 122 AC and SQ; 47 NSCLC FFPE samples Test set: 79 blinded cohort of NSCLC FFPE samples | miRNA-205 expression distinguishes SQ from AC |
| Bishop et al. (2010) [48] | 102 resected NSCLC: AC (50): grades: well (9), moderate (24), poor (17); SQ (52): grades: well (2), moderate (35), poor (15); 21 preoperative biopsies/aspirates | miRNAs such as miRNA-205 are reliable to classify NSCLC |
| Montani et al. (2015) [14] | COSMOS study with high-risk individuals (n = 1115): Calibration Set: lung cancer (12): stage: I (11), II-III (1); lung cancer deaths (1); no lung cancer (12) Validation Set: lung cancer (36): stage: I (31), II-III (5); lung cancer deaths (3); no lung cancer (972); PN+COPD (81) Specificity Set: no lung cancer (83); PN + COPD + NOD (78); Benign (5) All (CT screening): lung cancer (48): stage: I (42), II-III (6); lung cancer deaths (4); no lung cancer (1067); PN + COPD + NOD (159); Benign (5) | miR-Test using 13 miRNAs (miRNA-92a-3p, miRNA-30b-5p, miRNA-191-5p, miRNA-484, miRNA-328-3p, miRNA-30c-5p, miRNA-374a-5p, let-7d-5p, miRNA-331-3p, miRNA-29a-3p, miRNA-148a-3p, miRNA-223-3p, miRNA-140-5p) represent a useful tool for lung cancer screening in high-risk individuals |
| Sozzi et al. (2014) [13] | MILD trial study: 939 plasma samples: no lung cancer (870); lung cancer (69): stage: I (37); II to III (12); IV (19) | Plasma-based miRNA signatures from patients in two independent LDCT screening studies of 24 circulating miRNAs has diagnostic and prognostic performance |
| Hennessey et al. (2012) [49]) | Phase I/II serum biomarker study: Training set: 30 NSCLC patients: AC (20), SQ (10); stage: I (10), II (9), III (10), IV (0); 20 healthy controls Test set: 55 NSCLC patients: AC (30), SQ (25); stage: I (33), II (13), III (7), IV (2); 75 healthy controls | Combination of miRNA-15b and miRNA-27b discriminate NSCLC from healthy controls |
| Markou et al. (2013) [50] | 59 resected NSCLC and adjacent normal tissue: Training set: 19 tumor/normal tissues Test set: 40 tumor: AC (16), SCC (21), Other (3); stage: I (11), II-IV (29); lymph node: negative (23), positive (17); plasma samples from 37 NSCLC patients and 28 healthy donors | 8 circulating plasma miRNAs (miRNA-21, miRNA-30d, miRNA-451, miRNA-10a, miRNA-30e-5p, miRNA-126 *, miRNA-126, miRNA-145) were differential expressed in NSCLC; miRNA-520d, miRNA-489, miRNA-181b, miRNA-513, miRNA-26b, miRNA-189 and miRNA-520e were differentially expressed between AC and SQ |
| Wang et al. (2011) [51] | 88 NSCLC patients: AC (37), SQ (21), other (30); stage: I–II (47), III (41); lymph node metastasis: No (53), Yes (35); 17 healthy individual | Serum miRNA-21 expression useful as a prognostic marker for NSCLC patients |
| Study (Ref.) | Patient Cohort | Important Reported Findings |
|---|---|---|
| Zhu et al. (2015) [29] | Meta-Analysis: 8 studies with 845 patients: NSCLC (2), colorectal cancer (1), gastric cancer (1), pancreatic cancer (2), clear cell renal cell carcinoma (1), osteosarcoma (1) | MALAT-1 serve as a molecular marker for cancer metastasis and prognosis |
| Ji et al. (2003) [68] | NSCLC patients (70): AC (26), SQ (34), LCC (10); stage: I (37), II (13), IIIA (20) | MALAT-1 and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer |
| Weber et al. (2013) [22] | 45 NSCLC patients: AC (21): Stage: I/II (0),III/IV (21); SQ (24): I/II (3), III/IV (21); 25 controls | MALAT1 as complementary diagnostic biomarker in NSCLC |
| Yao et al. (2012) [12] | 65 NSCLC patient serum: biopanning stage: tissue and serum samples (25): AC (12), SQ (12), LCC (1); stage: I (6), II (5), III (9), IV (5) identification stage: NSCLC serum samples (40): AC (17), SQ (22), LCC (1); stage: I (11), II (8), III (17), IV (4); 41 normal controls | A four serum biomarker SMOX, NOLC1, MALAT1 and HMMR show a high diagnostic accuracy for detecting early stage NSCLC |
| Qiu et al. (2014) [69] | NSCLC tissues/paired adjacent normal tissues | CCAT2 is an AC-specific lncRNA and promotes invasion of NSCLC; biomarker for lymph node metastasis. |
| Chen et al. (2016) [70] | SCLC tissues and cell lines | CCAT2 serves as an oncogenic lncRNA, and an independent unfavorable prognostic factor in SCLC patients |
| Liu et al. (2013) [71] | Tissues from 42 NSCLC/adjacent non-tumor lung patients: stage: I/II (25), III/IV (17); 4 NSCLC cell lines: AC (3; A549, SPC-A1, NCI-H1975); SQ (1; SK-MES-1); normal human bronchial epithelial cell line (1; 16HBE) | HOTAIR represent diagnostic biomarker of poor prognosis in NSCLC |
| Yang et al. (2013) [72] | A549 cells and cisplatin resistant A549/CDDP cells (microarray profiling of mRNAs, lncRNAs and miRNAs) | 8 mRNAs (BMP4, CTSB, NKD2, BAG1, TGFB1, EGFR, JUN, CUL2), 8 lncRNAs (AK123263, CES1P1-001, RP3-508I15.14, AK126698, TP53TG1, AC090952.4.1, uc003bgl.1, NCRNA00210) and 5 miRNAs (miRNA-17, miRNA-21, let-7i, miRNA-138, miRNA-194) potentially play a key role in cisplatin resistance; AK126698 appears to confer cisplatin resistance by targeting the Wnt pathway |
| Sui et al. (2016) [73] | 465 AC patient RNA sequencing profiles (from TCGA); 53 AC patients | Correlation of AFAP1-AS1 and LINC00472 as potential biomarkers for diagnosis and prognosis |
| Tantai et al. (2015) [74] | 64 NSCLC tissues/matched adjacent non-tumor patient tissues; stage: I (15), II/III (17) | Combination of XIST and HIF1A-AS1 had a higher positive diagnostic efficiency of NSCLC patient screening |
| Gong et al. (2016) [75] | 498 lung cancer patients (467 patients at least two cycles of platinum-based chemotherapy); 213 healthy controls | HOTTIP, CCAT2, H19, HOTAIR, MALAT1 and ANRIL potential clinical biomarkers to predict lung cancer risk and platinum-based chemotherapy response |
| Yuan et al. (2016) [76] | Meta-analysis of eight published GWAS datasets with 17,153 cases and 239,337 controls | SNP rs114020893 of NEXN-AS1 at 1p31.1 may contribute to lung cancer susceptibility |
| Yang et al. (2014) [6] | 5 NSCLC gene expression datasets from GEO: Training set: GSE27262, GSE19804, GSE19188 and GSE30219 Test set: GSE18842 | 47 lncRNAs differentially expressed in NSCLC; 19 lncRNAs differed in expression between SCC and AC |
| White et al. (2014) [7] | Three lung RNA-Seq datasets: 72 AC/adjacent normal pairs 55 AC/adjacent normal pairs + 243 unmatched tumors from TCGA 34 SQ/adjacent normal pairs + 163 unmatched tumors from TCGA | 463 and 315 up- and down-regulated lncRNA in AC tumors relative to SQ; 27 lncRNAs differentially expressed between AC and SQ |
| Zhang et al. (2015) [2] | AC and SQ microarray | 1646 differentially expressed lncRNA LINC01133 showed the largest up-regulation in SQ but not in AC |
| Wei et al. (2016) [1] | Paired tissue samples of RNA sequencing or microarray data from TCGA and GEO | lncRNA expression is different in AC and SQ knockdown of the up-regulated lncRNA AFAP1-AS1 and LINC00511 impaired AC cell proliferation; knockdown of PVT1 inhibited SQ cell growth four 6-lncRNAs signature expression patterns were found to be significantly associated with AC and SQ patient overall and progression-free survival |
| Tool (Ref.) | Purpose | Website |
|---|---|---|
| miRBase [109] | miRNA database | http://www.mirbase.org/ |
| MiR2Disease [110] | Interactions/pathways | http://www.mir2disease.org/ |
| TargetScan [111] | Target prediction | http://www.targetscan.org/vert_71/ |
| PicTar [112] | Target prediction | http://www.pictar.org/ |
| PITA [113] | Target prediction | https://genie.weizmann.ac.il/pubs/mir07/mir07_data.html |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kunz, M.; Wolf, B.; Schulze, H.; Atlan, D.; Walles, T.; Walles, H.; Dandekar, T. Non-Coding RNAs in Lung Cancer: Contribution of Bioinformatics Analysis to the Development of Non-Invasive Diagnostic Tools. Genes 2017, 8, 8. https://doi.org/10.3390/genes8010008
Kunz M, Wolf B, Schulze H, Atlan D, Walles T, Walles H, Dandekar T. Non-Coding RNAs in Lung Cancer: Contribution of Bioinformatics Analysis to the Development of Non-Invasive Diagnostic Tools. Genes. 2017; 8(1):8. https://doi.org/10.3390/genes8010008
Chicago/Turabian StyleKunz, Meik, Beat Wolf, Harald Schulze, David Atlan, Thorsten Walles, Heike Walles, and Thomas Dandekar. 2017. "Non-Coding RNAs in Lung Cancer: Contribution of Bioinformatics Analysis to the Development of Non-Invasive Diagnostic Tools" Genes 8, no. 1: 8. https://doi.org/10.3390/genes8010008